

Abstract
Because obesity leads to a state of chronic, low-grade inflammation and oxidative stress, we hypothesized that the contribution of genes to variation in a biomarker of these two processes may be influenced by the degree of adiposity. We tested this hypothesis using samples from the San Antonio Family Heart Study that were assayed for activity of lipoprotein-associated phospholipase A2 (Lp-PLA2), a marker of inflammation and oxidative stress. Using an approach to model discrete genotype×environment (G×E) interaction, we assigned individuals to one of two discrete diagnostic states (or “adiposity environments”): nonobese or obese, according to criteria suggested by the World Health Organization. We found a genomewide maximum LOD of 3.39 at 153 cM on chromosome 1 for Lp-PLA2. Significant G×E interaction for Lp-PLA2 at the genomewide maximum (P=1.16×10-4) was also found. Microarray gene-expression data were analyzed within the 1-LOD interval of the linkage signal on chromosome 1. We found two transcripts—namely, for Fc gamma receptor IIA and heat-shock protein (70 kDa)—that were significantly associated with Lp-PLA2 (P<.001 for both) and showed evidence of cis-regulation with nominal LOD scores of 2.75 and 13.82, respectively. It would seem that there is a significant genetic response to the adiposity environment in this marker of inflammation and oxidative stress. Additionally, we conclude that G×E interaction analyses can improve our ability to identify and localize quantitative-trait loci.
From studies of humans and animal models, accumulating evidence has suggested a positive association between measures of adiposity and biomarkers of inflammation and oxidative stress.1–11 At the molecular level, this association seems to arise from the increased expression of adipokines in the white adipose tissue1,2 of central adipose depots.3–5 As a result of these advances, a unifying theory on the etiology of the metabolic syndrome posits that obesity leads to a state of chronic, low-grade inflammation and oxidative stress and that it is this pathological condition—of chronic inflammation and oxidative stress—that underlies most of the clinical sequelae associated with the metabolic syndrome.12–27
On the basis of these findings, we hypothesized that the contribution of genes to variation in biomarkers of inflammation and oxidative stress may be influenced by the degree of adiposity—that is, “adiposity environment”—in individuals in whom they are expressed. We sought to test this hypothesis in the San Antonio Family Heart Study (SAFHS), which is a study of the genetic determinants of cardiovascular disease (CVD) in Mexican American families of San Antonio. We used the modeling approach of discrete genotype×environment (G×E) interaction, where two discrete adiposity environments—obese and nonobese—were defined according to criteria suggested by the World Health Organization (WHO).28
The SAFHS population comprises large Mexican American extended families randomly ascertained with respect to CVD.29 The SAFHS protocols were approved by the Institutional Review Board at the University of Texas Health Science Center at San Antonio, and all study participants provided written informed consent. The pedigree relationships exhibited by the sample population are reported in table 1.
Table 1. .
Pedigree Relationship Types in the SAFHS
| Relationship | No. of Observed Pairs |
| Parent-offspring | 2,550 |
| Full siblings | 1,780 |
| Half siblings | 260 |
| Grandparent-grandchild | 2,234 |
| Avuncular | 3,583 |
| Half avuncular | 498 |
| First cousins | 3,365 |
Fasting blood samples were obtained from study participants at a clinic exam and were shipped the same day to Southwest Foundation for Biomedical Research (SFBR), San Antonio. Plasma and serum were isolated by low-speed centrifugation, and the buffy coat was harvested for DNA extraction.
We analyzed a biomarker of inflammation and oxidative stress in atherogenesis—namely, plasma activity of lipoprotein-associated phospholipase A2 (Lp-PLA2), which is also known as “platelet-activating factor acetylhydrolase” (PAF-AH).30–32 Plasma Lp-PLA2 activity was determined using a commercial colorimetric assay (Cayman Chemical) with 2-thio-PAF as substrate and according to the manufacturer’s directions. Samples were run in duplicate, with average coefficients of variation of 2.5%. Enzyme activity was expressed in units of μmol/min/ml. Residuals from a least-squares multiple linear regression—with use of age, sex, age squared, oral-contraceptive use, and menopause status as independent variables—were exactly normalized using an inverse Gaussian transformation in SOLAR (hereafter referred to as “rnLp-PLA2”).33 Anthropometrics, including height, weight, and waist and hip circumferences, were measured at a clinic exam as part of the SAFHS protocol. BMI, defined as the ratio of weight (kg) to height squared (m2), and waist-to-hip ratio (WHR), defined as the ratio of waist circumference to hip circumference, were determined from the anthropometric data.
It is by now well established that abdominal obesity is a major metabolic-syndrome risk factor.19,22,24,27 In an attempt to incorporate the abdominal-obesity component into a clinical definition of the metabolic syndrome, a WHO expert committee defined the abdominally obese as those with a combined BMI >30 kg×m-2 and a WHR >0.90 in men and >0.85 in women.28 We used these cutoffs to define a dichotomous adiposity environment, as given by the indicator variable
 |
The sample size of individuals with data for rnLp-PLA2, BMI, and WHR combined is 1,341.
DNA extracted from lymphocytes was used in PCRs for the amplification of individual DNA (N=1,339) at 432 dinucleotide-repeat microsatellite loci (i.e., STRs), spaced ∼10 cM apart across the 22 autosomes, with fluorescently labeled primers from the MapPairs Human Screening set, versions 6 and 8 (Research Genetics). PCRs were performed separately, according to manufacturer specifications, in Applied Biosystems 9700 thermocyclers. The products of separate PCRs, for each individual, were pooled using the Robbins Hydra-96 Microdispenser, and a labeled size standard was added to each pool. The pooled PCR products were loaded into an ABI PRISM 377 or 3100 Genetic Analyzer for laser-based automated genotyping. The STRs and standards were detected and quantified, and genotypes were scored using the Genotyper software package (Applied Biosystems).
