

Abstract
Receptors that endocytose high-density lipoproteins (HDL) have been elusive. Here yolk-sac endoderm-like cells were used to identify an endocytic receptor for HDL. The receptor was isolated by HDL affinity chromatography and identified as cubilin, the recently described endocytic receptor for intrinsic factor-vitamin B12. Cubilin antibodies inhibit HDL endocytosis by the endoderm-like cells and in mouse embryo yolk-sac endoderm, a prominent site of cubilin expression. Cubilin-mediated HDL endocytosis is inhibitable by HDL2, HDL3, apolipoprotein (apo)A-I, apoA-II, apoE, and RAP, but not by low-density lipoprotein (LDL), oxidized LDL, VLDL, apoC-I, apoC-III, or heparin. These findings, coupled with the fact that cubilin is expressed in kidney proximal tubules, suggest a role for this receptor in embryonic acquisition of maternal HDL and renal catabolism of filterable forms of HDL.
High-density lipoprotein (HDL) particles are known to facilitate the transport of cholesterol from extrahepatic tissues to the liver for repackaging into new lipoproteins, bile acid synthesis, or excretion into the bile (1, 2). This process has been proposed to be an important mechanism for the antiatherogenic effects of HDL. HDL also provides cholesterol to the ovaries, testes, placenta, and adrenal glands for steroid biosynthesis. Moreover, HDL is important in maternal-embryonic nutrition, providing cholesterol, triglycerides, and lipid-soluble vitamins to the placenta, yolk sac, and embryo (3, 4).
Characterization of factors involved in the regulation of HDL catabolism has been the focus of intensive research (5–7). As a result, it is known that the kidney and liver are major sites of HDL catabolism and that lipolytic modification of the particle and structural integrity of its apolipoprotein (apo) components are significant factors in determining the half life of HDL (5, 8–12). The mechanism by which HDL is catabolized in the kidney, liver, and other tissues such as the placenta and yolk sac is not fully understood. The scavenger receptor SR-BI has been shown to function as an HDL “docking” receptor that mediates selective cholesterol uptake (13–15). However, unlike catabolism of low-density lipoprotein (LDL) by the classic LDL receptor, HDL catabolism by SR-BI does not involve holoparticle uptake and lysosomal degradation. This conclusion was supported by the finding that transgenic mice deficient in SR-BI display elevated levels of plasma HDL cholesterol yet exhibit no change in the level of plasma apoA-I (15). Endocytosis and lysosomal degradation of holoparticle HDL is nevertheless known to occur (16), but endocytic HDL receptors have remained elusive. In the present study, we have identified a HDL receptor and characterized its capacity to mediate HDL holoparticle endocytosis leading to lysosomal degradation.
MATERIALS AND METHODS
Proteins.
Human apolipoproteins apoA-I, apoA-II, apoC-I, and apoC-III were purified as described previously (17, 18). Human apoE3 was purchased from Calbiochem, apoJ from Quidel (San Diego), and BSA, ovalbumin, and heparinase I from Sigma. Recombinant human RAP was purified as described (19).
Lipoproteins.
Human 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine (DiI)-HDL, 3,3′-dioctadecyloxacarbocyanine, perchlorate (DiO)-LDL, and rabbit βVLDL were purchased from Biomedical Technologies. Human HDL (density 1.063–1.21 g/ml), HDL2, HDL3, delipidated HDL (apoHDL), and LDL were prepared as described (20, 21). DiI-HDL, HDL, apoHDL, HDL2, and HDL3 were depleted of apoE-HDL and other heparin-binding particles according to Oram (22), dialyzed against 150 mM NaCl, 50 mM Tris pH 7.4 (TBS) containing 0.3 mM EDTA, and filter sterilized. Human lipoprotein (a) [Lp(a)] was obtained from Peter Harpel (Mount Sinai Medical Center, New York), oxidized LDL from Alicia Jenkins (Medical University of South Carolina, Charleston, SC), and human VLDL (Svedberg flotation rate 100–400) from David Chappell (University of Iowa College of Medicine, Iowa City, IA). Lipoprotein concentration was determined by BCA (bicinchoninic acid) protein assay (Pierce).
Antibodies.
IgGs from rabbit antimegalin (23, 24) were purified by protein-G-Sepharose chromatography. Rabbit anticubilin serum and IgG were provided by Pierre Verroust (Hospital Tenon, Paris, France). Horseradish-peroxidase-conjugated anti-rabbit IgG was obtained from Amersham Pharmacia.
Cells.
Mouse embryonal teratocarcinoma F9 cells (ATCC CRL1720) were differentiated by treatment with retinoic acid (RA) and dibutyryl cyclic AMP (Bt2cAMP) for 6 days as described (25).
