

Abstract
Only a single patient with 3-hydroxyisobutyryl-CoA hydrolase deficiency has been described in the literature, and the molecular basis of this inborn error of valine catabolism has remained unknown until now. Here, we present a second patient with 3-hydroxyisobutyryl-CoA hydrolase deficiency, who was identified through blood spot acylcarnitine analysis showing persistently increased levels of hydroxy-C4-carnitine. Both patients manifested hypotonia, poor feeding, motor delay, and subsequent neurological regression in infancy. Additional features in the newly identified patient included episodes of ketoacidosis and Leigh-like changes in the basal ganglia on a magnetic resonance imaging scan. In cultured skin fibroblasts from both patients, the 3-hydroxyisobutyryl-CoA hydrolase activity was deficient, and virtually no 3-hydroxyisobutyryl-CoA hydrolase protein could be detected by western blotting. Molecular analysis in both patients uncovered mutations in the HIBCH gene, including one missense mutation in a conserved part of the protein and two mutations affecting splicing. A carefully interpreted acylcarnitine profile will allow more patients with 3-hydroxyisobutyryl-CoA hydrolase deficiency to be diagnosed.
The catabolic pathway for the branched-chain amino acid valine is believed to be exceptional because part of the pathway between 3-hydroxyisobutyryl-CoA and propionyl-CoA proceeds via free acids, thus requiring a specific hydrolase. This is in marked contrast with the degradation pathways of the other branched-chain amino acids (i.e., leucine and isoleucine, in which the intermediates distal to the 2-oxo-acids are all CoA thioesters). Indeed, the hydrolysis of an activated acyl group in the heart of a catabolic pathway is not only uncommon but also energetically unfavorable, especially when subsequent steps of the pathway again involve CoA thioester intermediates. Although the evidence for the need of a specific hydrolase in the catabolic pathway of valine was poor, strong support for this hypothesis nevertheless came from one case report, published in 1982, documenting 3-hydroxyisobutyryl-CoA hydrolase (HIBCH) deficiency (MIM 250620).1 However, the molecular basis of HIBCH deficiency has remained unknown.
Brown and colleagues1 described a male infant (patient 1), born to consanguineous parents, who clinically manifested various physical malformations (dysmorphic facial features, multiple vertebral anomalies, tetralogy of Fallot, and—at post mortem examination—agenesis of the cingulate gyrus and corpus callosum), as well as failure to thrive and marked hypotonia. More importantly, however, the urine of this patient persistently demonstrated increased levels of S-2-carboxypropyl-cysteamine and S-2-carboxypropyl-cysteine.1,2 As the latter compound can be formed by the condensation between cysteine and methacrylyl-CoA, an intermediate in valine oxidation, the abnormal levels suggested a defect in the valine catabolic pathway. In valine catabolism (fig. 1), methacrylyl-CoA is converted to 3-hydroxyisobutyryl-CoA by crotonase, but fibroblast and liver homogenates of patient 1 showed normal hydratase activities towards methacrylyl-CoA (as well as crotonyl-CoA and tiglyl-CoA) when compared with controls. This finding suggested that, if a defect in the catabolic pathway of valine were to exist, it would be distal to this hydration reaction. Indeed, through a coupled enzyme assay using methacrylyl-CoA and an excess of the enzyme crotonase, Brown et al.1 demonstrated a 20% residual activity of HIBCH in cultured skin fibroblasts.1 No additional patients with HIBCH deficiency have been reported so far.
Figure 1. .

Catabolism of L-valine. The names of the intermediates in the pathway for catabolism of L-valine are shown on the left, with solid arrows indicating enzymatic reactions. The structures of methacrylyl-CoA, 3-hydroxyisubutyryl-CoA, and 3-hydroxyisobutyrate are depicted on the left. The names of the metabolites that are increased as result of HIBCH deficiency are shown by dashed arrows on the right.