Mistyping analyses were performed on the preliminary genotype-marker data with SimWalk2, following the recommendations of the program developers for accounting for mistyping error, by (1) blanking the errant called alleles, (2) recalling them conditional on the analysis (i.e., reassigning them under a different allele designation), or (3) retyping the mistyped marker or markers as resources permitted.34,35 Our overall rate of blanking mistyped markers was 1.37%. These mistyping analyses allow investigators to account for Mendelian errors and spurious double recombinants, both of which can severely reduce the power of a linkage analysis if not accounted for.35 After addressing mistyping errors (by blanking, recalling, or retyping), these genotype data were then used to compute maximum-likelihood estimates of allele frequencies in SOLAR.33 Empirical estimates of identity-by-descent (IBD) allele sharing at points throughout the genome for every relative pair were computed using the Loki package, which uses Markov chain–Monte Carlo methods.36 The multipoint IBD estimates are required under our variance-components modeling approach (see below). The SimWalk2 and Loki programs both require chromosomal maps. We used the set of high-resolution chromosomal maps provided by the research group at deCODE genetics, which are included in Web table E in the work of Kong et al.37
Total RNA was extracted from 1,000 lymphocyte samples by use of QIAGEN RNeasy 96 kits, and concentration was determined spectrophotometrically by use of a NanoDrop. Integrity of resuspended total RNA was determined by electrophoretic separation and subsequent laser-induced florescence detection by use of the RNA 6000 Nano Assay Chip Kit on the Bioanalyzer 2100 with the 2100 Expert software (Agilent Technologies). Antisense RNA (aRNA) was synthesized and purified using the Ambion MessageAmp II Amplification Kit, following the Illumina Sentrix Array Matrix 96-well expression protocol. Biotin-16-UTP (Roche)–labeled aRNA was hybridized to Illumina Sentrix Human Whole Genome (WG-6) Expression BeadChips. These BeadChips contain six arrays, each with 47,289 probes derived from human genes in the National Center for Bioinformatics Information (NCBI) Reference Sequence and UniGene databases. This system uses a “direct hybridization” assay, whereby gene-specific probes are used to detect labeled RNAs. Each bead in the array contains a 50-mer, sequence-specific oligonucleotide probe synthesized using Illumina’s Oligator in-house technology. Each array on a Human WG-6 BeadChip provides genomewide transcriptional coverage of well-characterized genes, gene candidates, and splice variants. The Human WG-6 Expression BeadChips were scanned on the Illumina BeadArray 500GX Reader, a two-channel, 0.8-μm–resolution confocal laser scanner, by use of Illumina BeadScan image data acquisition software (ver. 2.3.0.13). Illumina BeadStudio software (ver. 1.5.0.34) was used for data visualization and quality-control metrics.
To preclude confusion, we note that these data are expression levels of RNA transcripts and are not genotypic data. They are treated herein as phenotypic data. It should also be noted that lymphocytes may not fully reflect the expression of all genes influencing adiposity, oxidative stress, and inflammation. However, there is growing use of lymphocytes as surrogate models for other tissues, such as neural tissues,38–40 and such work has generated substantial new discoveries.40
The hypothesis of differential response to two environments is an example of the application of the theory of discrete G×E interaction.41 There is now a good number of published reports on the utility of this approach, both for understanding the relationship between genotype and environment in the process of phenotype determination and to aid in the identification and localization of QTLs.42–56 Under the theory of discrete G×E interaction, significant interaction arises for heterogeneity in the additive genetic variance (polygenic or QTL), an additive genetic correlation coefficient (polygenic or QTL) significantly different from unity, or both conditions.41 Therefore, to test for G×E interaction, we sought to falsify the null versions of the conditions that give rise to G×E interaction, which are a homogeneous additive genetic variance (polygenic or QTL) and/or an additive genetic correlation coefficient (polygenic or QTL) equal to 1.
For all possible pairwise combinations of values for the adiposity indicator variables fx and fz, where x and z are index individuals in the sample, the G×adiposity environment (G×A) interaction model covers three types of pairwise comparisons: within-obese, within-nonobese, and across-adiposity-environment comparisons:
 |
where y is any given phenotype; 2φxz gives the expected coefficient of relationship,
 |
where the κij are coefficients giving the jth locus-specific probability that a pair of relatives share i alleles IBD; 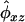 is the estimated kinship coefficient based on marker data; δxz is defined as 1 when individuals x and z are the same and 0 otherwise; σ2go, σ2gn, σ2qo, σ2qn, σ2eo, and σ2en are, respectively, the within-obese and within-nonobese additive polygenic, QTL, and environmental variances (the positive square roots of which give their corresponding SDs); and
is the estimated kinship coefficient based on marker data; δxz is defined as 1 when individuals x and z are the same and 0 otherwise; σ2go, σ2gn, σ2qo, σ2qn, σ2eo, and σ2en are, respectively, the within-obese and within-nonobese additive polygenic, QTL, and environmental variances (the positive square roots of which give their corresponding SDs); and 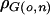 and
and 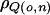 are the across-adiposity–environment additive polygenic and QTL correlation coefficients, respectively. We refer to this model as the “linkage interaction model.” It will be necessary at this point to define the polygenic interaction model as a constrained version of the linkage interaction model in which the following constraint holds: σ2qo=σ2qn=0.
are the across-adiposity–environment additive polygenic and QTL correlation coefficients, respectively. We refer to this model as the “linkage interaction model.” It will be necessary at this point to define the polygenic interaction model as a constrained version of the linkage interaction model in which the following constraint holds: σ2qo=σ2qn=0.
The top and middle cases on the right side of the previous equation are the within-adiposity–environment versions of the standard linkage model used by Almasy and Blangero,33 which hold for the obese and nonobese environments, respectively. The crucial part of the model is given by the bottommost case, which gives the covariance for the across-adiposity–environment comparison. Note that we are allowing for the possibility of heterogeneity in the residual environmental variance. This is necessary to preclude bias in detection of heterogeneity in the genetic-variance components. In all of our models, 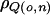 was constrained to equal 1, because the contribution to the model made by
was constrained to equal 1, because the contribution to the model made by  tends to be offset by the increase in degrees of freedom relative to the standard linkage model (results not shown).
tends to be offset by the increase in degrees of freedom relative to the standard linkage model (results not shown).
We used SOLAR to perform genome screens under standard linkage and G×A interaction models across all 22 autosomes. For the standard linkage case, the likelihood ratio statistic, denoted by Λ, is distributed as 1/2χ20+ 1/2χ21.57 It is important to note that SOLAR automatically corrects the LOD score for the standard case, to account for the above mixture distribution. The observed LOD scores under the G×A interaction model need to be further corrected because of the increase in degrees of freedom relative to the standard linkage model. Following Self and Liang,57 it can be shown that when the polygenic interaction and linkage interaction models are compared, Λ is distributed as 1/4χ20+ 1/2χ21+ 1/4χ22. We refer to this latter correction for increase in degrees of freedom as the corrected LOD score. Since the LOD score is equal to 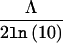 , we can obtain a corrected LOD score on the basis of the appropriate distribution. To test the null hypothesis of homogeneity in the QTL variance (i.e., σ2qo=σ2qn) at the genomewide maximum (the point along the genome that has the highest LOD score), we performed likelihood-ratio tests. For model comparisons in which the QTL variances are constrained to be equal under the null hypothesis and in which the QTL variances are free to vary under the alternative hypothesis, Λ is distributed as χ21.
, we can obtain a corrected LOD score on the basis of the appropriate distribution. To test the null hypothesis of homogeneity in the QTL variance (i.e., σ2qo=σ2qn) at the genomewide maximum (the point along the genome that has the highest LOD score), we performed likelihood-ratio tests. For model comparisons in which the QTL variances are constrained to be equal under the null hypothesis and in which the QTL variances are free to vary under the alternative hypothesis, Λ is distributed as χ21.
