

Abstract
Neurofibromatosis type 1 (NF1) is characterized by café-au-lait spots, skinfold freckling, and cutaneous neurofibromas. No obvious relationships between small mutations (<20 bp) of the NF1 gene and a specific phenotype have previously been demonstrated, which suggests that interaction with either unlinked modifying genes and/or the normal NF1 allele may be involved in the development of the particular clinical features associated with NF1. We identified 21 unrelated probands with NF1 (14 familial and 7 sporadic cases) who were all found to have the same c.2970-2972 delAAT (p.990delM) mutation but no cutaneous neurofibromas or clinically obvious plexiform neurofibromas. Molecular analysis identified the same 3-bp inframe deletion (c.2970-2972 delAAT) in exon 17 of the NF1 gene in all affected subjects. The ΔAAT mutation is predicted to result in the loss of one of two adjacent methionines (codon 991 or 992) (ΔMet991), in conjunction with silent ACA→ACG change of codon 990. These two methionine residues are located in a highly conserved region of neurofibromin and are expected, therefore, to have a functional role in the protein. Our data represent results from the first study to correlate a specific small mutation of the NF1 gene to the expression of a particular clinical phenotype. The biological mechanism that relates this specific mutation to the suppression of cutaneous neurofibroma development is unknown.
Neurofibromatosis type 1 (NF1 [MIM 162200]) is a complex disorder that affects many cell types and involves multiple body systems. Inheritance is autosomal dominant, with a prevalence of 1 in 4,0001 and with penetrance of the mutant gene essentially complete by age 5 years. This inherited disorder is due to germline mutations of the NF1 gene, a 17q11.2-located gene that spans 280 kb, is composed of 61 exons, and encodes a 12-kb mRNA transcript.2,3 Neurofibromin, the NF1 gene product, is a ubiquitously expressed protein that is usually present at low levels in essentially all tissues. This large protein (2,818 aa) exhibits structural and sequence similarity to the mammalian guanine triphosphatase–activating protein (GAP)–related protein family, whose members are evolutionarily conserved. The most highly conserved region of the protein is the neurofibromin GAP-related domain (GRD), which is encoded by exons 20–27a of the NF1 gene and functions by down-regulating Ras activity.4,5 Two other domains of neurofibromin have been described—a cysteine/serine–rich domain (CSRD) and a Sec14p domain.6,7
The major clinical features of NF1 are multiple café-au-lait (CAL) spots, skinfold freckling, benign neurofibromas, and Lisch nodules. The development of these features in patients with NF1 is age dependent, with six or more CAL spots usually present by the 2nd year of life, whereas cutaneous neurofibromas develop only later in childhood or in adulthood. Cutaneous neurofibromas are present in almost all patients with NF1 by age 20 years.1,8 They lie within the dermis or epidermis and are found to move with the skin when it is manipulated on examination. Neurofibromas may also develop at any point along the course of the peripheral nerves that arise from the spinal root outward. The neurofibromas that develop on the more peripheral nerves are usually palpable as discrete nodular subcutaneous swellings and are usually painful on palpation. These deeper lesions are usually referred to as “subcutaneous” or “nodular” neurofibromas, and they were reported to be present in 27% (81/298) of patients in one study.9 Only a small proportion of these neurofibromas cause symptoms sufficient to require their removal; only ∼5% (4/86) of adults with NF1 in a Welsh population study had had one removed because of localized paresthesia, whereas symptomatic spinal neurofibromas were removed in only 2% (2/86) of the affected individuals in the same study.1,10
The other main type of neurofibroma are those referred to as “plexiform” neurofibromas, which can be divided into two types—diffuse and nodular.11 Diffuse neurofibromas are the most common form; they are found on routine examination of the skin and usually present as diffuse subcutaneous swellings with ill-defined margins. They were present in 30% of patients in the Welsh series.1 The overlying dermis and epidermis are often affected by a combination of hypertrophy, pigmentation, and hypertrichosis. Most of these lesions are located on the trunk and are not associated with major problems. Those located on the face, however, may be associated with major disfigurement, and large lesions located elsewhere may be associated with significant bony and/or skin overgrowth. The nodular plexiform neurofibromas may not be obvious on routine examination because they are located more deeply within the body, involving either the major nerve plexuses or the spinal roots. The advent of computed tomography and magnetic resonance imaging scanning has given us the opportunity to study the frequency of these internal tumors in populations with NF1. These studies have shown a high frequency of asymptomatic spinal and internal plexiform lesions.12–14
Numerous other disease complications affecting various body systems can occur, and their development usually cannot be predicted, even within families. These include learning problems, various malignant tumors, scoliosis, long-bone pseudarthrosis, and cardiovascular disease in the form of arterial stenosis and pulmonary valve stenosis.1,15,16 There are a number of minor NF1 features that are not usually associated with significant morbidity but that are encountered at a much greater frequency in patients with NF1 than in the general population. These features include short stature, macrocephaly (both true and relative), and pectus excavatum or carinatum.1,17
Rare families have been reported in which the majority of affected individuals exhibit very similar NF1 features in two or more generations. The best-defined of these families are those with familial CAL spots alone, with or without skinfold freckling.18–20 Additional examples include families with Watson syndrome—characterized by pulmonary stenosis, learning problems, and paucity of cutaneous neurofibromas21–23—and families with multiple spinal neurofibromas.24,25 Of these various familial subtypes, only Watson syndrome appears to be genetically homogeneous but exhibits allelic heterogeneity. Families have been reported with both CAL spots and spinal neurofibromas that are linked or unlinked to the NF1 gene.18–20,24,25 In our clinical experience, such families represent <5% of an NF1 clinical population. The majority of families exhibit wide intrafamilial variation in both the number of major features and the occurrence of complications. Two studies quantitatively analyzed the familial variation in NF126,27 and found evidence for the involvement of modifying genes, and perhaps also the normal NF1 allele, in the development of particular disease features.
Subsequent to the cloning of the NF1 gene in 1990, numerous studies have looked at genotype-phenotype correlation in NF1.28,29 However, with the exception of those patients with the NF1 microdeletion syndrome who develop a large number of neurofibromas for their age, exhibit dysmorphic features, show a probable increased predisposition to the development of malignant peripheral nerve–sheath tumors (MPNST), and exhibit cardiac abnormalities and overgrowth,30–41 no other clinically significant findings have been reported.
The finding of the same ΔAAT mutation in exon 17 of the NF1 gene in three families with no neurofibromas prompted us to review our own mutation database for patients with the same mutation and to contact colleagues in other laboratories worldwide. We here report a cohort of 21 unrelated probands, along with an additional 26 affected relatives, all of whom have the same inframe AAT deletion but no cutaneous neurofibromas.
Material and Methods
Samples from the probands of the three original large multigeneration families (fig. 1A–1C) were referred to the DNA service laboratory in Cardiff because of their unusual phenotypes. Having identified the same mutation in these families, we examined the clinical phenotypes exhibited by four other patients whom we had previously identified with this specific genotype, and we also contacted relevant laboratories worldwide that had extensive experience of NF1 genotyping, to ascertain other patients with this genotype. This study was approved by the appropriate institutional review board.
Figure 1. .

Five unrelated multigeneration families with NF1, in which each clinically assessed affected individual (identified by family numbers) has the same 3-bp AAT deletion mutation but has not developed cutaneous neurofibromas.