In this report, we describe a second patient with HIBCH deficiency (patient 2), who was the first child of healthy, nonconsanguineous white parents and was born at term following an uneventful pregnancy. Initially, he fed poorly but otherwise appeared well, until 4 mo of age, when his parents noted head bobbing. This was followed by a delay in motor milestones and then by ataxia and a loss of skills. He was able to roll over at age 5 mo but lost this ability at age 10 mo. He was reaching out and grasping objects at 4 mo but gradually lost this skill at 13 mo. From age 9 mo, he started to have transient absences and episodes of eye rolling. At age 10 mo, he had a febrile illness, during which he became irritable and more wobbly. He lost the ability to finger feed. Examination at age 11 mo revealed an alert, interactive child who had no nystagmus but constant titubation of the head. He had marked truncal ataxia and was unable to sit unsupported. Tone, power, and reflexes were normal in the upper limbs; tone was reduced, particularly distally, in the lower limbs. He had marked dysmetria and tremor on reaching out. At age 14 mo, following 2 d of coryza and lethargy, he became acutely unwell, with a reduced level of consciousness and metabolic acidosis, and required intubation and ventilatory support. Blood pH was 7.29, with a base deficit of 15.8 mmol/L. Blood lactate and ammonia levels were normal. Echocardiography demonstrated no anomalies. Examination following recovery from this illness showed drowsiness, variable nystagmus, sluggish papillary responses, pooling of secretions in the mouth, fasciculation of the tongue, titubation of the head, and upper- and lower-limb hypotonia with preservation of reflexes. He showed no improvement on treatment with thiamine or a combination of vitamin C, vitamin E, and ubiquinone. As he recovered from his episode of acute encephalopathy, he developed dystonia, predominantly affecting the right arm and leg. A CT scan performed during the episode of acute encephalopathy revealed generalized edema of the brain, with loss of grey/white differentiation in the basal ganglia. In addition, a magnetic resonance imaging (MRI) scan (fig. 2) detected signal abnormalities in the globus pallidus and the midbrain, with asymmetrical involvement of the cerebral peduncles (right greater than left). No structural abnormalities of the brain were observed.
Figure 2. .

MRI scans made when patient 2 was 14 mo old (T2-weighted images). There is signal abnormality in the brain regions indicated by the white arrows: in the globi pallidi (upper panel) and in the midbrain with asymmetrical involvement of the cerebral peduncles (R→L) (lower panel). The appearances were considered likely to represent a neurometabolic disorder. No structural abnormality was noted.
As the neuroimaging was reminiscent of Leigh disease, we investigated the activities of pyruvate dehydrogenase and pyruvate carboxylase in cultured human skin fibroblasts and measured the activities of respiratory chain complexes in a muscle biopsy of patient 2, as described elsewhere.3–7 Normal activities were found for pyruvate carboxylase and pyruvate dehydrogenase. The activities of the respiratory chain complexes, expressed as a ratio to the activity of citrate synthase, revealed a marked reduction of complex I (patient 0.089, controls 0.104–0.268) and a borderline reduction of complex IV (patient 0.013, controls 0.014–0.034). These results were obtained on a muscle biopsy sample obtained 3 mo after the major episode of metabolic decompensation. A second muscle biopsy sample, obtained 3 mo later, showed normal activities of all four respiratory chain complexes. Urine organic acid analysis during ketotic episodes showed excessive excretion of 3-hydroxybutyrate and acetoacetate and moderate excretion of lactate, 2-hydroxyisovalerate, 2-oxoisocaproate, dicarboxylic acids (C6, C8, C10), and methylmalonate, with normal levels of methylcitrate. In contrast, he excreted normal levels of urinary organic acids when well. Normal values were found for nonfasting blood 3-hydroxybutyrate (<0.05 mM) and for blood and CSF lactate. Urine amino acid analysis showed normal results, except on one occasion, when it showed increased ratios to creatinine of several amino acids (including glycine, alanine, and taurine), as well as one unidentified ninhydrin positive compound.