We found that rnLp-PLA2 has a heritability of 0.55 (P=2.07×10-41). Using the G×A interaction model, we found a corrected, genomewide maximum LOD of 3.39 at 153 cM on chromosome 1 near marker D1S1595 (figs. 1 and 2). A LOD score of 3.0 is taken as indicative of genomewide significance.58 Moreover, using an approach based on the work of Feingold et al.59 and implemented in Gauss 6.0.17 (Aptech Systems), we can compute the genomewide P value that corresponds to our LOD score of 3.39. This approach takes into account the finite marker density in the linkage map used in the multipoint QTL screens and the mean recombination rate for the pedigreed population studied. The genomewide P value computed under said approach is .01477. In contrast, maximization of models lacking G×A interaction did not provide strong evidence of a QTL anywhere in the genome. There is, however, a suggestive LOD score of 2.46 at 140 cM on chromosome 1 under the standard linkage model, with a corresponding genomewide P value of .14402. To highlight the improvement given by the incorporation of interaction effects, the results under the standard linkage model for chromosome 1 are also displayed. It will be noted that the location of the maximum LOD score changes from 140 cM on chromosome 1 under the standard linkage model to 153 cM on chromosome 1 under the G×A interaction model. One plausible explanation is that the shift in peaks is simply the result of a model that affords a more precise signal location. In keeping with this view, Blangero et al. showed that G×E interaction models increase the power to detect linkage signals and precision of signal location.60 Another plausible explanation is that there is another gene located at the linkage peak under the standard linkage model and that the G×A interaction model recovers information that, in the aggregate, points to another location, harboring another gene, as the maximum while at the same time retaining the peak first observed under the standard linkage model. We cannot distinguish between these alternatives at the present time.
Figure 1. .

Lp-PLA2 linkage on chromosome 1. Comparison of the G×A interaction model under the WHO definition of adiposity status with the standard linkage model. The solid line indicates LOD plot under the G×A interaction model and the dashed line indicates LOD plot under the standard linkage model.
Figure 2. .

Multipoint genome scan of Lp-PLA2
We found significant QTL G×A interaction for rnLp-PLA2 at the genomewide maximum on chromosome 1 (P=2.32×10-4) (fig. 3). Specifically, the QTL additive genetic variance decreased from the nonobese to the obese environment. This finding may indicate a gene that is negatively regulated by adipose tissue. For instance, adiponectin is negatively associated with obesity,61,62 and a regulatory protein that both is related to the adiponectin gene and is itself negatively regulated by the proinflammatory cytokine, tumor necrosis factor-α (TNF-α), has been shown to be located in a region encompassing our linkage signal.63–65 Additionally, the gene for the adiponectin receptor AdipoR1 has been mapped to a broad region on chromosome 1 that encompasses our linkage signal.66 Interestingly, the polygenic additive genetic variance exhibited significant heterogeneity (i.e., polygenic G×A interaction) (P=3.54×10-4) (fig. 3) and increased from the nonobese to the obese environment. This observation is consistent with the knowledge that adiposity promotes the expression of proinflammatory cytokines, such as TNF-α.12–27 Our findings of decreasing QTL variance and increasing polygenic variance in response to the adiposity environment may be reflective of down-regulated and up-regulated signals related to the inflammation response. The additive environmental variance also significantly decreased from the nonobese to the obese environment (P=6.86×10-4) (fig. 3). This observation is consistent with the way the determinative system affecting Lp-PLA2 comes under relatively more genetic control when going from the nonobese to obese environment.
Figure 3. .

G×A interaction effects. The solid line with unblackened diamonds indicates genetic SD (gsd), the solid line with blackened circles indicates QTL SD (qsd), and the dashed line with blackened triangles indicates environmental SD (esd).
To our knowledge, there are at least three other genome-scan studies that have reported signals on chromosome 1 in the vicinity of our signals for traits related to obesity and/or the metabolic syndrome.67–69 Ng et al.,67 in the Hong Kong Family Diabetes Study, reported their genomewide maximum LOD of 4.5 at chromosome 1q near marker D1S1653 for the metabolic syndrome. Marker D1S1653 is located at 151.68 cM on the deCODE map and is very near our genomewide maximum for the G×A interaction analyses. In the Framingham Heart Study,68 Dupuis et al. reported a LOD of 3.86 at chromosome 1q near marker D1S1679 for monocyte-chemoattractant protein-1 (MCP-1). Marker D1S1679 is positioned at 160 cM in our chromosome 1 map, which is just at the outer margin of the 1-LOD interval in our G×A interaction analyses. Moreover, similar to Lp-PLA2, MCP-1 is another biomarker of vascular inflammation. In a study of nuclear families ascertained at the University of Pennsylvania,69 Reed et al. reported a LOD equivalent of 2.2 on chromosome 1 near marker D1S484 for plasma cholesterol levels. Marker D1S484 is at 157.51 cM on the deCODE map, which again is within the 1-LOD interval in our G×A interaction analyses.
We analyzed the gene-expression data to further characterize the area under the linkage peak. Because there are exactly 341 transcripts within the 1-LOD interval around the maximum, we employed a statistical methodology, based on the false-discovery rate (FDR) and Q Value concepts, to deal with the multiple-testing problem.70–74 The literature on the detection of differential gene expression in DNA microarray data—and similar such cases of data mining in statistical genetics and genomics—seems to point to methods that control the FDR rather than to the highly conservative method of controlling for the familywise error rate, such as the well-known Bonferroni correction.70–81 The Q value is mathematically defined as the minimum positive FDR (pFDR) observed for a set of significant results: Q value=min{pFDR}. It is a measure of the proportion of false-positive results expected on declaring a particular test to be significant at a given significance level, denoted by α. Following the recommendations of the developers of this method for preliminary investigations, we can threshold on the Q value, such that we obtain an FDR⩽α.
In linear models in which the transcript is the dependent variable and rnLp-PLA2 is the covariate, we can use the P value on the beta-coefficient for rnLp-PLA2 as a measure of the association of the transcript with rnLp-PLA2. By the Q Value method, we observed 10 transcripts under the Q value threshold for FDR⩽α⩽.05 (table 2 and fig. 4). To test for cis-regulation, which is defined as regulatory elements located at the gene,82–84 we computed the pointwise LOD score at the location reported in the NCBI and UniGene databases for each of the 341 transcripts within the 1-LOD interval of the rnLp-PLA2 linkage signal. We take as our significance level a LOD score of 1.44, which is equivalent to α⩽.01. Using the Q Value method for P values computed from the LOD scores, and thresholding for FDR⩽α⩽.01, we observed seven transcripts (table 3 and fig. 4). Two of these—namely, the gene encoding Fc gamma receptor IIA (FCGR2A [MIM 146790]) (LOD 2.75) and the gene encoding heat-shock protein (70 kDa) (HSP70) (LOD 13.82) (HSP70=HSPA6 [MIM 140555])—are presented in tables 2 and 3. We interpret these results to mean that these two genes—namely, FCGR2A and HSP70—are good candidates to pursue for further study.