A clinical questionnaire was completed for each patient. The clinical data on pulmonic stenosis were based on the clinician declaring lack of or presence of pulmonic stenosis. The significance of clinical findings was calculated using the Z test for proportions. The one-tailed probabilities associated with the resulting Z values were used to estimate the significance of the difference. Sporadic cases of NF1 who were younger than 6 years were excluded from analysis because, at their age, they would not be expected to have developed cutaneous neurofibromas.
NF1 mutation screening in these families involved a variety of complementary mutation-detection techniques, including SSCP, heteroduplex analysis, denaturing high-performance liquid chromatography, direct sequencing, RT-PCR, and multiplex ligation-dependent probe amplification (MLPA).42–47
Bioinformatic Sequence Analysis: Complexity Analysis
To assess the potential contribution that the local DNA sequence structure might have made to the molecular mechanism that produced this specific NF1 microdeletion, we performed complexity analysis of the immediate flanking DNA sequence.48,49
In essence, complexity analysis involves the partitioning of a given sequence S into r consecutive fragments: S=S(1:i1)S(i1+1:i2)…S(ir-1+1:ir=N), where each S(ik-1+1:ik) is the longest fragment downstream of position ik-1 for which a direct, inverted, or symmetric sequence copy is located upstream of position ik-1+1 and where N is the length of the sequence S. For example, if S=ACACCACTCC, then the partition of S computed with respect to direct repeats is A-C-AC-CAC-T-CC. The first, second, and fifth fragments are of length 1, since nucleotides A, C, and T do not occur upstream of their first-occurring positions. The third fragment (AC) is a copy of substring S(1:2), the fourth fragment (CAC) is a copy of substring S(2:4)=CAC, and the sixth fragment (CC) is a copy of substring S(4:5). The number of fragments in the partition, C(S)=r, is termed “the complexity of S.” Thus, for the sequence in the example given above, the complexity with respect to direct repeats is 6. In a similar way, sequence complexity can be calculated with respect to inverted repeats and symmetric elements.
A previous complexity analysis of ∼4,000 microdeletions and ∼2,000 microinsertions derived from the Human Gene Mutation Database (HGMD) demonstrated that, in 80%–90% of such mutations, repetitive elements identified as making the most significant contribution to the change in sequence complexity, consequent to a mutation, were of those sequence repeats that were also directly involved in mediating the mutational change itself. Hence, the most probable mechanism of mutation is that in which the change in complexity is maximized.49,50
Results
The study group consisted of 21 unrelated probands and 26 affected relatives. The NF1 clinical features exhibited by these 47 affected individuals are summarized in table 1, and pedigrees for the five families in which at least 2 affected individuals were studied are shown in figure 1. The ages of the 47 individuals (21 males and 26 females) examined ranged from 2 to 65 years: 2 were younger than 5 years, 3 were aged 6–10 years, 25 were aged 11–20 years, 2 were aged 21–30 years, 7 were aged 31–40 years, and 8 were older than 40 years.
Table 1. .
Clinical Details for 47 Affected Individuals from 21 Families with NF1 with c.2970-2972 delAAT[Note]
| Clinical Feature |
||||||||||
| Family and Individual |
Sex/ Age (years) |
Axillary/Inguinal Freckling | Lisch Nodules | Short Stature | Macrocephaly | Pectus Abnormality | Scoliosis | Pulmonary Stenosis | LDa | Other |
| A: | ||||||||||
| 1 | F/59 | Y | Y | N | N | N | N | N | N | N |
| 2 | M/65 | N | N | N | N | N | N | N | N | Multiple lipomas |
| 3 | F/56 | Y | N | N | N | N | N | N | N | N |
| 4 | F/35 | Y | N | N | N | N | N | N | N | Multiple lipomas |
| 5 | M/36 | Y | N | N | Y | N | N | N | N | N |
| 6 | M/5 | Y | N | Y | N | N | N | N | Y | N |
| 7 | M/2 | Y | Y | N | N | N | N | N | N | N |
| B: | ||||||||||
| 1 | M/50 | Y | N | N | Y | N | N | N | N | N |
| 2 | F/14 | Y | N | N | N | Y | Y | N | Y | Arterial stenosis |
| 3 | F/15 | Y | N | N | N | Y | Y | N | N | N |
| C: | ||||||||||
| 1 | F/63 | Y | N | N | ? | N | N | N | N | N |
| 2 | F/41 | Y | N | N | N | N | N | N | N | N |
| 3 | F/40 | Y | N | N | N | N | N | N | N | N |
| 4 | M/17 | Y | N | N | N | N | N | N | N | N |
| 5 | F/16 | Y | N | N | N | N | N | N | N | N |
| 6 | F/8 | Y | N | N | N | N | N | N | Y | Cerebellar cavernous hemangioma |
| 7 | F/16 | Y | N | N | N | N | N | N | N | N |
| 8 | F/14 | Y | N | N | N | N | N | N | N | N |
| 9 | F/13 | Y | N | N | N | N | N | N | N | N |
| 10 | F/8 | Y | N | N | N | N | N | N | N | N |
| D: | ||||||||||
| 1 | F/38 | N | N | N | N | N | N | N | N | N |
| 2 | F/11 | N | N | N | N | N | N | N | N | N |
| 3 | F/11 | N | N | N | N | N | N | N | N | N |
| 4 | M/17 | N | N | N | N | N | N | N | N | N |
| E: | ||||||||||
| 1 | F/36 | Y | ? | N | N | N | N | N | N | N |
| 2 | F/14 | N | ? | Y | N | Y | N | N | N | N |
| 3 | M/13 | Y | ? | Y | N | Y | N | Y | Y | N |
| 4 | M/10 | N | ? | N | N | Y | N | Y | Y | N |
| 5 | F/8 | N | ? | Y | N | Y | N | Y | N | N |
| F: | ||||||||||
| 1 | M/53 | N | ? | N | N | N | N | N | N | N |
| 2 | M/19 | Y | Y | N | N | N | N | N | Y | Atrial septal defect |
| G: | ||||||||||
| 1 | M/51 | Y | N | N | ? | ? | N | N | N | N |
| 2 | M/20 | Y | N | N | ? | ? | N | N | N | N |
| H: | ||||||||||
| 1b | M/15 | N | N | N | N | N | N | N | N | N |
| I: | ||||||||||
| 1 | M/34 | N | N | N | N | N | Y | N | N | N |
| J: | ||||||||||
| 1b | F/18 | Y | N | N | Y | N | N | N | N | N |
| K: | ||||||||||
| 1 | F/8 | Y | N | N | N | N | N | N | N | N |
| L: | ||||||||||
| 1 | M/12 | Y | N | N | N | N | N | N | N | N |
| M: | ||||||||||
| 1b | M/11 | N | N | Y | N | N | N | N | Y | Genitourinary abnormalities |
| N: | ||||||||||
| 1 | F/12 | N | ? | N | N | Y | N | Y | N | N |
| O: | ||||||||||
| 1 | M/17 | Y | ? | N | Y | N | N | N | Y | N |
| P: | ||||||||||
| 1b | M/15 | Y | N | N | N | N | N | N | N | N |
| Q: | ||||||||||
| 1b | M/13 | N | N | N | N | N | N | N | N | N |
| R: | ||||||||||
| 1 | M/36 | N | N | N | N | N | N | N | N | N |
| S: | ||||||||||
| 1 | F/10 | Y | N | N | N | N | N | N | N | N |
| T: | ||||||||||
| 1b | F/27 | Y | ? | N | N | N | Y | N | Y | Spinal neurofibroma |
| U: | ||||||||||
| 1b | F/25 | Y | N | N | N | N | N | N | N | N |
Note.— All individuals had no cutaneous, subcutaneous, or clinically obvious plexiform neurofibromas. All had six or more CAL macules, except for patient A3.