125I-HDL Binding, Internalization, and Degradation Assays. HDL internalization and degradation assays were performed as described for LDL and apoJ (23, 25, 26). ApoE-free HDL was labeled with [125I]-iodine by using the iodine monochloride method (27). RA/Bt2cAMP-treated and untreated F9 cells were seeded into gelatin-coated wells (Corning) at 4 × 104 cells/cm2 and allowed to grow for 18 h in serum-free medium (SFM) [DMEM containing ITS (insulin 5 μg/ml, transferrin 5 μg/ml, sodium selenite 5 ng/ml, Boehringer Mannheim)], and penicillin/streptomycin). Before addition of 125I-HDL, the cells were washed with SFM and incubated in the assay medium (DMEM/20 mM Hepes/ITS/penicillin/streptomycin/1.5% ovalbumin) alone or in the presence of HDL (80 μg/ml), RAP (1 μM), chloroquine (50 μM), BSA (1 μM), anticubilin IgG (150 μg/ml) or control IgG (150 μg/ml) and incubated for 30 min at 37°C, 5% CO2. The medium was then removed and apoE-free 125I-HDL (2 μg/ml) in assay medium alone or in assay medium containing HDL, RAP, chloroquine, BSA, or IgGs at the aforementioned concentrations was added and incubated with the cells for 6.5 h. The conditioned medium was treated with trichloroacetic acid (10% final) and centrifuged at 10 K × g for 10 min. The amount of radioactivity in the supernatant was taken to represent the amount of degraded HDL (28). The cell layer was washed three times with ice-cold Dulbecco’s phosphate buffered saline (dPBS) and then treated with 0.5 mg/ml trypsin, 50 μg/ml proteinase K, 0.53 mM EDTA in dPBS (trypsin-K-EDTA) for 3 min at 4°C. The released cells were pelleted, and the amount of radioactivity in the pellet was measured and taken to represent the amount of internalized HDL.
To study HDL binding to RA/Bt2cAMP-treated F9 cells, a filter assay described previously (29) for measuring binding of [125I]-LDL to precipitated oocyte membranes was adopted. Aliquots of membrane precipitates containing 45 μg of protein were combined with the indicated amounts of 125I-HDL and unlabeled HDL in 100 μl (25 mM NaCl/2 mM CaCl2/16 mg/ml BSA/12.5 mM Tris⋅HCL, pH 8.0 final buffer composition) and incubated for 2 h at 24°C. Receptor-bound 125I-HDL was collected on 0.45-μm cellulose acetate filters (Micron Separations) as described (29). Binding data were fit to a single class site model by using ligand (30) and assuming a molecular mass for HDL of 200 kDa.
Measurement of DiI-HDL and DiO-LDL Uptake by Flow Cytometry.
RA/Bt2cAMP-treated and untreated F9 cells (1–1.5 × 105 cells/cm2) were grown for 18 h in SFM. After the cell layers were washed with SFM, DiI-HDL or DiO-LDL was added to a final concentration of 1 μg/ml and incubated for 2 h at 37°C, 5% CO2. For competition experiments, competitors (HDL, HDL2, HDL3, LDL, oxidized LDL, VLDL, β-VLDL, Lp(a) (100 μg/ml), apoA-I (28.3 μg/ml, 1 μM), apoA-II (17.3 μg/ml, 1 μM), apoC-I (6.6 μg/ml, 1 μM), apoC-III (8.8 μg/ml, 1 μM), apoJ (70 μg/ml, 1 μM), ovalbumin (45 μg/ml, 1 μM), RAP (39 μg/ml, 1 μM), apoE (8.5 μg/ml, 0.25 μM), heparin (80 μM, 250 units/ml) or IgGs (200 μg/ml) were added in SFM and incubated for 45 min before the addition of the fluorescent lipoproteins. After the incubation, the medium was removed, cell layers washed with SFM, and the cells were released with trypsin-K-EDTA. The cells were washed once with DMEM, twice with dPBS, then subjected to flow cytometry (FACStar Plus, Becton Dickinson). Plotted data are from gated cells having fluorescence intensity values > the autofluorescence values of 99% of unlabeled cells. For some experiments, cell layers were treated with heparinase I (10 units/ml) for 2 h before the addition of the DiI-HDL. This treatment reduced the levels of cell layer-associated [35SO4] by 50% that of controls.
HDL-Sepharose Affinity Chromatography. HDL-Sepharose affinity chromatography was performed by using detergent extracts of RA/Bt2cAMP-treated and untreated F9 cells that were either cell surface [125I]-labeled or unlabeled. For radiolabeling, cells were washed with dPBS and detached by using 2 mM EDTA/dPBS. The released cells were washed twice with DMEM, then twice with dPBS. Cells (1 × 108) were radioiodinated by using the lactoperoxidase/glucose oxidase method (31). After labeling, the cells were resuspended in 50 mM octyl-β--d-glucoside (Calbiochem) in TBS containing 1 mM CaCl2/mM MgCl2/2 mM PMSF (OG buffer) and passed repeatedly through a 21-gauge needle. The extract was clarified by centrifugation at 100 K × g for 1 h. Extracts were applied to columns of Sepharose-CL4B (5 ml) equilibrated with the OG buffer. Sepharose CL4B-absorbed extracts were applied to columns of apoHDL (apoE-free) coupled to cyanogen bromide-activated-Sepharose (10 mg protein/ml resin). The columns were washed with OG buffer and the bound proteins eluted with sequential applications of 1, 4, and 8 M urea/50 mM Tris, pH 7.4. Peak fractions were pooled and dialyzed against TBS, 1 mM PMSF and absorbed on wheat germ lectin-agarose (Amersham Pharmacia). Bound proteins were eluted with 0.5 M N-acetyl-glucosamine in OG buffer, separated by SDS/PAGE, and analyzed by Coomassie staining and autoradiography.
Nonradiolabeled cells (1 × 109) were released and washed as described above, then suspended in 0.25 M sucrose/10 mM Hepes, pH 7.4, containing an EDTA-free protease inhibitor mixture (Boehringer Mannheim). The cells were homogenized on ice by using a Polytron homogenizer (Kinematica, Lucerne, Switzerland) and the homogenates centrifuged at 2 K × g for 10 min. The resulting supernatants were centrifuged at 100 K × g for 1 h. The pellets were resuspended in 2 ml of 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS) buffer (20 mM CHAPS/1 mM PMSF/TBS) by repeated passage through a 21-gauge needle. The extract was clarified by centrifugation at 100 K × g for 1 h. The concentration of protein in the supernatants was determined, and equal amounts of protein from each cell type were applied to columns of Sepharose-CL4B (Amersham Pharmacia) equilibrated with CHAPS buffer. Sepharose CL4B-absorbed extracts were applied to columns of apoHDL-Sepharose and incubated 18 h at 4°C with nutational movement. Bound proteins were eluted with 8 M urea/50 mM Tris, pH 7.4, separated by SDS/PAGE, and analyzed by silver staining and immunoblotting.