Table 2. .
Transcripts under the 1-LOD Interval of the Linkage Signal on Chromosome 1 that are Significantly Associated with Lp-PLA2
| Symbol | Name | Function | Locationa (cM) |
Pb | Q |
| CTMP | C-terminal modulator protein | Protein kinase B regulation | 147.18 | .00174 | .04653 |
| CTSS | Cathepsin S | Elastase activity | 147.64 | 5.15×10-4 | .01738 |
| SNX27 | Sorting nexin, family member 27 | Intracellular sorting | 148.52 | 4.81×10-4 | .01738 |
| FLJ23221 | Chromosome 1 ORF 54 | ORF | 148.78 | .00164 | .04653 |
| PBXIP1 | Pre-B-cell leukemia transcription factor interacting protein 1 | Transcriptional regulation | 151.85 | 2.77×10-4 | .01738 |
| SYT11 | Synaptotagmin, isoform 11 | Mast-cell regulation | 152.77 | 1.30×10-4 | .01738 |
| FCER1A | Fc epsilon receptor 1A | Mast-cell activation | 156.21 | 3.03×10-4 | .01738 |
| Hmm8932 | Hmm8932 | Gnomon predicted gene | 157.95 | 5.21×10-4 | .01738 |
| FCGR2A | Fc gamma receptor 2A | C-reactive–protein receptor | 158.42 | 1.52×10-4 | .01738 |
| HSPA6=HSP70 | Heat-shock protein (70 kDa) | Chaperone-protein folding | 158.44 | 4.18×10-4 | .01738 |
Locations are averaged interpolations against our map, with use of physical distances obtained from the University of California–Santa Cruz (UCSC) genome browser (Human [Homo sapiens] Genome Browser Gateway) for the two markers flanking the linkage peak at 153 cM.
P value of the beta coefficient for Lp-PLA2 in a linear model in which the transcript is the dependent variable.
Figure 4. .

Transcripts under the linkage signal; transcripts within the 1-LOD interval of the linkage peak at 153 cM. Top, Negative logarithm (base 10) of the Q values for the test of association between the transcript and Lp-PLA2 plotted against chromosomal location. The dashed line corresponds to the Q value threshold of .05 for the beta coefficients. Bottom, Negative logarithm (base 10) of the Q values computed from the pointwise LOD scores plotted against chromosomal location. The grid line at 2.0 corresponds to the Q value threshold of .01 for the pointwise LOD scores. Location is expressed in terms of megabases along the ordinate, for better separation. To plot all values on a scale at which the Q value threshold could be easily discerned, the three highest −log Q values for MGC31963, HSP70, and ATF6 were arbitrarily given a −log Q value of 3.0. Their real corresponding Q values are in table 3 (HSPA6=HSP70).
Table 3. .
Transcripts under the 1-LOD Interval of the Linkage Signal on Chromosome 1 that Show cis-Regulation
| Symbol | Name | Function | Locationa (cM) |
Pb | Q |
| ARNT | Aryl hydrocarbon receptor nuclear translocator | Xenobiotic metabolism | 147.72 | 8.46×10-5 | .00421 |
| MGC31963 | Chromosome 1 ORF 85 | ORF | 153.20 | 4.75×10-19 | 0 |
| FCRH3 | Fc receptor–like protein 3 | Immunoglobulin receptor | 154.59 | 2.98×10-5 | .00178 |
| FCGR2A | Fc gamma receptor 2A | C-reactive–protein receptor | 158.42 | .00019 | .00693 |
| HSPA6=HSP70 | Heat-shock protein (70 kDa) | Chaperone-protein folding | 158.44 | 7.52×10-16 | 0 |
| FCGR2B | Fc fragment of immunoglobulin | Mast-cell activation | 158.49 | 2.23×10-5 | .00167 |
| ATF6 | Activating transcription factor 6 | Transcription factor | 158.60 | 3.41×10-11 | 0 |
Locations are averaged interpolations against our map, with use of physical distances obtained from the UCSC genome browser (Human [Homo sapiens] Genome Browser Gateway) for the two markers flanking the linkage peak at 153 cM.
P values are computed from the nominal LOD score at the location given in the NCBI and UniGene databases.
FCGR2A is the receptor for C-reactive protein (CRP), a well-known acute-phase protein of the inflammation process involved in vascular dysfunction.85–87 Immunocytochemical work has shown that FCGR2A is highly expressed in the proliferative zones of atherosclerotic lesions.88 Moreover, it has been shown in human monocytes that have the H131 mutation at the FCGR2A gene that FCGR2A has a significantly decreased binding to CRP87 and that this mutation seems to confer protection against peripheral atherosclerosis.89 The increased expression of heat-shock proteins, including HSP70, is known to be associated with an induced inflammatory response.90–93 Consistent with this knowledge, it has been noted that HSP70 is overexpressed in several cell types, including monocytes, macrophages, and smooth-muscle cells, in advanced atherosclerotic lesions.93
It should be noted that the beta-coefficients indicating the relationship of the transcript to rnLp-PLA2 were negative in sign for both transcripts (for FCGR2A, β=-0.13; for HSP70, β=-0.12). This is consistent with our observation of a decreasing QTL variance in rnLp-PLA2 from the nonobese to the obese-adiposity environment. To further characterize these transcripts, we performed multiple linear-regression analyses of the transcript expression levels as the independent variable and of age, sex, their second-order terms, and a relevant clinical trait as the dependent predictors. The analyzed clinical traits were diabetes status, impaired glucose tolerance, insulin resistance, adiposity status as defined above, dyslipidemia status, hypertension status, history of heart attack, history of heart surgery, and common and internal carotid artery intima-media thicknesses. For definitions of diabetes status, impaired glucose tolerance, insulin resistance, dyslipidemia status, and hypertension status, we again referred to the criteria suggested by the WHO.28 There were significant associations between FCGR2A-expression levels and adiposity status (P=.01636), insulin resistance (P=.00255), and dyslipidemia status (P=1.3×10-5). There was one clearly significant association between HSP70-expression levels and dyslipidemia status (P=.02578). The association between HSP70-expression levels and insulin resistance was barely nonsignificant (P=.05613). All other associations for both transcripts were nonsignificant. Taken together, the linkage and gene expression analyses indicate that we have identified a gene that is involved in the inflammation process and is negatively regulated by the adiposity environment.
It is notable that, more than a decade ago, Després and colleagues proposed a hypothesis similar to the one addressed herein.94–96 It was suggested that visceral adiposity was capable of modulating the genetic susceptibility to coronary heart disease. Our results are supportive of that original hypothesis. In particular, we conclude that there is a significant polygenic and QTL genetic response to the adiposity environment in Lp-PLA2, which is an important biomarker of inflammation and oxidative stress; that G×E interaction analyses can improve our ability to identify and localize QTLs; and that there are at least two strong candidate genes underlying the QTL that we identified. Identification of genes and their variants involved in the inflammatory and oxidative processes in the metabolic syndrome is of high significance not only for the understanding of its metabolic pathogenesis but also for the development of therapeutic strategies to reduce the mortality and morbidity of this 21st-century epidemic.