LD = learning difficulty.
Sporadic cases of NF1. All other individuals were familial cases.
The individuals in families A–G were all examined at the time of data collection for the study. Family E was recently reported because of its NF1 and Noonan syndrome (NS) phenotypes.51 The DNA samples from the remaining probands had been previously genotyped in various NF1 research laboratories, and only a few of these patients were subsequently followed up for this study. The proband in family N was originally referred to our laboratory with a diagnosis of Watson syndrome, and this individual was included in a previous study.29
All affected individuals analyzed in this study were confirmed as having the same NF1 mutation: a 3-bp deletion in exon 17 (c.2970-2972delAAT).6,42,44,52–54 The mutation is predicted to lead to the loss of one of a pair of methionine residues, 991 and 992, of the neurofibromin protein. This mutation was not found in any of the eight clinically assessed unaffected relatives analyzed from families A–C.
Any possibility that this 3-bp deletion represents a polymorphism was excluded, since this mutation completely segregated with the disease in 29 affected individuals from the five unrelated multigeneration families. Additionally, in two sporadic cases of NF1 with this mutation (from families H and T), no such sequence change was found in either set of the unaffected parents. This sequence alteration was not detected in >900 normal chromosomes 17 analyzed. Finally, this 3-bp deletion was the only sequence change identified in five unrelated patients with NF1 for whom comprehensive NF1 mutation analysis was conducted; this included the direct sequencing of the entire coding region, as well as analysis to detect the presence of either a total gene deletion or a multiexon deletion/duplication by MLPA.47
In table 2, the frequencies of NF1 features in this cohort are compared with those found in previous large NF1 studies.1,9,10,17,55 The common clinical features in all affected individuals in this cohort are six or more CAL spots, with 30 of 47 individuals having associated skinfold freckling. None of the individuals examined had cutaneous neurofibromas, and no clinically obvious plexiform lesions were reported. One patient in family T had had a symptomatic spinal neurofibroma removed. In family D, only one branch of the family was available for examination by the clinical geneticist, and none of these affected subjects had any neurofibromas. Cutaneous neurofibromas were, however, reported to be present in the remaining affected adults in this family, but this could not be formally assessed, nor could the mutation be analyzed in this branch of the family.
Table 2. .
NF1 Clinical Features in 47 Individuals with the 3-bp AAT Deletion Compared with Clinical Data Derived from Previous Large-Scale Patient-Cohort NF1 Studies
| Frequency |
|||
| NF1 Feature | Present Patient Cohort | Previous Patient Cohorts1,9,10,14,55 |
P |
| Cutaneous neurofibromasa | 0/18 | 99/991 | .0000 |
| Major external plexiform neurofibromasb | 0/41 | 7/1151 | .0000 |
| Axillary freckling | 30/47 | 95/1341 | .1835 |
| Macrocephaly | 4/45 | 39/1151 | .0007 |
| Short stature | 5/47 | 39/1241 | .0027 |
| Pectus anomalies | 7/45 | 36/12714 | .0443 |
| Learning problems | 8/47 | 41/1241 | .0192 |
| Scoliosisc | 2/20 | 11/961 | .4254 |
| Pulmonary stenosis | 4/47 | 25/2,33255 | <.0001 |
| Symptomatic spinal neurofibroma | 1/47 | 2/961 | .4932 |
In patients aged ⩾20 years.
In patients aged ⩾10 years.
In patients aged ⩾18 years.
In table 2, we present comparisons of the different kinds of neurofibromas in this study with those in previous large studies and provide statistical analysis of the data. For cutaneous neurofibromas, we looked at the frequency in those aged ⩾20 years, since the majority of patients with NF1 in previous studies have had these lesions by that age. We did not analyze subcutaneous lesions because these could be missed on routine examination. For plexiform lesions, we only analyzed the frequency of lesions that would be clinically obvious, as some smaller lesions may be missed on casual examination. We therefore analyzed these lesions only in patients aged ⩾10 years and compared their frequency with that of the major plexiform neurofibromas found in a previous study (in which they were defined as “large head and neck lesions” and “limb/trunk lesions associated with significant skin/bone hypertrophy”).1,10
With regard to the occurrence of NF1 complications, it is often difficult to make definite conclusions, since the frequency of the majority of them is <5% in large NF1 cohorts. However, the data suggest a significantly lower frequency (at a 5% level [8/47]) of learning problems (table 2), although it was not always possible to use the rigorous criteria often used for defining “learning disability.” Scoliosis occurred at a frequency similar to that in previous studies. Pulmonary stenosis, however, occurred in apparent excess, but with three of the four cases in one family. The minor disease features were present at a frequency much lower than normal, with only 4 of 45 having macrocephaly (i.e., head circumference at or above the 97th percentile), 5 of 47 having short stature (i.e., height at or below the 3rd percentile), and 7 of 45 having a pectus abnormality.
Of the other medical problems, vascular disease presenting as arterial stenosis is a rare but definite NF1 complication (e.g., individual B2 in table 1). Two probands had additional medical problems that we felt were coincidental—a cerebellar cavernous hemangioma in the proband of family C (individual C6) and posterior urethral valves in the patient with sporadic NF1 in family M.
Sequence Analysis
The maximum change in complexity was observed on the measure with respect to inverted repeats; therefore, it was hypothesized that the molecular mechanism most likely to have produced this particular mutation is the process of quasi-palindrome correction. Examination of the partition of the nucleotide sequences flanking this deletion identified two almost-perfect inverted repeats, TAGCATTG and CaatGATGTTA (deleted nucleotides are underlined, and perfect Watson-Crick base-pairing nucleotides are shown in bold). These two inverted repeats could have mediated the hairpin-loop structure formation (fig. 2). In addition, G-T pairing might also have contributed to stabilizing the hairpin-loop, since the overall shape of G-T pairing is similar to the Watson-Crick pairing.56 It is the deletion of the aat sequence, which bulges out of the hairpin-loop structure, that is predicted to lead to a quasi-palindrome correction and to the stabilization of the hairpin loop.
Figure 2. .
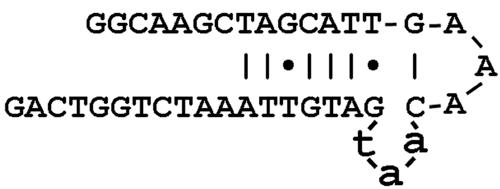
An AAT microdeletion within exon 17 of the NF1 gene mediated by palindrome correction and the formation of a more prominent hairpin-loop structure.
Possible Effect of c.2970-2972delAAT on RNA Splicing
cDNA-based mutation analysis in blood lymphocytes showed normal splicing of exon 17, hence refuting the hypothesis that this 3-bp deletion might exert its pathogenicity by causing splicing abnormalities43,53; this was the only mutation identified. Analysis of normal and mutant DNA sequence was undertaken using both splice-site prediction software57,58 and exon splice-enhancer software (ESE-Finder).59 In agreement with cDNA work, neither of the splice-site tools predicted any change in the identified splice site resulting from this mutation. Analysis of the mutant sequence with the ESE-Finder program, however, demonstrated that two new potential Ser-Arg–rich protein binding sites were created by the 3-bp deletion.