Confocal Microscopic Analysis of DiI-HDL Uptake by Cultured Cells and Mouse Embryos. Cells were plated into gelatin-coated wells of plastic chamber slides (Nalge Nunc) (4 × 104 cells/cm2) and incubated for 2 h at 37°C. The cells were washed and incubated in SFM for 18 h. Before addition of fluorescent lipoproteins, the cells were preincubated with SFM containing HDL, LDL (40 μg/ml), or RAP (1 μM) for 45 min. DiI-HDL (1 μg/ml) and DiO-LDL (1 μg/ml) were then added in SFM alone or in the presence of HDL, LDL (40 μg/ml), or RAP (1 μM) and incubated with the cells for 2 h. The cells were washed with dPBS, fixed in 3% formaldehyde in dPBS for 20 min, and analyzed by using a confocal microscope (Bio-Rad).
Embryos (8–8.5 days postcoitum) were removed from timed pregnant ICR mice so as to leave visceral yolk-sac endoderm intact and placed in DMEM. The embryos were incubated in DMEM containing RAP (1 μM), ovalbumin (1 μM), rabbit anticubilin IgG (400 μg/ml), or normal rabbit IgG (400 μg/ml) for 0.5 h at 37°C, 5% CO2. DiI-HDL was added to a final concentration of 1 μg/ml and incubated for 1 h. The medium was removed and the embryos washed with dPBS and fixed with 3% paraformaldehyde for 15 min. Fixed embryos were placed in dPBS and examined by confocal microscopy. The intensity of yolk-sac fluorescence in mouse embryos was evaluated by seven persons in a double-blinded fashion. The assessments of relative intensity were analyzed by a nonparametric statistical test by using rank and anova procedures (SAS Institute, Cary, NC).
For immunohistological localization of cubilin, embryos were fixed as above and permeabilized for 30 min in 0.02% TritonX-100 in dPBS, 0.1% azide and then blocked for 12 h at 4°C in 1% goat serum, dPBS, azide. The embryos were incubated with anticubilin IgG (10 μg/ml) in dPBS/goat serum/azide for 18 h at 4°C, washed in dPBS/azide for 18 h at 4°C, and incubated 18 h with dichlorotriazinyl aminofluorescein-labeled goat anti-rabbit IgG (Jackson ImmunoResearch) in 1% goat serum. The embryos were washed and examined by confocal microscopy.
RESULTS
F9 cells that were differentiated to yolk-sac endoderm-like cells with RA/Bt2cAMP treatment were found to internalize and degrade [125I]-labeled HDL (Fig. 1). The degradation of 125I-HDL occurred in lysosomes, as evidenced by the fact that chloroquine, a drug that inhibits lysosomal proteinase activity, effectively blocked the degradation (Fig. 1 a and b). In addition, RAP, a protein that inhibits ligand binding to LDL receptor (LDLR) family members, inhibited the endocytosis and degradation of 125I-HDL (Fig. 1 a and b). Undifferentiated F9 cells were unable to mediate HDL internalization and degradation (Fig. 1 c and d). When differentiated F9 cell membranes were analyzed for their ability to bind 125I-HDL, saturable and high-affinity (Kd = 77 ± 7 nM, n = 2) binding was observed (Fig. 1 e and f).
Figure 1.

RA/Bt2cAMP-treated F9 cells internalize and degrade HDL. (a–d) Amounts of apoE-free 125I-HDL internalized and degraded by RA/Bt2cAMP-treated (a and b) and untreated F9 cells (c and d) in the presence of unlabeled HDL, RAP, chloroquine, or BSA. Plotted values are means ±SD of triplicate values. Data in a–d are representative of five experiments. (e) Saturation curve for the binding of 125I-HDL to precipitated CHAPS extract of RA/Bt2cAMP-treated cell membranes. Data in e have been corrected for the amount of nonspecific binding obtained for each concentration of 125I-HDL in the presence of 2.1 μM/425 μg/ml unlabeled HDL. (f) Homologous ligand displacement assay in which 125I-HDL (12 nM/464 cpm/ng) was incubated with varying concentrations of unlabeled HDL (9.87–2,400 nM) and 45 μg of protein precipitated from a CHAPS extract of RA/Bt2cAMP-treated F9 cell membranes. (Inset) Scatchard plot of binding data shown in f. Each data point in e and f represents the mean of duplicate determinations.
Incubation of the RA/Bt2cAMP-treated F9 cells with HDL labeled with DiI (a fluorescent lipid) produced a punctate subcellular staining pattern consistent with a vesicular localization (Fig. 2a). The cell staining was blocked by addition of excess unlabeled HDL or RAP (Fig. 2 b and c). An overlapping pattern of punctate fluorescent staining was observed when the cells were incubated with both DiO-labeled LDL and DiI-HDL (Fig. 2 d–f). The colocalization of the two labeled lipoproteins is an indication that HDL is trafficked through the same endocytic compartments as LDL. Untreated F9 cells did not show staining after incubation with DiI-HDL but did show punctate staining after incubation with DiO-LDL (data not shown). Based on the results obtained by using radiolabeled and fluorescent lipid-labeled HDL, it can be concluded that the differentiated F9 cells express a receptor capable of mediating HDL endocytosis and lysosomal degradation. Furthermore, the fact that RAP can inhibit HDL endocytosis and degradation suggests that the HDL-endocytosis receptor is either a RAP-binding protein, or that a RAP-binding member of the LDLR family is indirectly involved.