Acknowledgments
We thank the Mexican American families of San Antonio who participated in the SAFHS. This research was funded by National Institutes of Health (NIH) grants P01 HL45522 and MH 59490 and was conducted in facilities constructed with support from NIH Research Facilities Improvement Program grants C06 RR013556 and C06 RR017515 and from SBC Communications (now AT&T).
Web Resources
The URLs for data presented herein are as follows:
- Human (Homo sapiens) Genome Browser Gateway, http://genome.ucsc.edu/cgi-bin/hgGateway (for physical distances)
- NCBI, http://www.ncbi.nlm.nih.gov/ (for the physical location of the transcripts)
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for FCGR2A and HSPA6)
- Q Value, http://faculty.washington.edu/~jstorey/qvalue/ (for a free software download)
- SOLAR, http://www.sfbr.org/solar/index.html (for a free software download)
References
- 1.Hotamisligil GS, Shargill NS, Spiegelman BM (1993) Adipose expression of tumor necrosis factor-α: direct role in obesity-linked insulin resistance. Science 259:87–91 10.1126/science.7678183 [DOI] [PubMed] [Google Scholar]
- 2.Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, Nakayama O, Makishima M, Matsuda M, Shimomura I (2004) Increased oxidative stress in obesity and its impact on the metabolic syndrome. J Clin Invest 114:1752–1761 10.1172/JCI200421625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Couillard C, Ruel G, Archer WR, Pomerleau S, Bergeron J, Couture P, Lamarche B, Bergeron N (2005) Circulating levels of oxidative stress markers and endothelial adhesion molecules in men with abdominal obesity. J Clin Endocrinol Metab 90:6454–6459 10.1210/jc.2004-2438 [DOI] [PubMed] [Google Scholar]
- 4.Panagiotakos DB, Pitsavos C, Yannakoulia M, Chrysohoou C, Stefanadis C (2005) The implication of obesity and central fat on markers of chronic inflammation: the ATTICA study. Atherosclerosis 183:308–315 10.1016/j.atherosclerosis.2005.03.010 [DOI] [PubMed] [Google Scholar]
- 5.Pihl E, Zilmer K, Kullisaar T, Kairane C, Mägi A, Zilmer M (2006) Atherogenic inflammatory and oxidative stress markers in relation to overweight values in male former athletes. Int J Obes 30:141–146 10.1038/sj.ijo.0803068 [DOI] [PubMed] [Google Scholar]
- 6.Dandona P, Weinstock R, Thusu K, Abdel-Rahman E, Aljada A, Wadden T (1998) Tumor necrosis factor-α in sera of obese patients: fall with weight loss. J Clin Endocrinol Metab 83:2907–2910 10.1210/jc.83.8.2907 [DOI] [PubMed] [Google Scholar]
- 7.Yudkin JS, Stehouwer CDA, Emeis JJ, Coppack SW (1999) C-reactive protein in healthy subjects: associations with obesity, insulin resistance, and endothelial dysfunction: a potential role for cytokines originating from adipose tissue? Arterioscler Thromb Vasc Biol 19:972–978 [DOI] [PubMed] [Google Scholar]
- 8.Kern PA, Ranganathan S, Li C, Wood L, Ranganathan G (2001) Adipose tissue tumor necrosis factor and interleukin-6 expression in human obesity and insulin resistance. Am J Physiol Endocrinol Metab 280:E745–E751 [DOI] [PubMed] [Google Scholar]
- 9.Vozarova B, Weyer C, Hanson K, Tataranni PA, Bogardus C, Pratley RE (2001) Circulating interleukin-6 in relation to adiposity, insulin action, and insulin secretion. Obes Res 9:414–417 [DOI] [PubMed] [Google Scholar]
- 10.Suzuki K, Ito Y, Ochiai J, Kusuhara Y, Hashimoto S, Tokudome S, Kojima M, Wakai K, Toyoshima H, Tamakoshi K, et al (2003) Relationship between obesity and serum markers of oxidative stress and inflammation in Japanese. Asian Pac J Cancer Prev 4:259–266 [PubMed] [Google Scholar]
- 11.Dandona P, Aljada A, Ghanim H, Mohanty P, Tripathy C, Hofmeyer D, Chaudhuri A (2004) Increased plasma concentration of macrophage migration inhibitory factor (MIF) and MIF mRNA in mononuclear cells in the obese and the suppressive action of metformin. J Clin Endocrinol Metab 89:5043–5047 10.1210/jc.2004-0436 [DOI] [PubMed] [Google Scholar]
- 12.Caballero AE (2003) Endothelial dysfunction in obesity and insulin resistance: a road to diabetes and heart disease. Obes Res 11:1278–1289 [DOI] [PubMed] [Google Scholar]
- 13.Lyon CJ, Law RE, Hsueh WA (2003) Minireview: adiposity, inflammation, and atherogenesis. Endocrinology 144:2195–2200 10.1210/en.2003-0285 [DOI] [PubMed] [Google Scholar]
- 14.Rajala MW, Scherer PE (2003) Minireview: the adipocyte—at the crossroads of energy homeostasis, inflammation, and atherosclerosis. Endocrinology 144:3765–3773 10.1210/en.2003-0580 [DOI] [PubMed] [Google Scholar]
- 15.Yudkin JS (2003) Adipose tissue, insulin action and vascular disease: inflammatory signals. Int J Obes Relat Metab Disord 27:S25–S28 10.1038/sj.ijo.0802496 [DOI] [PubMed] [Google Scholar]
- 16.Dandona P, Aljada A, Bandyopadhyay A (2004) Inflammation: the link between insulin resistance, obesity and diabetes. Trends Immunol 25:4–7 10.1016/j.it.2003.10.013 [DOI] [PubMed] [Google Scholar]
- 17.Ferroni P, Basili S, Falco A, Davì G (2004) Inflammation, insulin resistance, and obesity. Curr Atheroscler Rep 6:424–431 [DOI] [PubMed] [Google Scholar]
- 18.Trayhurn P, Wood IS (2004) Adipokines: inflammation and the pleiotropic role of white adipose tissue. Br J Nutr 92:347–355 10.1079/BJN20041213 [DOI] [PubMed] [Google Scholar]
- 19.Vega GL (2004) Obesity and the metabolic syndrome. Minerva Endocrinologica 29:47–54 [PubMed] [Google Scholar]
- 20.Avogaro A, de Kreutzenberg SV (2005) Mechanisms of endothelial dysfunction in obesity. Clin Chim Acta 360:9–26 10.1016/j.cccn.2005.04.020 [DOI] [PubMed] [Google Scholar]
- 21.Berg AH, Scherer PE (2005) Adipose tissue, inflammation, and cardiovascular disease. Circ Res 96:939–949 10.1161/01.RES.0000163635.62927.34 [DOI] [PubMed] [Google Scholar]