Discussion
We present data demonstrating that a 3-bp AAT mutation in exon 17 of the NF1 gene is associated with a very mild NF1 phenotype in the majority of cases. The most striking finding is the paucity of cutaneous, subcutaneous, and superficial plexiform neurofibromas in these patients. Cutaneous neurofibromas, being one of the hallmark clinical features of NF1, are present in almost all adult patients with NF1, in whom they often represent a major cause of morbidity. These data not only have immediate clinical application but, in a longer term, may also provide a better understanding of the biological reasons why patients with NF1 with the specific c.2970-2972delAAT mutation do not develop this kind of neurofibroma.
The 3-bp inframe deletion is predicted to produce a neurofibromin protein that lacks only a single methionine residue. The amino acid sequence surrounding this deleted methionine is highly conserved, and this sequence would therefore be expected to have a crucial functional role in the protein. Interestingly, this region of neurofibromin has been identified as a CSRD of the protein,60 a domain that contains a number of potential cyclic adenosine monophosphate (cAMP)–dependent protein kinase A binding sites, indicating a possible role in cAMP signaling.61 However, no suitable functional assay with which to monitor the overall activity of this CSRD domain is currently available. Indeed, the only confirmed biochemical test applicable to monitoring of neurofibromin activity involves the assay that measures the intrinsic activity of the GRD.36,62,63 The increased activation of Ras that is observed in neurofibromin-deficient cells often manifests itself by the increased activation of several downstream Ras effectors, including RAF, ERK1/2, PI3K, and p21-activation kinase. The activation of these various signaling pathways is likely to contribute to the increased proliferation observed in Nf1−/− Schwann cells and astrocytes, as well as to the increased proliferation and motility of the heterozygous Nf1+/− mast cells present in neurofibromas.63
Determination of the consequences of the methionine deletion on the overall physical structure of the mutant protein might reveal some information on the potential function of this region of neurofibromin.64 The lack of a suitable soluble polypeptide for this region of the protein does, however, preclude any immediate study of the possible effects of this mutation on the protein’s three-dimensional structure (K. Scheffzeck, personal communication).
It is possible that the mutant neurofibromin functions as a hypomorphic allele, in which the loss of the single methionine at either 991 or 992 results in a reduction in the overall level of neurofibromin in cells, but not to such an extent that it results in the development of neurofibromas. Assessment of the effect of this mutation at the protein level is, therefore, required. RT-PCR–based mutation analysis of mRNA from several patients with the ΔAAT mutation appears to indicate that this mutation is unlikely to have any effect on the overall splicing of the NF1 transcript, since sequencing of the mutant mRNA demonstrated that the mutant transcript lacked only one of the two AUG trinucleotides at codons 991 and 992.53
It is not known, however, whether the shortened mRNA is translated into a correspondingly shortened neurofibromin. Since this mutation is located on the border of the CSRD domain of neurofibromin, it is possible that the loss of methionine within this region may induce the aberrant phosphorylation of the threonine residue located immediately adjacent to the deleted methionine. It is important to ascertain whether this mutant neurofibromin exhibits aberrant interactions with other proteins. In an attempt to address some of these questions, we propose to undertake microarray analyses to assess changes in both gene expression and genomic copy-number variation, to identify potential modifying loci.
A total of 1,255 inframe deletions, ranging in size from 3 to 21 bp, are identified in the HGMD. Whereas the vast majority of these mutations are predicted to have arisen via a slipped-mispairing process,65 ∼3% of these mutations exhibit evidence for definite palindrome correction as the mechanism likely to underlie such inframe deletions.66 Notably, this ΔAAT mutation in exon 17 is predicted to be the result of a deletion of an aat sequence that looped out of a predicted hairpin structure, producing a palindrome sequence correction and leading to the increased stabilization of the hairpin loop.
Neurofibromas are heterogeneous at the cellular level, being composed of Schwann cells—along with fibroblasts, mast cells, and perineurial cells—and it is only the Schwann cells that carry somatic mutations.67,68 In a murine Nf1 model system, it has been shown that it is the interaction of haploinsufficient mast cells with Schwann cells, which are completely deficient of neurofibromin, that induces the development of neurofibromas.69 It is, therefore, possible that, in patients with the ΔAAT mutation, this cellular interaction between mast cells and Schwann cells is disrupted, resulting in the absence of neurofibroma formation.
Before this report, there has been no confirmed correlation, to our knowledge, between a specific small NF1 gene mutation and the NF1 phenotype. We reported a possible association between missense mutations and a lower frequency of Lisch nodules, but this finding needs confirmation in a larger cohort.29 Until now, the only consistent genotype-phenotype correlation has been in those patients with an NF1 microdeletion. These patients have specific dysmorphic features, a heavy burden of cutaneous neurofibromas, and, most likely, an elevated risk of MPNST.30,38,39,70 It has been suggested that the codeletion of the NF1 gene, along with some as-yet-unknown modifying gene(s), may jointly function in potentiating neurofibromagenesis. This may occur, either by the increase in the frequency of NF1 somatic mutations or by the selection for increased growth of neurofibromin-deficient Schwann cells.71 Our data now indicate that specific intragenic mutations may also be associated with the expression of specific clinical features.
High levels of intra- and interfamilial clinical variability are observed in many patients with NF1, even those with identical NF1 mutations.16,72 Attempts at making genotype-phenotype correlations in NF1 are likely to be confounded by the strong effect of age on a patient’s phenotype73 and the lack of independence of many of these clinical features.27,74 Furthermore, the great variety of pathogenic NF1 mutations scattered all over the gene,6,42–44,46 combined with the challenges to correctly classify the mutations according to their expected effect on protein function, greatly hampers these studies.75,76
In the present study, we were alerted to the significance of the ΔAAT mutation because of the absence of clinically detectable neurofibromas in affected individuals in the initial three families. In the Welsh population study, cutaneous neurofibromas were present in all affected individuals older than 20 years,1 yet none of the adults in this study had any lesions. In one of the families (fig. 1D), one branch of the family was unavailable for study but was reported to have neurofibromas. Modifying genes could be a possible explanation. An alternative would be that this family has more than one NF1 mutation.77 Indeed, we have identified three separate NF1 mutations in the affected members of a Portuguese family,78 and a further three families, each with two different NF1 mutations, have recently been found (M. Upadhyaya and L. Messiaen, unpublished data). With regard to other kinds of neurofibromas, we must be more cautious, since subcutaneous neurofibromas and small superficial plexiform lesions could be missed on routine examination. However, the data presented suggest that the ΔAAT mutation is also associated with a major decrease in the frequency of these lesions. This is important clinically, because these lesions have been shown to be associated with an increased risk of MPNST.38 We cannot comment on asymptomatic internal neurofibromas, since no body or spinal imaging has been undertaken. One patient (in family T) had a symptomatic spinal lesion removed, which indicates that such lesions may also occur with this mutation.
The absence of any form of neurofibroma has previously been observed in families with familial CAL spots alone and also in individuals with Watson syndrome, who generally have few, if any, cutaneous neurofibromas. In the absence of specific associated mutations, these diagnoses can be made only in multigenerational families. Of the reported families with CAL spots alone, only one has been linked to the NF1 locus.20 The affected family members each had six or more CAL spots only and no other NF1 features, although minor disease features are not specifically reported.