Remarkably, elevated levels of hydroxy-C4-carnitine were consistently found in blood spots of patient 2. Its concentration ranged from 0.45–1.73 μM, controls <0.4 μM. Elevation of hydroxy-C4-carnitine may occur in ketosis, but the elevated blood concentration persisted in this patient even when he was not ketotic. Tandem mass spectrometric analysis does not distinguish between 3-hydroxy-n-butyryl-carnitine and 3-hydroxyisobutyryl-carnitine. Either of these hydroxy-C4-carnitine species could be increased as a consequence of a specific enzyme defect, such as short chain 3-hydroxyacyl-CoA dehydrogenase deficiency (SCHAD [MIM 300256]) or HIBCH deficiency, respectively. Patients with SCHAD deficiency uniformly suffer from hyperinsulinism,8 a phenotype very different from that observed in patient 2. As expected, a normal activity of short chain 3-hydroxyacyl-CoA dehydrogenase was found in the fibroblasts of this patient, as well as in patient 1 (data not shown), suggesting HIBCH deficiency.
Therefore, we investigated fibroblasts of patients 1 and 2 for HIBCH activity, by use of a direct enzyme assay based on the use of the physiological substrate S-3-hydroxyisobutyryl-CoA. The thioester was synthesized from methyl S-3-hydroxyisobutyrate as described elsewhere.9,10 HIBCH activity was subsequently measured spectrophotometrically. The reaction mixture contained in a total volume of 250 μL, 100 mM Tris.HCl pH 8.0, 1 mM EDTA, 1 g/L Triton X-100, 0.1 mM, DTNB and 0.2–0.4 mg/ml fibroblast homogenate. After a preincubation period of 10 min, the reaction was started by the addition of 3-hydroxyisobutyryl-CoA at a final concentration of 0.2 mM. The reduction of DTNB was followed in time on a Cobas-Fara centrifugal analyzer (Roche) at 412 nm, with use of a molar extinction coefficient of 13,700 L/mol/cm. By use of this newly developed assay, a mean ± SD activity of 6.4 ± 1.6 nmol/min/mg protein was found in control fibroblasts (n=8), whereas, in fibroblasts from both patients, no HIBCH activity could be detected.
A human cDNA (HIBCH) encoding a protein with HIBCH activity has been described elsewhere.11 This HIBCH gene maps to chromosome 2q32.2 and has an ORF of 1,161 bp, encoding a protein of 386 aa residues with a calculated molecular weight of 43 kDa. Immunoblot analysis, performed using an antibody against HIBCH, revealed the absence of the HIBCH protein in patient 1 (fig. 3). Fibroblast lysates of patient 2 demonstrated an apparently lower expression of the HIBCH protein than was seen in controls.
Figure 3. .
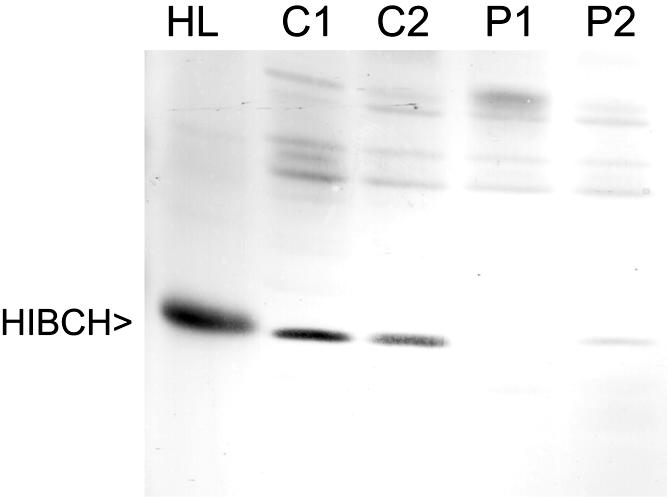
Immunoblot analysis of HIBCH in fibroblast lysates and human liver homogenates. 25 μg of human liver (HL) protein and equal amounts of fibroblast protein (100 μg) of two control subjects (C1 and C2), the index patient (P1), and the newly identified patient (P2) were subjected to SDS-PAGE and transferred onto nitrocellulose by semidry blotting. Polyclonal antibodies raised against purified rat liver HIBCH were used at a dilution of 1:5,000. Antigen-antibody complexes were visualized with goat anti-rabbit IgG-alkaline phosphatase conjugate. As a control for transfer of protein, each blot was reversibly stained with Ponceau S before the incubation with antibodies.