- 22.Dandona P, Aljada A, Chaudhuri A, Mohanty P, Garg R (2005) Metabolic syndrome: a comprehensive perspective based on interactions between obesity, diabetes, and inflammation. Circulation 111:1448–1454 10.1161/01.CIR.0000158483.13093.9D [DOI] [PubMed] [Google Scholar]
- 23.Fantuzzi A (2005) Adipose tissue, adipokines, and inflammation. J Allergy Clin Immunol 115:911–919 10.1016/j.jaci.2005.02.023 [DOI] [PubMed] [Google Scholar]
- 24.Hutley L, Prins JB (2005) Fat as an endocrine organ: relationship to the metabolic syndrome. Am J Med Sci 330:280–289 10.1097/00000441-200512000-00005 [DOI] [PubMed] [Google Scholar]
- 25.Lau DCW, Dhillon B, Yan H, Szmitko PE, Verma S (2005) Adipokines: molecular links between obesity and atherosclerosis. Am J Physiol Heart Circ Physiol 288:H2031–H2041 10.1152/ajpheart.01058.2004 [DOI] [PubMed] [Google Scholar]
- 26.Vincent HK, Taylor AG (2006) Biomarkers and potential mechanisms of obesity-induced oxidant stress in humans. Int J Obes (Lond) 30:400–418 [DOI] [PubMed] [Google Scholar]
- 27.Després J-P (2006) Is visceral obesity the cause of the metabolic syndrome? Ann Med 38:52–63 10.1080/07853890500383895 [DOI] [PubMed] [Google Scholar]
- 28.WHO Consultation (1999) Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: diagnosis and classification of diabetes mellitus. World Health Organization, Department of Noncommunicable Disease Surveillance, Geneva [Google Scholar]
- 29.MacCluer JW, Stern MP, Almasy L, Atwood LA, Blangero J, Comuzzie AG, Dyke B, Haffner SM, Henkel RD, Hixson JE, et al (1999) Genetics of atherosclerosis risk factors in Mexican Americans. Nutr Rev 57:S59–S65 [DOI] [PubMed] [Google Scholar]
- 30.Tselepsis AD, Chapman MJ (2002) Inflammation, bioactive lipids and atherosclerosis: potential roles of a lipoprotein-associated phospholipase A2, platelet activating factor-acetylhydrolase. Atherosclerosis Suppl 3:57–68 10.1016/S1567-5688(02)00045-4 [DOI] [PubMed] [Google Scholar]
- 31.Chait A, Han CY, Oram JF, Heinecke JW (2005) Lipoprotein-associated inflammatory proteins: markers or mediators of cardiovascular disease? J Lipid Res 46:389–403 10.1194/jlr.R400017-JLR200 [DOI] [PubMed] [Google Scholar]
- 32.Zalewski A, Macphee C (2005) Role of lipoprotein-associated phospholipase A2 in atherosclerosis: biology, epidemiology, and possible therapeutic targets. Arterioscler Thromb Vasc Biol 25:923–931 10.1161/01.ATV.0000160551.21962.a7 [DOI] [PubMed] [Google Scholar]
- 33.Almasy L, Blangero J (1998) Multipoint quantitative-trait linkage analysis in general pedigrees. Am J Hum Genet 62:1198–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sobel E, Lange K (1996) Descent graphs in pedigree analysis: applications to haplotyping, location scores, and marker sharing statistics. Am J Hum Genet 58:1323–1337 [PMC free article] [PubMed] [Google Scholar]
- 35.Sobel E, Papp JC, Lange K (2002) Detection and integration of genotyping errors in statistical genetics. Am J Hum Genet 70:496–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heath SC (1997) Markov chain Monte Carlo segregation and linkage analysis for oligogenic models. Am J Hum Genet 61:748–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kong A, Gudbjartsson DF, Sainz J, Jonsdottir GM, Gudjonsson SA, Richardsson B, Sigurdardottir S, Barnard J, Hallbeck B, Masson G, et al (2002) A high-resolution recombination map of the human genome. Nat Genet 31:241–247 [DOI] [PubMed] [Google Scholar]
- 38.Gladkevich A, Kauffman HF, Korf J (2004) Lymphocytes as a neural probe: potential for studying psychiatric disorders. Prog Neuropsychopharmacol Biol Psychiatry 28:559–576 10.1016/j.pnpbp.2004.01.009 [DOI] [PubMed] [Google Scholar]
- 39.Tsuang MT, Nossoya N, Yager T, Tsuang M-M, Guo S-C, Shyu KG, Glatt SJ, Liew CC (2005) Assessing the validity of blood-based gene expression profiles for the classification of schizophrenia and bipolar disorder: a preliminary report. Am J Med Genet B Neuropsychiatr Genet 133:1–5 [DOI] [PubMed] [Google Scholar]
- 40.Borovecki F, Lovrecic L, Zhou J, Jeong H, Then F, Rosas HD, Hersch SM, Hogarth P, Bouzou B, Jensen RV, et al (2005) Genome-wide expression profiling of human blood reveals biomarkers for Huntington’s disease. Proc Nat Acad Sci USA 102:11023–11028 10.1073/pnas.0504921102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blangero J (1993) Statistical genetic approaches to human adaptability. Hum Biol 65:941–966 [PubMed] [Google Scholar]
- 42.Leips J, Mackay TFC (2000) Quantitative trait loci for life span in Drosophila melanogaster: interactions with genetic background and larval density. Genetics 155:1773–1788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Madrid GA, MacMurray J, Lee JW, Anderson BA, Comings DE (2001) Stress as a mediating factor in the association between the DRD2 TaqI polymorphism and alcoholism. Alcohol 23:117–122 10.1016/S0741-8329(00)00138-5 [DOI] [PubMed] [Google Scholar]
- 44.Orwoll ES, Belknap JK, Klein RF (2001) Gender specificity in the genetic determinants of peak bone mass. J Bone Miner Res 16:1962–1971 10.1359/jbmr.2001.16.11.1962 [DOI] [PubMed] [Google Scholar]
- 45.Wang XL, Rainwater DL, VandeBerg JF, Mitchell BD, Mahaney MC (2001) Genetic contributions to plasma total antioxidant activity. Arterioscler Thromb Vasc Biol 21:1190–1195 [DOI] [PubMed] [Google Scholar]
- 46.Dilda CL, Mackay TFC (2002) The genetic architecture of Drosophila sensory bristle number. Genetics 162:1655–1674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Klein RF, Turner RJ, Skinner LD, Vartanian KA, Serang M, Carlos AS, Shea M, Belknap JK, Orwoll ES (2002) Mapping quantitative trait loci that influence femoral cross-sectional area in mice. J Bone Miner Res 17:1752–1760 10.1359/jbmr.2002.17.10.1752 [DOI] [PubMed] [Google Scholar]