The majority of individuals in our cohort had associated skinfold freckling, which was not a feature in the family reported elsewhere.20 However, there is a possibility that the ΔAAT mutation may be the causative mutation in families linked to chromosome 17.20 Three probands (of families H, M, and Q) in the present study do not fulfill the diagnostic criteria for NF1, since they have sporadic cases with CAL spots only. That each of these individuals exhibits the same 3-bp AAT deletion—already shown to completely segregate with the disease in five large unrelated families with NF1—and in the absence of any other detected NF1 mutation, suggests that this deletion is also highly likely to be the disease-causing mutation in these three individuals. We therefore suggest that this 3-bp deletion may be the causative mutation in the families with rare chromosome 17–linked autosomal dominant CAL. We therefore suggest that patients who present only with CAL spots should initially be tested for the ΔAAT mutation.
Molecular studies of Watson syndrome21–23 have shown it to be linked to the NF1 locus; however, several different mutations have been found, including an 80-kb deletion22 and an inframe tandem duplication in exon 28.23 Of the present cohort, the proband in family N was also examined as part of that previous study21 and was thought to exhibit a phenotype similar to those of families with a Watson-like syndrome in which 17q11.2 linkage was demonstrated. The identification of another different mutation in a patient with Watson syndrome in the present study would suggest that the NF1 mutation alone is unlikely to be the sole cause of the observed phenotype. Three affected individuals (from family E) all presented with pulmonary valve stenosis, and this family was reported elsewhere, since they exhibit several other NS features. It has been suggested that NF1-NS (NFNS), as characterized by the overlapping features of NF1 and NS, exists as a separate entity still needs to be ascertained, but, with current evidence, this now seems unlikely.79,80 Molecular studies of cohorts of individuals with NFNS have identified a variety of NF1 mutations, several of which were also found in patients with NF1 who did not exhibit any obvious NS-like features.54,81 NFNS features were reported only in the individuals in family E in the present study, although the ΔAAT mutation was previously found in one of the affected individuals in an NFNS study.81 This suggests that, although the AAT-deletion mutation alone may be insufficient to produce the NFNS phenotype, this mutation may still confer an increased predisposition to this phenotype. Given the recent findings of mutations in other components of the Ras–mitogen-activated protein kinase pathway in several syndromes with features that overlap with NF1 and NS, one possibility to explain the variable NFNS phenotype in people with NF1 gene mutations is the interaction with functional polymorphisms in other genes in the pathway.82
The clinical data from our cohort (tables 1 and 2) show a significant reduction of many NF1 features. Quantitative studies of families with NF1 have suggested that the development of the disease features is significantly associated with unlinked modifying genes, and these may be different for each disease feature.26 Szudek et al.27 suggested that the normal NF1 allele may also play a role. Our data give support to the role of other factors, since the phenotype varies even in this cohort. Pulmonary stenosis is the only NF1 complication that appeared in excess in our study; since it is also a feature in the patients with Watson syndrome, there may be a link between the paucity of neurofibromas and a predisposition to pulmonary stenosis.
It is the physical structure of a disease-associated protein, in conjunction with its interactions with other proteins, that is usually considered to be responsible for or to contribute to the observed variation in the clinical phenotype of a particular disorder. Therefore, to better interpret any potential genotype-phenotype relationships, one needs to consider the combinatorial effects of multiple different mutations and polymorphisms, whether allelic or not. A number of good examples are now known, in which different sequence variants, even within the same gene, may exert quite different effects on both the structure and function of the encoded protein and, hence, result in the observed interindividual variation in the clinical phenotype, or, indeed, in which different mutation types of a gene may result in completely different clinical phenotypes. For example, mutations of the lamin A/C gene (LMNA) are known to be associated with at least 12 different inherited disorders. These include a variety of muscular dystrophies, several variant lipodystrophies, and at least two progeria-like aging syndromes. However, close examination of the spectrum of LMNA mutations found in individuals affected with these various disorders has failed to find any direct association between the observed clinical phenotype and either the mutation type or its location within the gene.83,84
Three additional unrelated patients have been described with this specific mutation, but the required clinical details are not currently available.6,81 Fahsold and colleagues, in their large mutation study,6 identified two unrelated patients with NF1 with the ΔAAT mutation but, in addition, found another patient with NF1 who had a different 3-bp inframe deletion that resulted in the loss of an ATG in exon 21 of the NF1 gene. Interestingly, this mutation was also found to delete one of a pair of methionine residues (codons 1214 and 1215) from the mutant neurofibromin; however, no detailed clinical information is available for this patient.
In conclusion, the present study provides the first confirmed molecular evidence implicating the role of a specific 3-bp inframe deletion of the NF1 gene in the determination of a particular clinical phenotype—namely, the almost complete lack of the development of cutaneous, subcutaneous, and superficial plexiform neurofibromas. This compelling evidence for a defined genotype-phenotype correlation should prove invaluable in diagnosis and genetics counseling for future patients with NF1.
Acknowledgments
We thank the patients and their families for their support and Nicholas Sanek for his technical expertise. We are grateful to Prof. Julian Sampson for the clinical assessment of one of the families and to Dr. Ian Frayling for his support. We thank Cancer Research UK, the Wales Gene Park, NF Association UK, and Hayward Foundation, for their financial support. We thank the Wilderman family for their donation (to L.M.) to help support NF1 research for children with CAL spots only. C.L. was supported by Generalitat de Catalunya grant 2005SGR00018 and by Spanish government grants SAF2005-00833, SAF2006-05399, and ISC 111 CO3/07.