Figure 2.

(a) Punctate pattern of fluorescence observed in RA/Bt2cAMP-treated F9 cells incubated with DiI-HDL. RA/Bt2cAMP-treated F9 cells were incubated for 2 h with apoE-free DiI-HDL alone or in the presence of unlabeled HDL (b) or RAP (c) and examined by confocal microscopy. Incubation of RA/Bt2cAMP-treated F9 cells with both DiI-HDL (d) and DiO-LDL (e) reveals an overlapping vesicular pattern (f). (Bar = 25 μm).
A flow cytometry assay by using DiI-HDL was developed to evaluate uptake of DiI-HDL by RA/Bt2cAMP-treated F9 cells. DiI-HDL uptake by treated F9 cells was saturable, with half-maximal saturation occurring at 25 nM (Fig. 3). This value is in good agreement with the Kd of 77 nM obtained from Scatchard analysis (Fig. 1f). Little binding of DiI-HDL to untreated F9 cells was observed (Fig. 3). The assay was also used to evaluate lipoprotein and apolipoprotein specificity. HDL subclasses HDL2, HDL3, and their apoE-free forms competed for DiI-HDL uptake (Fig. 4 a and b). HDL3 was more effective than HDL2, irrespective of apoE content. The major HDL apolipoprotein constituents, apoA-I and apoA-II, as well as apoE competed effectively for DiI-HDL uptake, whereas apoC-I, apoC-III and apoJ displayed little or no ability to compete (Fig. 4 c and d). When lipoprotein specificity was evaluated, LDL, VLDL, βVLDL, and Lp(a) showed little or no effect on HDL uptake (Fig. 4 e and f). Similarly, oxidized LDL was unable to compete for HDL uptake (Fig. 4 g and h). Heparinase treatment has been shown to block apoE- and hepatic lipase-mediated uptake of HDL by cultured hepatocarcinoma cells (32). However, neither heparin nor heparinase I pretreatment of RA/Bt2cAMP-treated F9 cells inhibited DiI-HDL uptake (Fig. 4 g and h). Taken together, the results demonstrate that the process of HDL uptake mediated by differentiated F9 cells is highly specific, apparently involving interactions with apoA-I and/or apoA-II moieties.
Figure 3.

Flow cytometric analysis showing saturability of DiI-HDL uptake by RA/Bt2cAMP-treated F9 cells. Treated and untreated F9 cells were incubated with varying concentrations of DiI-HDL (1–500 nM) for 2 h then stripped of surface-associated material and analyzed. Each data point represents the mean ± range of duplicate determinations (1 × 104 cells/determination).
Figure 4.

Flow cytometric analysis of DiI-HDL uptake by RA/Bt2cAMP-treated F9 cells in the presence of competitors. (a and b) Cells were incubated with apoE-free DiI-HDL (1 μg/ml) alone or with unlabeled HDL, HDL2, HDL3, apoE-free HDL2, and apoE-free HDL3 (100 μg/ml). (c and d) Cells were incubated with DiI-HDL alone or with apolipoproteins apoA-I, apoA-II, apoC-I, apoC-III, apoJ, apoE, or ovalbumin. (e and f) Cells were incubated with DiI-HDL alone or with unlabeled HDL, and LDL, VLDL, βVLDL, or Lp(a). (g and h) Cells were incubated with DiI-HDL alone or with unlabeled HDL, LDL, oxidized LDL, heparin, or heparinase. (a, c, e and g) Percentage of cells that internalized DiI-HDL in the presence or absence of the indicated agents. (b, d, f, and h) Mean fluorescence intensity values of the cells that internalized DiI-HDL in the presence or absence of the indicated agents. Cells (1 × 104) were analyzed for each plotted value. A mean of 24% of the cells in the RA/Bt2cAMP-treated F9 cell cultures show measurable levels of internalized DiI-HDL. Data presented is representative of three experiments.
HDL-Sepharose chromatography was used to isolate the HDL holoparticle-endocytosis receptor from extracts of surface-radiolabeled RA/Bt2cAMP-treated F9 cells. A major Coomassie-stainable radiolabeled polypeptide of ≈500 kDa and several minor polypeptides of ≈400, 140, and 45 kDa were present in the HDL-Sepharose eluates derived from the treated F9 cell extracts but were not in eluates from the untreated cells (Fig. 5 a and b).
Figure 5.

HDL-Sepharose affinity chromatography of extracts of RA/Bt2cAMP-treated and untreated F9 cells. Profiles shown in a and b were from [125I]-surface-labeled cells, whereas c–e were from unlabeled cells. (a) Coomassie-stained gel of proteins eluted from HDL-Sepharose by using 1, 4, and 8 M urea-containing buffer (lanes 3–8). Lanes 1 and 10 contain aliquots of detergent extracts from each cell type. Molecular weight standards are in lanes 2 and 9. (b) Autoradiograph of the gel shown in a. (c) Immunoblot analysis of detergent extracts of RA/Bt2cAMP-treated (lane 1) and untreated (lane 2) F9 cells by using megalin and cubilin antibodies. (d) Silver-stained SDS/PAGE profile of sequential fractions eluted from HDL-Sepharose by using 8 M urea-containing buffer. Lane 1 contains molecular weight standards, lanes 2–5 correspond to HDL-Sepharose eluted fractions from extracts of RA/Bt2cAMP-treated F9 cells, lane 6 is blank, and lanes 7–10 are HDL-Sepharose eluted fractions from untreated F9 cells. (d) Anticubilin immunoblot analysis of the same fractions shown in c.