- 48.Leips J, Mackay TFC (2002) The complex genetic architecture of Drosophila life span. Exp Aging Res 28:361–390 10.1080/03610730290080399 [DOI] [PubMed] [Google Scholar]
- 49.Martin LJ, Mahaney MC, Almasy L, MacCluer JW, Blangero J, Jaquish CE, Comuzzie AG (2002) Leptin’s sexual dimorphism results from genotype by sex interactions mediated by testosterone. Obes Res 10:14–21 [DOI] [PubMed] [Google Scholar]
- 50.Martin LJ, Cole SA, Hixson JE, Mahaney MC, Czerwinski SA, Almasy L, Blangero J, Comuzzie AG (2002) Genotype by smoking interaction for leptin levels in the San Antonio Family Heart Study. Genet Epidemiol 22:105–115 10.1002/gepi.0135 [DOI] [PubMed] [Google Scholar]
- 51.Martin LJ, Kissebah AH, Sonnenberg GE, Blangero J, Comuzzie AG (2003) Genotype-by-smoking interaction for leptin levels in the Metabolic Risk Complications of Obesity Genes project. Int J Obes 27:334–340 10.1038/sj.ijo.0802232 [DOI] [PubMed] [Google Scholar]
- 52.Cole SA, Martin LJ, Peebles KW, Leland MM, Rice K, VandeBerg JL, Blangero J, Comuzzie AG (2003) Genetics of leptin expression in baboons. Int J Obes Relat Metab Disord 27:778–783 10.1038/sj.ijo.0802310 [DOI] [PubMed] [Google Scholar]
- 53.North KE, Martin LJ, Dyer T, Comuzzie AG, Williams JT (2003) HDL cholesterol in females in the Framingham Heart Study is linked to a region of chromosome 2q. BMC Genet 4:S98 10.1186/1471-2156-4-S1-S98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Czerwinski SA, Mahaney MC, Rainwater DL, VandeBerg JL, MacCluer JW, Stern MP, Blangero J (2004) Gene by smoking interaction: evidence for effects on low-density lipoprotein size and plasma levels of triglyceride and high-density lipoprotein cholesterol. Hum Biol 76:863–876 10.1353/hub.2005.0014 [DOI] [PubMed] [Google Scholar]
- 55.Hoffjan S, Nicolae D, Ostrovnaya I, Roberg K, Evans M, Mirel DB, Steiner L, Walker K, Shult P, Gangnon RE, et al (2005) Gene-environment interaction effects on the development of immune system responses in the 1st year of life. Am J Hum Genet 76:696–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lewis CE, North KE, Arnett D, Borecki IB, Coon H, Ellison RC, Hunt SC, Obermann A, Rich SS, Province MA, et al (2005) Sex-specific findings from a genome-wide linkage analysis of human fatness in non-Hispanic whites and African Americans: the HyperGEN Study. Int J Obes (Lond) 29:639–649 [DOI] [PubMed] [Google Scholar]
- 57.Self SG, Liang K-Y (1987) Asymptotic properties of maximum likelihood estimators and likelihood ratio tests under nonstandard conditions. J Am Stat Assoc 82:605–610 10.2307/2289471 [DOI] [Google Scholar]
- 58.Ott J (1999) Analysis of human genetic linkage, 3rd ed. Johns Hopkins University Press, Baltimore [Google Scholar]
- 59.Feingold E, Brown PO, Siegmund D(1993) Gaussian models for genetic linkage analysis using complete high-resolution maps of identity by descent.Am J Hum Genet 53:234–251 [PMC free article] [PubMed] [Google Scholar]
- 60.Blangero J, Williams JT, Almasy L (2000) Quantitative trait locus mapping using human pedigrees. Hum Biol 72:35–62 [PubMed] [Google Scholar]
- 61.Matsuzawa Y, Funahashi T, Kihara S, Shimomura I (2004) Adiponectin and metabolic syndrome. Arterioscler Thromb Vasc Biol 24:29–33 10.1161/01.ATV.0000099786.99623.EF [DOI] [PubMed] [Google Scholar]
- 62.Matsuzawa Y (2005) Adiponectin: identification, physiology and clinical relevance in metabolic and vascular disease. Atherosclerosis Suppl 6:7–14 10.1016/j.atherosclerosissup.2005.02.003 [DOI] [PubMed] [Google Scholar]
- 63.Schäffler A, Langman T, Palitzsch K-D, Schölmerich J, Schmitz G (1998) Identification and characterization of the human adipocyte apM-1 promoter. Biochim Biophys Acta 1399:187–197 [DOI] [PubMed] [Google Scholar]
- 64.Schäffler A, Orsó E, Palitzsch K-D, Büchler C, Drobnik W, Fürst A, Schölmerich J, Schmitz G (1999) The human apM-1, an adipocyte-specific gene linked to the family of TNF’s and to genes expressed in activated T cells, is mapped to chromosome 1q21.3-q23, a susceptibility locus identified for familial combined hyperlipidaemia (FCH). Biochem Biophys Res Comm 260:416–425 10.1006/bbrc.1999.0865 [DOI] [PubMed] [Google Scholar]
- 65.Barth N, Langmann T, Schölmerich J, Schmitz G, Schäffler A (2002) Identification of regulatory elements in the human adipose most abundant gene transcript-1 (apM-1) promoter: role of SP1/SP3 and TNF-α as regulatory pathways. Diabetologia 45:1425–1433 [DOI] [PubMed] [Google Scholar]
- 66.Yamauchi T, Kamon J, Ito Y, Tsuchida A, Yokomizo T, Kita S, Suglyama T, Miyagishi M, Hara K, Tsunoda M, et al (2003) Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature 423:762–769 (erratum 431:1123) 10.1038/nature01705 [DOI] [PubMed] [Google Scholar]
- 67.Ng CYM, So W-Y, Lam VKL, Cockram CS, Bell GI, Cox NJ, Chan JCN (2004) Genome-wide scan for metabolic syndrome and related quantitative traits in Hong Kong Chinese and confirmation of a susceptibility locus on chromosome 1q21-q25. Diabetes 53:2676–2683 [DOI] [PubMed] [Google Scholar]
- 68.Dupuis J, Larson MG, Vasan RS, Massaro JM, Wilson PWF, Lipinska I, Corey D, Vita JA, Keaney JF Jr, Benjamin EJ (2005) Genome scan of systemic biomarkers of vascular inflammation in the Framingham Heart Study: evidence for susceptibility loci on 1q. Atherosclerosis 182:307–314 10.1016/j.atherosclerosis.2005.02.015 [DOI] [PubMed] [Google Scholar]