Web Resources
The URLs for data presented herein are as follows:
- HGMD, http://www.hgmd.org/
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for NF1) [PubMed]
References
- 1.Huson SM, Harper PS, Compston DAS (1988) Von Recklinghausen neurofibromatosis: clinical and population study in south-east Wales. Brain 111:1355–1381 [DOI] [PubMed] [Google Scholar]
- 2.Upadhyaya M, Cooper DN (eds) (1998) Neurofibromatosis type 1: from genotype to phenotype. BIOS Publishers, Oxford [Google Scholar]
- 3.Viskochil DH (1998) Gene structure and function. In: Upadhyaya M, Cooper DN (eds) Neurofibromatosis type 1: from genotype to phenotype. BIOS Publishers, Oxford, pp 39–56 [Google Scholar]
- 4.Martin G, Viskochil D, Bollag G, McCabe PC, Crosier WJ, Haubruck H, Conroy L, Clark R, O’Connell P, Cawthon RM, et al (1990) The GAP-related domain of the neurofibromatosis type 1 gene product interacts with ras p21. Cell 63:843–849 10.1016/0092-8674(90)90150-D [DOI] [PubMed] [Google Scholar]
- 5.Cichowski K, Jacks T (2001) NF1 tumor suppressor gene function: narrowing the GAP. Cell 104:593–604 10.1016/S0092-8674(01)00245-8 [DOI] [PubMed] [Google Scholar]
- 6.Fahsold R, Hoffmeyer S, Mischung C, Gille C, Ehlers C, Kucukceylan N, Abdel-Nour M, Gewies A, Peters H, Kaufmann D, et al (2000) Minor lesion mutational spectrum of the entire NF1 gene does not explain its high mutability but points to a functional domain upstream of the GAP-related domain. Am J Hum Genet 66:790–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.D’Angelo I, Welti S, Bonneau, Scheffzek K (2006) A novel bipartite phospholipids-binding module in neurofibromatosis type 1 protein. EMBO Rep 7:174–179 10.1038/sj.embor.7400602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Friedman JM, Birch PH (1997) Type 1 neurofibromatosis: a descriptive analysis of the disorder in 1728 patients. Am J Med Genet 70:138–143 [DOI] [PubMed] [Google Scholar]
- 9.Khosrotehrani K, Bastji-Garin S, Riccardi VM, Birch P, Friedman JM, Wolkenstein P (2005) Subcutaneous neurofibromas are associated with mortality in neurofibromatosis type one: a cohort study of 703 patients. Am J Med Genet A 132:49–53 [DOI] [PubMed] [Google Scholar]
- 10.Huson SM (1989) Clinical and genetic studies of von Recklinghausen neurofibromatosis. MD thesis, University of Edinburgh, Edinburgh [Google Scholar]
- 11.Korf B (1999) Plexiform neurofibromas. Am J Med Genet 89:31–37 [DOI] [PubMed] [Google Scholar]
- 12.Thakkar SD, Feigen U, Mautner VF (1999) Spinal tumours in neurofibromatosis type one: an MRI study of frequency, multiplicity and variety. Neuroradiology 41:625–629 10.1007/s002340050814 [DOI] [PubMed] [Google Scholar]
- 13.Schorry EK, Crawford AH, Egelhoff JC, Lovell AM, Saal HM (1997) Thoracic tumours in children with neurofibromatosis-1. Am J Med Genet 74:533–537 [DOI] [PubMed] [Google Scholar]
- 14.Tonsgard JH, Kwak SM, Short PM, Dachman AH (1998) CT imaging in adults with neurofibromatosis-1: frequent asymptomatic plexiform lesions. Neurology 50:1755–1760 [DOI] [PubMed] [Google Scholar]
- 15.Friedman JM, Gutmann DH, MacCollin M, Riccardi VM (1999) Neurofibromatosis: phenotype, natural history, and pathogenesis, 3rd ed. The Johns Hopkins University Press, Baltimore and London [Google Scholar]
- 16.Rasmussen SJ, Friedman JM (2000) NF1 gene and neurofibromatosis 1. Am J Epidemiol 151:33–40 [DOI] [PubMed] [Google Scholar]
- 17.Cnossen MH, Moons KGM, Garssen MPJ, Pasmans NMT, de Goede-Bolder A, Niermeijer MF, Grobbee DE (1998) Minor disease features in neurofibromatosis type 1 (NF1) and their possible value in the diagnosis of NF1 in children ⩽6 years and clinically suspected of having NF1. J Med Genet 35:624–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Charrow J, Listernick R, Ward K (1993) Autosomal dominant multiple café-au-lait spots and neurofibromatosis 1: evidence of non-linkage. Am J Med Genet 45:606–608 10.1002/ajmg.1320450518 [DOI] [PubMed] [Google Scholar]
- 19.Brunner HG, Hulsebos T, Steijlen PM, der Kinderen DJ, vd Steen A, Hamel BC (1993) Exclusion of the neurofibromatosis 1 locus in a family with inherited café-au-lait spots. Am J Med Genet 46:472–474 10.1002/ajmg.1320460428 [DOI] [PubMed] [Google Scholar]
- 20.Abeliovich D, Gelman-Kohan Z, Silverstein S, Lerer I, Chemke J, Merin S, Zlotogora J (1995) Familial café-au-lait spots: a variant of neurofibromatosis type 1. J Med Genet 32:985–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Allanson JE, Upadhyaya M, Watson GH, Partingon M, Mackenzie A, Lahey D, Macleod H, Sarfarazi M, Broadhead W, Harper PS (1991) Watson syndrome: is it a subtype of type 1 neurofibromatosis? J Med Genet 28:752–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Upadhyaya M, Shen M, Cherryson A, Farnham J, Maynard J, Huson SM, Harper PS (1992) Analysis of mutations in neurofibromatosis type 1. Hum Mol Genet 1:735–740 [DOI] [PubMed] [Google Scholar]
- 23.Tassabehji M, Strachan T, Sharland M, Colley A, Donnai D, Harris R, Thakker N (1993) Tandem duplication within a neurofibromatosis type 1 (NF1) gene exon in a family with features of Watson syndrome and Noonan syndrome. Am J Hum Genet 53:90–95 [PMC free article] [PubMed] [Google Scholar]
- 24.Pulst SM, Riccardi VM, Fain P, Korenberg JR (1991) Familial spinal neurofibromatosis: clinical and DNA linkage analysis. Neurology 41:1923–1927 [DOI] [PubMed] [Google Scholar]
- 25.Poyhonen M, Leisti E-L, Kytola S, Leisti J (1997) Hereditary spinal neurofibromatosis: a rare form of NF1? J Med Genet 34:184–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Easton DF, Ponder MA, Huson SM, Ponder BA (1993) An analysis of variation in expression of neurofibromatosis type 1 (NF1): evidence for modifying genes. Am J Hum Genet 53:305–313 [PMC free article] [PubMed] [Google Scholar]
- 27.Szudek J, Joe H, Friedman JM (2002) Analysis of intrafamilial phenotypic variation in neurofibromatosis type 1 (NF1). Genet Epidemiol 23:150–164 10.1002/gepi.1129 [DOI] [PubMed] [Google Scholar]
- 28.Easton DF (1998) Genotype-phenotype relationships in neurofibromatosis type 1. In: Upadhyaya M, Cooper DN (eds) Neurofibromatosis type 1: from genotype to phenotype. BIOS Publishers, Oxford, pp 167–171 [Google Scholar]
- 29.Castle B, Baser ME, Huson SM, Cooper DN, Upadhyaya M (2003) Evaluation of genotype-phenotype correlations in neurofibromatosis type 1. J Med Genet 40:e109 10.1136/jmg.40.10.e109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kayes LM, Burke W, Riccardi VM, Bennett R, Ehrlich P, Rubenstein A, Stephens K (1994) Deletions spanning the neurofibromatosis 1 gene: identification and phenotype of five patients. Am J Hum Genet 54:424–436 [PMC free article] [PubMed] [Google Scholar]