Megalin/LRP-2 represented a candidate for the ≈500-kDa HDL-Sepharose binding protein, given its similar size and that its expression is augmented by RA/Bt2cAMP treatment of F9 cells (25) (Fig. 5c). However, immunoblot analysis showed that the 500-kDa HDL-Sepharose-binding protein was not reactive with antibodies capable of detecting mouse megalin (data not shown). Another candidate was cubilin, a recently identified 460-kDa RAP-binding endocytic receptor for intrinsic factor-vitamin B12 (33, 34). In addition, cubilin had been shown to be expressed by yolk-sac endoderm cells (35), the phenotype of RA/Bt2cAMP-treated F9 cells. Immunoblotting with cubilin antibodies showed that cubilin was expressed by RA/Bt2cAMP-treated F9 cells but not by untreated F9 cells (Fig. 5c). Furthermore, the 500-kDa HDL-Sepharose binding protein reacted with cubilin antibodies (Fig. 5 d and e). The antibodies also reacted with an ≈800-kDa polypeptide present in the HDL-Sepharose eluate that corresponds to the previously described 800-kDa cubilin dimer (33). Cubilin antibodies did not react with polypeptides present in the HDL-Sepharose eluate derived from the untreated F9 cells. The results indicate that cubilin is the major constituent present in the profile of HDL-Sepharose binding proteins isolated from RA/Bt2cAMP-treated F9 cells.
To directly test the role of cubilin as an endocytic receptor for HDL, cubilin antibodies were evaluated for their ability to block DiI-HDL and 125I-HDL internalization mediated by the RA/Bt2cAMP-treated F9 cells. As shown in Fig. 6, cubilin antibodies effectively inhibited cellular uptake of DiI-HDL (a and b) but not DiO-LDL (c). Similarly, cubilin antibodies were found to effectively inhibit both the internalization and lysosomal degradation of 125I-HDL (Fig. 6 e and f).
Figure 6.

Cubilin antibodies inhibit HDL internalization and degradation. (a–c) RA/Bt2cAMP-treated F9 cells were incubated with apoE-free DiI-HDL alone or in the presence of unlabeled HDL, RAP, anticubilin IgG, antimegalin/LRP-2, or control IgG. (d) Treated F9 cells were incubated with DiO-LDL alone or in the presence of unlabeled LDL, HDL, RAP, antimegalin/LRP-2 IgG, or control IgG. (a, c, and d) Percentage of cells that internalized DiI-HDL or DiO-LDL in the presence or absence of the indicated agents. (b) Mean fluorescence intensity values of the cells (in a) that internalized DiI-HDL in the presence or absence of the indicated agents. Cells (1 × 104) were analyzed for each value plotted in a–d. Data in a–d are representative of three experiments. (e and f) Amounts of 125I-HDL internalized (e) and degraded (f) by RA/Bt2cAMP-treated F9 cells incubated with apoE-free 125I-HDL in assay medium alone or in assay medium containing unlabeled HDL, RAP, chloroquine, BSA, anticubilin IgG, or control IgG. Values in e and f are means ± SD of triplicate values.
The yolk-sac endoderm mediates uptake of maternal-derived HDL (3, 4) and is a major site of cubilin expression (35). To determine whether yolk-sac uptake of HDL is mediated by cubilin, mouse embryos having intact visceral yolk sacs were incubated in medium containing DiI-HDL with or without antagonists of cubilin activity, RAP, or anticubilin IgG. In the absence of an antagonist, DiI-HDL became incorporated exclusively within the visceral extraembryonic yolk-sac endoderm (Fig. 7 a, b, and d). Little or no DiI-HDL was detected within the intraembryonic endoderm. Consistent with these findings, immunostaining of embryos with cubilin antibodies revealed that cubilin is expressed by the visceral extraembryonic endoderm but not the intraembryonic endoderm (Fig. 7f). When RAP was coincubated with DiI-HDL (Fig. 7c), compared with the control protein ovalbumin (Fig. 7 a and b), there was a significant reduction (P < 0.01) in the incorporation of the DiI-HDL into extraembryonic endoderm, as indicated by fluorescence intensity rank scores 9.77 ± 0.22 SD and 4.16 ± 0.15 SD, respectively. When cubilin antibodies were coincubated with DiI-HDL (Fig. 7e), there was also a significant decrease (P < 0.01) in the incorporation of the labeled HDL into extraembryonic endoderm compared with coincubation of DiI-HDL with control IgG (Fig. 7d), as indicated by rank scores of 9.16 ± 0.41 SD and 3.83 ± 0.41 SD, respectively.
Figure 7.