- 69.Reed DR, Nanthakumar E, North M, Bell C, Price RA (2001) A genome-wide scan suggests a locus on chromosome 1q21-q23 contributes to normal variation in plasma cholesterol concentration. J Mol Med 79:262–269 10.1007/s001090100212 [DOI] [PubMed] [Google Scholar]
- 70.Storey JD (2002) A direct approach to false discovery rates. J R Statist Soc B 64:479–498 10.1111/1467-9868.00346 [DOI] [Google Scholar]
- 71.Storey JD, Tibshirani R (2003) Statistical significance for genomewide studies. Proc Nat Acad Sci USA 100:9440–9445 10.1073/pnas.1530509100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Storey JD, Tibshirani R (2003) Statistical methods for identifying differentially expressed genes in DNA microarrays. Methods Mol Biol 224:149–157 [DOI] [PubMed] [Google Scholar]
- 73.Storey JD (2003) The positive false discovery rate: a Bayesian interpretation and the q-value. Ann Stat 31:2013–2035 10.1214/aos/1074290335 [DOI] [Google Scholar]
- 74.Storey JD, Taylor JE, Siegmund D (2004) Strong control, conservative point estimation and simultaneous consistency of false discovery rates: a unified approach. J R Statist Soc B 66:187–205 10.1111/j.1467-9868.2004.00439.x [DOI] [Google Scholar]
- 75.van den Oord JCG, Sullivan PF (2003) False discoveries and models for gene discovery. Trends Genet 19:537–542 10.1016/j.tig.2003.08.003 [DOI] [PubMed] [Google Scholar]
- 76.van den Oord JCG, Sullivan PF (2003) A framework for controlling false discovery rates and minimizing the amount of genotyping in the search for disease mutations. Hum Hered 56:188–199 10.1159/000076393 [DOI] [PubMed] [Google Scholar]
- 77.Ewens WJ, Grant GR (2005) Statistical methods in bioinformatics: an introduction, 2nd ed. Springer Verlag, New York [Google Scholar]
- 78.Nguyen DV (2004) On estimating the proportion of true null hypotheses for false discovery rate controlling procedures in exploratory DNA microarray studies. Comp Stat Data Anal 47:611–637 10.1016/j.csda.2004.01.006 [DOI] [Google Scholar]
- 79.Li SS, Bigler J, Lampe JW, Potter JD, Feng Z (2005) FDR-controlling testing procedures and sample size determination for microarrays. Stat Med 24:2267–2280 10.1002/sim.2119 [DOI] [PubMed] [Google Scholar]
- 80.Pawitan Y, Michiels S, Koscielny S, Gusnato A, Ploner A (2005) False discovery rate, sensitivity and sample size for microarray studies. Bioinformatics 21:3017–3024 10.1093/bioinformatics/bti448 [DOI] [PubMed] [Google Scholar]
- 81.Sabatti C (2006) False discovery rate and multiple comparison procedures. In: Allison DB, Page GP, Beasley TM, Edwards JW (eds) DNA microarrays and related genomics techniques: design, analysis, and interpretation of experiments. Chapman & Hall/CRC, Boca Raton, FL, pp 289–304 [Google Scholar]
- 82.de Konig D-J, Haley CS (2005) Genetical genomics in humans and model organisms. Trends Genet 21:377–381 10.1016/j.tig.2005.05.004 [DOI] [PubMed] [Google Scholar]
- 83.Gibson G, Weir B (2005) The quantitative genetics of transcription. Trends Genet 21:616–623 10.1016/j.tig.2005.08.010 [DOI] [PubMed] [Google Scholar]
- 84.Li J, Burmeister M (2005) Genetical genomics: combining genetics with gene expression analysis. Hum Mol Genet 14:R163–R169 10.1093/hmg/ddi267 [DOI] [PubMed] [Google Scholar]
- 85.Hansson GK, Robertson A-KL, Söderberg-Nauclér C (2006) Inflammation and atherosclerosis. Annu Rev Pathol 1:297–329 [DOI] [PubMed] [Google Scholar]
- 86.Bharadwaj D, Stein M-P, Volzer M, Mold C, Du Clos TW (1999) The major receptor for C-reactive protein on leukocytes is Fcγ receptor II. J Exp Med 190:585–590 10.1084/jem.190.4.585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Stein M-P, Edberg JC, Kimberly RP, Mangan EK, Bharadwaj D, Mold C, Du Clos TW (2000) C-reactive protein binding to FcγRIIa on human monocytes and neutrophils is allele-specific. J Clin Invest 105:369–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ratcliffe NR, Kennedy SM, Morganelli PM (2001) Immunocytochemical detection of Fcγ receptors in human atherosclerotic lesions. Immunol Lett 77:169–174 10.1016/S0165-2478(01)00217-6 [DOI] [PubMed] [Google Scholar]
- 89.van der Meer IM, Witteman JCM, Hofman A, Kluft C, de Maat MPM (2004) Genetic variation in Fcγ receptor IIa protects against advanced peripheral atherosclerosis. Thromb Haemost 92:1273–1276 [DOI] [PubMed] [Google Scholar]
- 90.Snoeckx LH, Cornelussen RN, Van Nieuwenhoven FA, Reneman RS, Van Der Vusse GJ (2001) Heat shock proteins and cardiovascular pathophysiology. Physiol Rev 81:1461–1497 [DOI] [PubMed] [Google Scholar]
- 91.Pockley AG (2002) Heat shock proteins, inflammation, and cardiovascular disease. Circulation 105:1012–1017 10.1161/hc0802.103729 [DOI] [PubMed] [Google Scholar]
- 92.Xu Q (2002) Role of heat shock proteins in atherosclerosis. Arterioscler Thromb Vasc Biol 22:1547–1559 10.1161/01.ATV.0000029720.59649.50 [DOI] [PubMed] [Google Scholar]
- 93.Mehta TA, Ettelaie JGC, Venkatasubramaniam A, Chetter IC, McCollum PT (2005) Heat shock proteins in vascular disease—a review. Eur J Vasc Endovasc Surg 29:395–402 [DOI] [PubMed] [Google Scholar]
- 94.Després J-P, Moorjani S, Lupien PJ, Tremblay A, Nadeau A, Bouchard C (1992) Genetic aspects of susceptibility to obesity and related dyslipidemias. Mol Cell Biochem 113:151–169 10.1007/BF00231535 [DOI] [PubMed] [Google Scholar]
- 95.Després J-P (1994) Dyslipidaemia and obesity. Baillieres Clin Endocrinol Metab 8:629–660 10.1016/S0950-351X(05)80289-7 [DOI] [PubMed] [Google Scholar]
- 96.Després J-P (1996) Visceral obesity and dyslipidaemia: contribution of insulin resistance and genetic susceptibility. In: Angel A, Anderson H, Bouchard C, Lau D, Leiter L, Mendelson R (eds) Proceedings of the 7th International Congress on Obesity. John Libbey, London, pp 525–532 [Google Scholar]