- 31.Wu BL, Austin MA, Schneider GH, Boles RG, Korf BR (1995) Deletion of the entire NF1 gene detected by FISH: four deletion patients associated with severe manifestations. Am J Med Genet 59:528–535 10.1002/ajmg.1320590427 [DOI] [PubMed] [Google Scholar]
- 32.Cnossen MH, van der Est MN, Breuning MH (1997) Deletions spanning the neurofibromatosis type 1 gene: correlations in neurofibromatosis type 1? Hum Mut 9:458–464 [DOI] [PubMed] [Google Scholar]
- 33.Leppig KA, Kaplan P, Viskochil D, Weaver M, Ortenberg J, Stephens K (1997) Familial neurofibromatosis 1 microdeletions: cosegregation with distinct facial phenotype and early onset of cutaneous neurofibromata. Am J Med Genet 73:197–204 [DOI] [PubMed] [Google Scholar]
- 34.Tonsgard JH, Yelavarthi KK, Cushner S, Short MP, Lindgren V (1997) Do NF1 gene deletions result in a characteristic phenotype? Am J Med Genet 73:80–86 [DOI] [PubMed] [Google Scholar]
- 35.Lopez-Correa C, Dorscher M, Brems H, Lazaro C, Clementi M, Upadhyaya M, Dooijes D, Moog U, Kehrer-Sawatzki H, Rutkowski JL, et al (2001) Recombination hotspot in NF1 microdeletion patients. Hum Mol Genet 10:1387–1392 10.1093/hmg/10.13.1387 [DOI] [PubMed] [Google Scholar]
- 36.Upadhyaya M, Osborn M, Maynard J, Kim MR, Tamanoi F, Cooper DN (1997) Mutational and functional analysis of NF1. Hum Genet 99:88–92 10.1007/s004390050317 [DOI] [PubMed] [Google Scholar]
- 37.Upadhyaya M, Ruggieri M, Maynard J, Osborn M, Hartog C, Mudd S, Penttinen M, Cordeiro I, Ponder M, Ponder BA, et al (1998) Gross deletions of the neurofibromatosis type 1 (NF1) gene are predominantly of maternal origin and commonly associated with a learning disability, dysmorphic features and developmental delay. Hum Genet 102:591–597 10.1007/s004390050746 [DOI] [PubMed] [Google Scholar]
- 38.De Raedt T, Brems H, Wolkenstein P, Vidaud D, Pilotti S, Perrone F, Mautner V, Frahm S, Sciot R, Legius E (2003) Elevated risk for MPNST in NF1 microdeletion patients. Am J Hum Genet 72:1288–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Upadhyaya M, Spurlock G, Majounie E, Griffiths S, Forrester N, Baser M, Huson SM, Evans G, Ferner R (2006) The heterogeneous nature of germline NF1 gene mutations in NF1 patients with malignant peripheral nerve sheath tumours (MPNSTs). Hum Mut 27:716 10.1002/humu.9429 [DOI] [PubMed] [Google Scholar]
- 40.Venturin M, Guarnieri P, Natacci F, Stabile M, Tenconi R, Clementin M, Hernandez C, Thompson P, Upadhyaya M, Larizza L, et al (2004) Mental retardation and cardiovascular malformations in NF1 microdeleted patients point to candidate genes in 17q11.2. J Med Genet 41:35–41 10.1136/jmg.2003.014761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Spiegel M, Oexle K, Horn D, Windt E, Buske A, Albrecht B, Prott E, Seemanova E, Seidel J, Rosenbaum T, et al (2005) Childhood overgrowth in patients with common NF1 microdeletions. Eur J Hum Genet 13:883–888 10.1038/sj.ejhg.5201419 [DOI] [PubMed] [Google Scholar]
- 42.Ars E, Kruyer H, Morell M, Pros E, Serra E, Ravella A, Estivill X, Lazaro C (2003) Recurrent mutations in the NF1 gene are common among neurofibromatosis type 1 patients. J Med Genet 40:e82 10.1136/jmg.40.6.e82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Messiaen L, Callens T, Mortier G, Beysen D, Vandenbroucke I, Van Roy N, Speleman F, Paepe AD (2000) Exhaustive mutation analysis of the NF1 gene allows identification of 955 of mutations and reveals a high frequency of unusual splicing defects. Hum Mut 15:541–555 [DOI] [PubMed] [Google Scholar]
- 44.Mattocks C, Baralle D, Tarpey P, ffrench-Constant C, Bobrow M, Whittaker J (2004) Automated comparative sequence analysis identifies mutations in 89% of NF1 patients and confirms a mutation cluster in exons 11–17 distinct from the GAP-related domain. J Med Genet 41:e48 10.1136/jmg.2003.011890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Upadhyaya M, Han S, Consoli C, Majounie E, Horan M, Thomas NS, Potts C, Griffiths S, Ruggieri M, von Deimling A, et al (2004) Characterisation of the somatic mutational spectrum of the neurofibromatosis type 1 (NF1) gene in neurofibromatosis patients with benign and malignant tumours. Hum Mut 23:134–146 10.1002/humu.10305 [DOI] [PubMed] [Google Scholar]
- 46.Griffiths S, Thompson P, Frayling I, Upadhyaya M (2006) Molecular diagnosis of neurofibromatosis type 1: 2 years experience. Fam Cancer (http://www.springerlink.com/content/vq717jw5u0x25344/?p=1cc7aaa399944ac9a599b5cba9839c46&pi=8) (electronically published August 31, 2006; accessed November 21, 2006) [DOI] [PubMed]
- 47.Wimmer K, Yao S, Claes K, Kehrer-Sawatzki H, Tinschert S, De Raedt T, Legius E, Callens T, Beiglbock H, Maertens O, et al (2006) Spectrum of single and multi-exon NF1 copy number changes in a cohort of 1,100 unselected NF1 patients. Genes Chrom Cancer 45:265–276 10.1002/gcc.20289 [DOI] [PubMed] [Google Scholar]
- 48.Gusev VD, Nemytikova LA, Chuzhanova NA (1999) On the complexity measures of genetic sequences. Bioinformatics 15:994–999 10.1093/bioinformatics/15.12.994 [DOI] [PubMed] [Google Scholar]
- 49.Chuzhanova NA, Anassis EJ, Ball E, Krawczak M, Cooper DN (2003) Meta-analysis of indels causing human genetic disease: mechanisms of mutagenesis and the role of local DNA sequence complexity. Hum Mut 21:28–44 10.1002/humu.10146 [DOI] [PubMed] [Google Scholar]
- 50.Ball EV, Stenson PD, Krawczak M, Cooper DN, Chuzhanova NA (2005) Micro-deletions and micro-insertions causing human genetic disease: common mechanisms of mutagenesis and the role of local DNA sequence complexity. Hum Mut 26:205–213 10.1002/humu.20212 [DOI] [PubMed] [Google Scholar]
- 51.Stevenson DA, Viskochil DH, Rope AF, Carey JC (2006) Clinical and molecular aspects of an informative family with neurofibromatosis type 1 and Noonan phenotype. Clin Genet 69:246–253 10.1111/j.1399-0004.2006.00576.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shen MH, Harper PS, Upadhyaya M (1993) Neurofibromatosis type 1 (NF1): the search for mutations by PCR-heteroduplex analysis on Hydrolink gels. Hum Mol Genet 2:1861–1864 [DOI] [PubMed] [Google Scholar]
- 53.Ars E, Serra E, Garcia J, Kruyer H, Gaona A, Lazaro C, Estivill X (2000) Mutations affecting mRNA splicing are the most common molecular defects in patients with neurofibromatosis type 1. Hum Mol Genet 9:237–247 10.1093/hmg/9.2.237 [DOI] [PubMed] [Google Scholar]
- 54.Baralle D, Mattocks C, Kalidas K, Elmslie F, Whittaker J, Lees M, Ragge N, Patton MA, Winter RM, ffrench-Constant C (2003) Different mutations in the NF1 gene are associated with neurofibromatosis-Noonan syndrome (NFNS). Am J Med Genet A 119:1–8 10.1002/ajmg.a.20023 [DOI] [PubMed] [Google Scholar]