To establish that HIBCH deficiency is caused by mutations in HIBCH, we analyzed the gene at the genomic level and the mRNA level in both patients. We identified a homozygous IVS3-9T→G mutation in patient 1, consistent with parental consanguinity. The IVS3-9T→G mutation was absent from 210 control chromosomes. However, this T→G transversion only slightly weakens the consensus sequence for a splice acceptor site, since a pyrimidine is preferred over a purine at this position in the consensus sequence. Hence, the consequence of this missense mutation on the splicing efficiency was investigated by analysis of HIBCH cDNA obtained by RT-PCR from skin fibroblast RNA. Sequence analysis of the amplicon revealed a homozygous 8-bp insertion (c.219_220insTTGAATAG), after the last base of exon 3, causing a frameshift (K73fsX86). The 8-bp insertion resulted from retention of the 3′ end of intron 3. Close examination of the intronic DNA sequence adjacent to the IVS3-9T→G mutation revealed a stronger homology to the consensus sequence of a splice acceptor site than present in the wild-type intronic sequence, with identity at 9 of 10 intronic bases instead of 7 of 10 (fig. 4). Both the wild-type and mutant sequences are preceded by a DNA sequence with strong homology to the branch point consensus sequence, thus constituting a rather strong alternative cryptic splice acceptor site (fig. 4). Missplicing was evident, because only the aberrant spliced transcript could be detected.
Figure 4. .

Cryptic splice acceptor site in patient 1. A, Sequence analysis of the genomic DNA region from which the 8-bp insertion originates and amplified by PCR from control subjects identified an intron of 2,183 bp (small lettering) with consensus splice donor– and splice acceptor–site sequences (italics and underlined) and branch point (underlined). The complete nucleotide sequence of the intron can be obtained from GenBank (accession number NT_005403). B, In patient 1, a T→G mutation (*) at the −9 position of the authentic splice acceptor site (underlined) results in alternative splicing at a cryptic splice acceptor site located 9 bp 5′ of the authentic splice acceptor site and preceded by a consensus branch point sequence. The 8-bp intron sequence, which is retained as a result of the aberrant splicing, is indicated in bold. Original exon sequences are indicated in capitals. Consensus sequences are defined as follows: splice donor site, [exon]..(C/A)AG↓gt(a/g)agt..[intron]; branch point, (t/c)n(t/c)t(g/a)ac (18–40 bp 5′ of splice acceptor site); splice acceptor site, [intron]..(t/c)6n(t/c)ag ↓ G(G/T)..[exon].
Sequence analysis revealed that patient 2 was compound heterozygous for a missense mutation, c365A→G (Y122C), and a splice acceptor site mutation, IVS2-3C→G. The latter mutation was paternal, whereas the mother was a heterozygous carrier of the c365A→G mutation. The missense mutation predicts the substitution of the bulky amino acid residue tyrosine, which is conserved among different species, including Mus musculus (mouse), Rattus norvegicus (rat), Arabidopsis thaliana (plant), Caenorhabditis elegans (nematode), Gallus gallus (chicken), Xenopus tropicalis (frog), Bos taurus (cow), Canis familiaris (dog), Pongo pygmaeus (orangutan), Danio rerio (zebrafish), Saccharomyces cerevisiae (budding yeast) and Schizosaccharomyces pombe (fission yeast). The IVS2-3C→G transversion disrupts the consensus sequence for a splice acceptor site, given that a pyrimidine (cytosine or thymine) is preferred over guanine at this position in the consensus sequence. Thus, the significance of this particular mutation on splicing was investigated by analysis of HIBCH cDNA. Remarkably, sequence analysis revealed a heterozygous 2-bp insertion resulting from retention of the 3′ end of intron 2, causing a frameshift (R27fsX50). Indeed, the intronic DNA sequence adjacent to the IVS2-3C→G mutation presented a strong homology to the consensus sequence of a splice acceptor site, with identity at 9 of 10 intronic bases. Because this sequence is preceded by a branch point consensus sequence, it constitutes an alternative splice acceptor site. Compound heterozygosity for the Y122C and the IVS2-3C→G mutations resulted in a complete absence of HIBCH activity in the patient, indicative of a pathogenetic effect of both mutations. However, to exclude that the Y122C and the IVS2-3C→G mutations present polymorphic variants, we analyzed 210 control chromosomes for both mutations, assuming a general polymorphic variant frequency of 0.01, but we did not detect either of these mutations. On the basis of these data, we conclude that HIBCH deficiency is caused by mutations in the HIBCH gene.