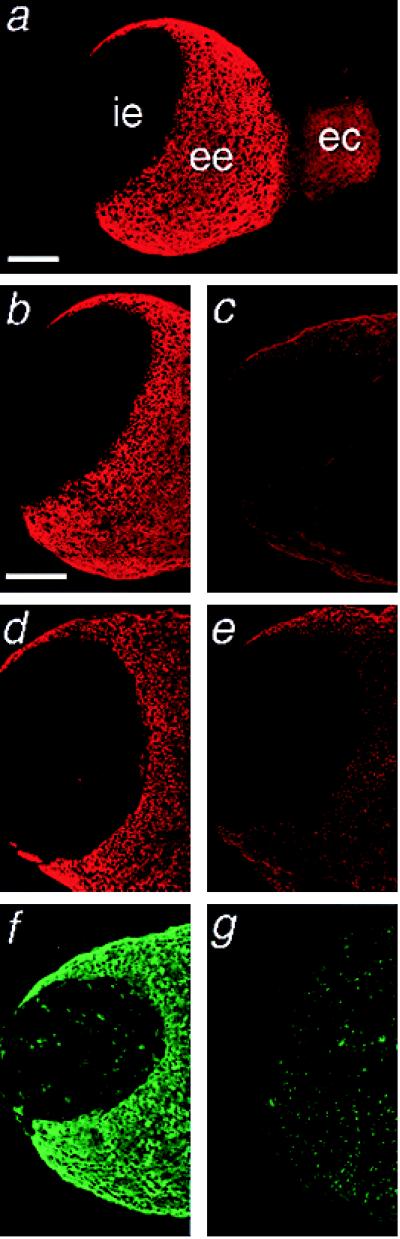
Cubilin antagonists inhibit incorporation of DiI-HDL into the extraembryonic visceral endoderm of mouse embryos. (a–e) Confocal images of mouse embryos (8 days postcoitum) incubated for 1 h ex utero in DMEM containing DiI-HDL plus ovalbumin (a and b), RAP (c), normal rabbit IgG (d), or rabbit anticubilin IgG (e). (f and g) Mouse embryos stained with rabbit anticubilin IgG (f) or normal rabbit IgG (g). i.e., intraembryonic endoderm; ee, extraembryonic endoderm; ec, ectoplacental cone. (Bar = 200 μm.)
DISCUSSION
Here we report the identification of cubilin as a cell-surface receptor that mediates endocytosis and lysosomal degradation of holoparticle HDL. A key to this identification was the use of an in vitro model of holoparticle HDL uptake that permitted isolation of the receptor and characterization of salient aspects of the holoparticle uptake process, including lipoprotein and apolipoprotein specificity. The role of cubilin as a holoparticle HDL endocytosis receptor was confirmed through the use of cubilin antibodies to inhibit internalization of HDL labeled on either its apolipoprotein or lipid constituents. The endocytic activity of cubilin reported here is consistent with recent studies showing cubilin to be an endocytic receptor for intrinsic factor-vitamin B12 and Ig light chain (33, 36).
Given the lack of a membrane spanning element, cubilin is considered to be a peripheral membrane protein (34). The absence of a transmembrane and cytoplasmic domain in cubilin raises the question of how it can mediate HDL endocytosis. An answer could be that cubilin-mediated clearance requires a coreceptor. Such is the case with the endocytosis of urokinase type plasminogen activator (uPA), which involves its phosphoinositol-linked receptor uPAR and coreceptors of the LDL receptor family (37). Megalin is an obvious candidate for a cubilin coreceptor because of its reported interaction with cubilin (34). Whereas megalin antibodies capable of inhibiting endocytic functions of megalin (23, 25, 26), including DiO-LDL uptake (Fig. 6c), had little or no inhibitory effect on DiI-HDL uptake (Fig. 6d), a megalin antiserum obtained from Pierre Verroust inhibited HDL uptake by 25% (data not shown). The variability in these results leaves open the possibility that megalin plays a role as a coreceptor in cubilin-mediated uptake of HDL. Moreover, the fact that RAP and apoE are potent competitors of HDL uptake is consistent with possible involvement of members of the LDL receptor family. However, RAP has also been shown to bind cubilin (33). The role of other minor polypeptides present in the HDL-Sepharose chromatography elution profile must also be considered.
The extraembryonic visceral endoderm of the yolk sac is a major site of cubilin expression (35). Studies have shown this endoderm mediates uptake of maternal HDL (3, 4, 38); however, the significance of maternal HDL to embryonic development is not fully understood. The vital role that yolk-sac cubilin plays in embryonic development is indicated by the teratogenic effects observed when cubilin antibodies are administered to pregnant rats (35). In light of our findings, we speculate that the cubilin antibody-induced defects may be the result of inhibition of cubilin-mediated transport of maternal HDL by the extraembryonic visceral endoderm, perhaps leading to embryonic deficiency in HDL-associated cholesterol, lipid-soluble vitamins (A, E, and K), phospholipids, and/or triglycerides. Consistent with this hypothesis, developmental abnormalities induced by the cubilin antibodies, including neurodevelopmental/craniofacial defects, are similar to developmental defects caused by mutations in genes involved in cholesterol biosynthesis (7-dehydrocholesterol-Δ7 -reductase), lipoprotein trafficking (apoB and megalin), and in the cholesterol-modified signaling protein, sonic hedgehog (39).