- 55.Lin AE, Birch PH, Korf BR, Tenconi R, Niimura M, Poyhonen M, Uhas KA, Sigorini M, Virdis R, Romano C, Bonioli E, et al (2000) Cardiocascular malformations and other cardiovascular abnormalities in neurofibromatosis 1. Am J Med Genet 95:108–117 [DOI] [PubMed] [Google Scholar]
- 56.Calladine CR, Drew HR, Luisi F, Travers AA (2004) Understanding DNA: the molecule and how it works, 3rd ed. Elsevier Academic Press, Amsterdam, pp 334 [Google Scholar]
- 57.Reese MG, Eeckman FH, Kulp D, Haussler D (1997) Improved splice site detection in Genie. J Comput Biol 4:311–323 [DOI] [PubMed] [Google Scholar]
- 58.Hebsgaard SM, Korning PG, Tolstrup N, Engelbrecht J, Rouze P, Brunak S (1996) Splice site prediction in Arabidopsis thaliana DNA by combining local and global sequence information. Nucleic Acids Res 24:3439–3452 10.1093/nar/24.17.3439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cartegni L, Wang J, Zhu Z, Zhang MQ, Krainer AR (2003) ESEfinder: a Web resource to identify exonic splicing enhancers. Nucleic Acids Res 31:3568–3571 10.1093/nar/gkg616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Izawa I, Tamaki N, Saya H (1996) Phosphorylation of the neurofibromatosis type 1 gene product (neurofibromin) by cAMP-dependent protein kinase. FEBS Lett 382:53–59 10.1016/0014-5793(96)00137-8 [DOI] [PubMed] [Google Scholar]
- 61.Marchuk DA, Saulino AM, Tavakkol R, Swaroop M, Wallace MR, Andersen LB, Mitchell AL, Gutmann DH, Boguski M, Collins FS (1991) cDNA cloning of the type 1 neurofibromatosis gene: complete sequence of NF1 gene product. Genomics 11:931–940 10.1016/0888-7543(91)90017-9 [DOI] [PubMed] [Google Scholar]
- 62.Basu TN, Gutmann DH, Fletcher JA, Glover TW, Collins FS, Downward J (1992) Aberrant regulation of ras proteins in malignant tumor cells from neurofibromatosis type 1 patients. Nature 356:713–715 10.1038/356713a0 [DOI] [PubMed] [Google Scholar]
- 63.Arun D, Gutmann D (2004) Recent advances in neurofibromatosis type 1. Curr Opin Neurol 17:101–105 10.1097/00019052-200404000-00004 [DOI] [PubMed] [Google Scholar]
- 64.Scheffzek K, Lautwein A, Kabsch W, Ahmadian MR, Wittinghofer A (1996) Crystal structure of the GTPase-activating domain of human p120GAP and implications for the interaction with Ras. Nature 384:591–596 10.1038/384591a0 [DOI] [PubMed] [Google Scholar]
- 65.Streisinger G, Okada Y, Emrich J, Newton J, Tsugita A, Terzaghi E, Inouye M (1966) Frameshift mutations and the genetic code. Cold Spring Harb Symp Quant Biol 31:77–84 [DOI] [PubMed] [Google Scholar]
- 66.Ripley LS (1982) Model for the participation of quasi-palindromic DNA sequences in frameshift mutations. Proc Natl Acad Sci USA 79:4128–4132 10.1073/pnas.79.13.4128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Serra E, Rosenbaum T, Winner U, Aldeo R, Ars E, Estivill X, Lenard HG, Lazaro C (2000) Schwann cells harbor the somatic NF1 mutation in neurofibromas: evidence of two Schwann cells subpopulations. Hum Mol Genet 9:3055–3084 10.1093/hmg/9.20.3055 [DOI] [PubMed] [Google Scholar]
- 68.Maertens O, Brems H, Vandesompele J, De Raedt T, Heyns I, Rosenbaum T, De Schepper S, De Paepe A, Mortier G, Janssens S, et al (2006) Comprehensive NF1 screening on the cultured Schwann cells from neurofibromas. Hum Mut 27:1030–1040 10.1002/humu.20389 [DOI] [PubMed] [Google Scholar]
- 69.Zhu Y, Ghosh P, Charnay P, Burns DK, Parada LF (2002) Neurofibromas in NF1: Schwann cell origin and role of tumor environment. Science 296:920–922 10.1126/science.1068452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kluwe L, Friedrich RE, Peiper M, Friedman J, Mautner VF (2003) Constitutional NF1 mutations in neurofibromatosis 1 patients with malignant peripheral nerve sheath tumors. Hum Mutat 22:420 10.1002/humu.9193 [DOI] [PubMed] [Google Scholar]
- 71.Dorschner MO, Sybert VP, Weaver M, Pletcher BA, Stephens K (2000) NF1 microdeletion breakpoints are clustered at flanking repetitive sequences. Hum Mol Genet 9:35–46 10.1093/hmg/9.1.35 [DOI] [PubMed] [Google Scholar]
- 72.Upadhyaya M, Cooper DN (1998) The mutational spectrum in neurofibromatosis type 1 and its underlying mechanisms. In: Upadhyaya M, Cooper DN (eds) Neurofibromatosis type 1: from genotype to phenotype. BIOS Publishers, Oxford, pp 65–88 [Google Scholar]
- 73.DeBella K, Szudek J, Friedman JM (2000) Use of the NIH criteria for diagnosis of NF1 in children. Pediatrics 105:608–614 10.1542/peds.105.3.608 [DOI] [PubMed] [Google Scholar]
- 74.Szudek J, Birch SJ, Friedman JM (2000) Growth in North American white children with neurofibromatosis 1 (NF1). J Med Genet 37:933–938 10.1136/jmg.37.12.933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Messiaen LM, Wimmer K (2005) Pitfalls of automated comparative sequence analysis as a single platform for routine clinical testing for NF1. J Med Genet 42:e25 [PMC free article] [PubMed] [Google Scholar]
- 76.Zatkova A, Messiaen L, Vandenbroucke I, Wieser R, Fonatsch C, Krainer AR, Wimmer K (2004) Disruption of exonic splicing enhancer elements is the principal cause of exon skipping associated with seven nonsense or missense alleles of NF1. Hum Mutat 24:491–501 10.1002/humu.20103 [DOI] [PubMed] [Google Scholar]
- 77.Klose A, Peters H, Hoffmeyer S, Buske A, Luder A, Heb D, Lehmann R, Nurnberg P, Tinscgert S (1999) Two independent mutations in a family with neurofibromatosis type 1 (NF1). Am J Med Genet 83:6–12 [DOI] [PubMed] [Google Scholar]
- 78.Upadhyaya M, Majounie E, Thompson P, Han S, Consoli C, Krawczak M, Cordeiro I, Cooper DN (2003) Three different pathological lesions in the NF1 gene originating de novo in a family with neurofibromatosis type 1. Hum Genet 112:12–17 10.1007/s00439-002-0840-1 [DOI] [PubMed] [Google Scholar]
- 79.Carey JC (1998) Editorial comment: neurofibromatosis-Noonan syndrome. Am J Med Genet 75:263–264 [DOI] [PubMed] [Google Scholar]
- 80.Colley A, Donnai D, Evans DGR (1996) Neurofibromatosis/Noonan phenotype: a variable feature of type 1 neurofibromatosis. Clin Genet 49:59–64 [DOI] [PubMed] [Google Scholar]
- 81.DeLuca A, Bottillo I, Sarkozy A, Carta C, Neri C, Bellacchio E, Schirinzi A, Conti E, Zampino G, Battaglia A, et al (2005) NF1 gene mutations represent the major molecular event underlying neurofibromatosis-Noonan syndrome. Am J Hum Genet 77:1092–1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bentires-Alj M, Kontardis MI, Neel BG (2006) Stops along the RAS pathway in human genetic disease. Nat Med 12:283–285 10.1038/nm0306-283 [DOI] [PubMed] [Google Scholar]
- 83.Mattout A, Dechat T, Adam SA, Goldman RD, Gruenbaum Y (2006) Nuclear lamins, disease and aging. Curr Opin Cell Biol 18:335–341 10.1016/j.ceb.2006.03.007 [DOI] [PubMed] [Google Scholar]
- 84.Burke B, Stewart CL (2006) The laminopathies: the functional architecture of the nucleus and its contribution to disease. Annu Rev Genomics Hum Genet 7:369–405 10.1146/annurev.genom.7.080505.115732 [DOI] [PubMed] [Google Scholar]