Until now, only one patient with HIBCH deficiency had been described. The urine of this patient showed abnormal levels of S-2-carboxypropyl-cysteine and S-2-carboxypropyl-cysteamine, which were found using a combination of high-voltage electrophoresis, paper chromatography, and ninhydrin staining.2 However, in many present-day laboratories, routine analysis of plasma and urinary amino acids is performed by liquid chromatography using lithium citrate buffers for elution.12 Unfortunately, S-2-carboxypropyl-cysteine was not available as a reference substance, which precluded a correct identification in patient 2. The concentration of S-2-carboxypropyl-cysteine and S-2-carboxypropyl-cysteamine in patient 1’s urine2 was estimated to be <10 μM. Furthermore, S-2-carboxypropyl-cysteine has a retention time close to those of glycine, alanine, and citrulline.12 Because these amino acids are commonly present in the urine of children, they can easily obscure the presence of the cysteine adducts. As a consequence, additional patients with HIBCH deficiency may easily have remained undetected by this amino acid–analyzer approach. This is exemplified by patient 2, in whom only one out of four urine samples showed an unidentified ninhydrin positive compound. However, we were unable to determine if this compound was S-2-carboxypropyl-cysteine or S-2-carboxypropyl-cysteamine. Fortunately, it was the detection of persistently elevated hydroxy-C4-carnitine in the blood of this patient that led us to determine the activity of HIBCH.
Both patients with HIBCH deficiency demonstrated delayed development of motor skills, hypotonia, initial poor feeding, and a deterioration in neurological function during the first stages of life. However, the neuropathology of patient 1 included agenesis of the cingulate gyrus and the corpus callosum, whereas no structural brain abnormalities were observed in patient 2. Moreover, the brain anomalies of patient 2, as indicated on the CT and MRI scans, were predominantly in the basal ganglia—as is commonly seen in respiratory chain disorders (Leigh disease), glutaric aciduria type I, methylmalonic acidaemia, and priopionic acidaemia. It is also important to note that, in contrast with patient 1, patient 2 manifested no dysmorphic (facial) features and no congenital heart disease (tetralogy of Fallot) and is still alive today.
Treatment of patients with HIBCH deficiency should be aimed at prevention of an increased flux through the catabolic pathway of L-valine. Hence, it is advisable to reduce the protein intake and maintain a high carbohydrate intake, especially during episodes of ketosis (as is done for other disorders of branched chain amino acid catabolism). Other possible therapeutic approaches include administration of carnitine to release coenzyme A and increase the elimination of 3-hydroxyisobutyryl-CoA as urinary 3-hydroxyisobutyrylcarnitine, together with administration of pantothenate (vitamin B5) to compensate for the fact that at least part of intracellular CoA will be trapped in the form 3-hydroxyisobutyryl-CoA.