The expression of cubilin along the apical surfaces of kidney proximal tubule cells (35) highlights its potential to clear HDL from the glomerular filtrate. The process of glomerular filtration of HDL, although acknowledged to take place, is poorly characterized. Neutral substances with diameters >8 nm and anionic substances >6 nm are not filtered by the glomerulus (40). HDL particles range in diameter from 7.2–12.9 nm, with HDL3 ranging from 7.2–8.8 nm and HDL2 ranging from 8.8–12.9 nm (41). Thus, HDL3 is a likely HDL subclass for cubilin to encounter in the postglomerular filtrate. In addition, lipid-poor apoA-I, which represents as much as 8% of total serum apoA-I (42), may also be cleared from the filtrate by cubilin. Factors such as selective cholesterol uptake by SR-BI and lypolysis act to reduce HDL particle size and lead to accelerated HDL clearance presumably via the kidney (14, 43, 44). An expected consequence of renal clearance of HDL3 or lipid-poor apoA-I by cubilin is catabolism of these particles. Catabolism is consistent with our results and with results from nephron microperfusion studies showing that 125I-HDL3 is lysosomally degraded within proximal tubule cells (45). Another possible consequence could be recycling of HDL/HDL apolipoproteins, perhaps through a process in which HDL particles and/or apolipoprotein constituents bypass degradation and are delivered to the blood for reutilization. Such a consequence may be related to our finding that chloroquine treatment inhibited 125I-HDL degradation yet did not lead to intracellular accumulation of internalized HDL (Fig. 1a). Typically, chloroquine treatment leads to intracellular accumulation of ligands targeted for lysosomal degradation such as LDL (25). It is therefore possible that HDL/HDL apolipoproteins can be trafficked via at least two pathways after cubilin-mediated endocytosis, one that leads to the lysosomal degradation and another that leads out of the cell (e.g., transcytosis). This latter process may be similar to “retroendocytosis” of HDL, in which internalization is followed by unloading of the cholesteryl ester and retrograde secretion of the lipid-depleted particle (46). It also may be similar to transintestinal epithelium transport of IgG and A (47) and transcytosis of LDL across the blood-brain barrier (48).
In summary, RA/Bt2cAMP-treatment of a mouse teratocarcinoma cell line induces the formation of yolk-sac endoderm-like cells capable of endocytosing and lysosomally degrading HDL. HDL binding to these cells is saturable and of high affinity. The receptor responsible for mediating the binding and endocytosis of HDL was identified as cubilin. Uptake of HDL by mouse embryo yolk-sac endoderm was shown to be mediated by cubilin. The significance of cubilin-mediated uptake of HDL to the processes of maternal-embryonic lipoprotein transport and embryonic development remains to be established, as does the potential of cubilin to mediate renal uptake of plasma-derived HDL and consequences of its activity on HDL homeostasis.
Acknowledgments
We thank Dr. Pierre Verroust for providing cubilin antibodies. This work was supported by an American Heart Association Grant to W.S.A. S.M.H. and W.O.T. are recipients of fellowships from the American Heart Association.
ABBREVIATIONS
- HDL
high-density lipoprotein
- LDL
low-density lipoprotein
- RA
retinoic acid
- SFM
serum-free medium
- CHAPS
2 ml of 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate
- DiI
1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine
- Bt2cAMP
dibutyryl cyclic AMP
- dPBS
Dulbecco’s phosphate-buffered saline
- apo
apolipoprotein
- Lp(a)
lipoprotein (a)
References
- 1.Eisenberg S. J Lipid Res. 1984;25:1017–1058. [PubMed] [Google Scholar]
- 2.Tall A R. J Clin Invest. 1990;86:379–384. doi: 10.1172/JCI114722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Woollett L A. J Lipid Res. 1996;37:1246–1257. [PubMed] [Google Scholar]
- 4.Wyne K L, Woollett L A. J Lipid Res. 1998;39:518–530. [PubMed] [Google Scholar]
- 5.Glass C, Pittman R C, Civen M, Steinberg D. J Biol Chem. 1985;260:744–750. [PubMed] [Google Scholar]
- 6.Horowitz B S, Goldberg I J, Merab J, Vanni T M, Ramakrishnan R, Ginsberg H N. J Clin Invest. 1993;91:1743–1752. doi: 10.1172/JCI116384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Silver D L, Jiang X C, Tall A R. J Biol Chem. 1999;274:4140–4146. doi: 10.1074/jbc.274.7.4140. [DOI] [PubMed] [Google Scholar]
- 8.Le N A, Ginsberg H N. Metabolism. 1988;37:614–617. doi: 10.1016/0026-0495(88)90077-7. [DOI] [PubMed] [Google Scholar]
- 9.Brinton E A, Eisenberg S, Breslow J L. J Clin Invest. 1991;87:536–544. doi: 10.1172/JCI115028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deeb S S, Cheung M C, Peng R L, Wolf A C, Stern R, Albers J J, Knopp R H. J Biol Chem. 1991;266:13654–13660. [PubMed] [Google Scholar]
- 11.Rader D J, Castro G, Zech L A, Fruchart J C, Brewer H B. J Lipid Res. 1991;32:1849–1859. [PubMed] [Google Scholar]