In conclusion, we have resolved the molecular basis of HIBCH deficiency. By doing so, we also provide conclusive evidence that, in humans, part of the catabolic pathway of valine does indeed proceed via free acids, in contrast with the degradation of the other branched-chain amino acids leucine and isoleucine. Furthermore, we have identified the second patient with HIBCH deficiency. Both patients with HIBCH deficiency demonstrated progressive neurodegeneration. Our data suggest that analysis of blood spot acylcarnitines may circumvent the problematic analysis of the amino acids S-2-carboxypropyl-cysteine and S-2-carboxypropyl-cysteamine, which previously led to the diagnosis of HIBCH deficiency. Moreover, because hydroxy-C4-carnitine could appear in routine acyl-carnitine analysis, we predict that more patients with HIBCH deficiency will be diagnosed as this technique becomes more widely available. Until we have more experience with the natural history of this disorder and the effect of therapeutic interventions, it is probably wise to regard HIBCH deficiency as a disorder associated with progressive neurological damage, ultimately leading to early death. Fortunately, the full elucidation of the molecular and metabolic basis of the disorder will allow prenatal diagnosis.
Web Resources
Accession numbers and URLs for data presented herein are as follows:
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ (for HIBCH [accession numbers NT_005403])
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/OMIM/
References
- 1.Brown GK, Hunt SM, Scholem R, Fowler K, Grimes A, Mercer JF, Truscott RM, Cotton RG, Rogers JG, Danks DM (1982) β-hydroxyisobutyryl coenzyme A deacylase deficiency: a defect in valine metabolism associated with physical malformations. Pediatrics 70:532–538 [PubMed] [Google Scholar]
- 2.Truscott RJ, Malegan D, McCairns E, Halpern B, Hammond J, Cotton RG, Mercer JF, Hunt S, Rogers JG, Danks DM (1981) Two new sulphur-containing amino acids in man. Biomed Mass Spectrom 8:99–104 10.1002/bms.1200080304 [DOI] [PubMed] [Google Scholar]
- 3.Ragan CI, Wilson MY, Darley-Usman VM (1988) Subfractionation of mitochondria and isolation of proteins of oxidative phosphorylation. In: Darley VM, Rickwood D, Wilson MT (eds) Mitochondria: a practical approach. IRL Press, Oxford, pp 79–113 [Google Scholar]
- 4.King TE (1967) Preparation of succinate cytochrome c reductase and cytochrome b-c1 particle and reconstruction of Succinate cytochrome c reductase. Methods Enzymol 10:446–451 [Google Scholar]
- 5.Wharton DC, Tzagoloff A (1967) Cytochrome oxidase from beef heart mitochondria. Methods Enzymol 10:245–250 [Google Scholar]
- 6.Shepherd JA, Garland PB (1969) Citrate synthase activity from rat liver. Methods Enzymol 13:11–19 [Google Scholar]
- 7.Heales SJR, Hargreaves IP, Olpin SE (1996) Diagnosis of mitochondrial electron transport chain defects in small muscle biopsies. J Inher Metab Dis 19:P151 [Google Scholar]
- 8.Clayton PT, Eaton S, Aynsley-Green A, Edginton M, Hussain K, Krywawych S, DattaV, Malingre HE, Berger R, van de Berg I (2001) Hyperinsulinism in short-chain L-3-hydroxyacyl-CoA dehydrogenase deficiency reveals the importance of β-oxidation in insulin secretion. J Clin Invest 108:457–465 10.1172/JCI200111294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rasmussen JT, Borchers T, Knudsen J (1990) Comparison of the binding affinities of acyl-CoA-binding protein and fatty-acid-binding protein for long-chain acyl-CoA esters. Biochem J 265:849–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rougraff PM, Paxton R, Kuntz MJ, Crabb DW, Harris RA (1988) Purification and characterization of 3-hydroxyisobutyrate dehydrogenase from rabbit liver. J Biol Chem 263:327–331 [PubMed] [Google Scholar]
- 11.Hawes JW, Jaskiewicz J, Shimomura Y, Huang B, Bunting J, Harper ET, Harris RA (1996) Primary structure and tissue-specific expression of human β-hydroxyisobutyryl-coenzyme A hydrolase. J Biol Chem 271:26430–26434 10.1074/jbc.271.42.26430 [DOI] [PubMed] [Google Scholar]
- 12.Moore S, Spackman DH, Stein WH (1958) Automatic recording apparatus for use in the chromatography of amino acids. Fed Proc 17:1107–1115 [PubMed] [Google Scholar]