- 12.Jackle S, Rinninger F, Lorenzen T, Greten H, Windler E. Hepatology. 1993;17:455–465. doi: 10.1002/hep.1840170316. [DOI] [PubMed] [Google Scholar]
- 13.Acton S, Rigotti A, Landschulz K T, Xu S, Hobbs H H, Krieger M. Science. 1996;271:518–520. doi: 10.1126/science.271.5248.518. [DOI] [PubMed] [Google Scholar]
- 14.Kozarsky K F, Donahee M H, Rigotti A, Iqbal S N, Edelman E R, Krieger M. Nature (London) 1997;387:414–417. doi: 10.1038/387414a0. [DOI] [PubMed] [Google Scholar]
- 15.Rigotti A, Trigatti B L, Penman M, Rayburn H, Herz J, Krieger M. Proc Natl Acad Sci USA. 1997;94:12610–12615. doi: 10.1073/pnas.94.23.12610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Steinberg D. Science. 1996;271:460–461. doi: 10.1126/science.271.5248.460. [DOI] [PubMed] [Google Scholar]
- 17.Brewer H B, Ronan R, Meng M, Bishop C. Methods Enzymol. 1986;128:223–246. doi: 10.1016/0076-6879(86)28070-2. [DOI] [PubMed] [Google Scholar]
- 18.Jackson R L, Holdsworth G. Methods Enzymol. 1986;128:288–296. doi: 10.1016/0076-6879(86)28074-x. [DOI] [PubMed] [Google Scholar]
- 19.Williams S E, Ashcom J D, Argraves W S, Strickland D K. J Biol Chem. 1992;267:9035–9040. [PubMed] [Google Scholar]
- 20.Osborne J C. Methods Enzymol. 1986;128:213–222. doi: 10.1016/0076-6879(86)28069-6. [DOI] [PubMed] [Google Scholar]
- 21.Kelly J L, Kruski A W. Methods Enzymol. 1986;128:170–180. doi: 10.1016/0076-6879(86)28067-2. [DOI] [PubMed] [Google Scholar]
- 22.Oram J F. Methods Enzymol. 1986;129:645–659. doi: 10.1016/0076-6879(86)29096-5. [DOI] [PubMed] [Google Scholar]
- 23.Hammad S M, Ranganathan S, Loukinova E, Twal W O, Argraves W S. J Biol Chem. 1997;272:18644–18649. doi: 10.1074/jbc.272.30.18644. [DOI] [PubMed] [Google Scholar]
- 24.Kounnas M Z, Haudenschild C C, Strickland D K, Argraves W S. In Vivo. 1994;8:343–351. [PubMed] [Google Scholar]
- 25.Stefansson S, Chappell D A, Argraves K M, Strickland D K, Argraves W S. J Biol Chem. 1995;270:19417–19421. doi: 10.1074/jbc.270.33.19417. [DOI] [PubMed] [Google Scholar]
- 26.Kounnas M Z, Loukinova E B, Stefansson S, Harmony J A, Brewer B H, Strickland D K, Argraves W S. J Biol Chem. 1995;270:13070–13075. doi: 10.1074/jbc.270.22.13070. [DOI] [PubMed] [Google Scholar]
- 27.Goldstein J L, Basu S K, Brown M, S. Methods Enzymol. 1983;98:241–260. doi: 10.1016/0076-6879(83)98152-1. [DOI] [PubMed] [Google Scholar]
- 28.Goldstein J L, Brown M S. J Biol Chem. 1974;249:5153–5162. [PubMed] [Google Scholar]
- 29.George R, Barber D L, Schneider W J. J Biol Chem. 1987;262:16838–16847. [PubMed] [Google Scholar]
- 30.Munson P J, Rodbard D. Anal Biochem. 1980;107:220–239. doi: 10.1016/0003-2697(80)90515-1. [DOI] [PubMed] [Google Scholar]
- 31.Ashcom J D, Tiller S E, Dickerson K, Cravens J L, Argraves W S, Strickland D K. J Cell Biol. 1990;110:1041–1048. doi: 10.1083/jcb.110.4.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhong-Sheng J, Dichek H L, Miranda R D, Mahley R W. J Biol Chem. 1997;272:31285–31292. doi: 10.1074/jbc.272.50.31285. [DOI] [PubMed] [Google Scholar]
- 33.Birn H, Verroust P J, Nexo E, Hager H, Jacobsen C, Christensen E I, Moestrup S K. J Biol Chem. 1997;272:26497–26504. doi: 10.1074/jbc.272.42.26497. [DOI] [PubMed] [Google Scholar]
- 34.Moestrup S K, Kozyraki R, Kristiansen M, Kaysen J H, Rasmussen H H, Brault D, Pontillon F, Goda F O, Christensen E I, Hammond T G, et al. J Biol Chem. 1998;273:5235–5242. doi: 10.1074/jbc.273.9.5235. [DOI] [PubMed] [Google Scholar]
- 35.Sahali D, Mulliez N, Chatelet F, Dupuis R, Ronco P, Verroust P. J Exp Med. 1988;167:213–218. doi: 10.1084/jem.167.1.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Batuman V, Verroust P J, Navar G L, Kaysen J H, Goda F O, Campbell W C, Simon E, Pontillon F, Lyles M, Bruno J, et al. Am J Physiol. 1998;275:F246–54. doi: 10.1152/ajprenal.1998.275.2.F246. [DOI] [PubMed] [Google Scholar]
- 37.Strickland D K, Kounnas M Z, Argraves W S. FASEB J. 1995;9:890–898. doi: 10.1096/fasebj.9.10.7615159. [DOI] [PubMed] [Google Scholar]
- 38.Hatzopoulos A K, Rigotti A, Rosenberg R D, Krieger M. J Lipid Res. 1998;39:495–508. [PubMed] [Google Scholar]
- 39.Farese R V, Jr, Herz J. Trends Genet. 1998;14:115–120. doi: 10.1016/s0168-9525(97)01377-2. [DOI] [PubMed] [Google Scholar]
- 40.Bohrer M P, Baylis C, Humes H D, Glassock R J, Robertson C R, Brenner B M. J Clin Invest. 1978;61:72–78. doi: 10.1172/JCI108927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nichols A V, Krauss R M, Musliner T A. Methods Enzymol. 1986;128:417–431. doi: 10.1016/0076-6879(86)28084-2. [DOI] [PubMed] [Google Scholar]
- 42.Neary R H, Gowland E. Clin Chem. 1987;33:1163–1169. [PubMed] [Google Scholar]
- 43.Levak-Frank S, Weinstock P H, Hayek T, Verdery R, Hofmann W, Ramakrishnan R, Sattler W, Breslow J L, Zechner R. J Biol Chem. 1997;272:17182–17190. doi: 10.1074/jbc.272.27.17182. [DOI] [PubMed] [Google Scholar]
- 44.Ueda Y, Royer L, Gong E, Zhang J, Cooper P N, Francone O, Rubin E M. J Biol Chem. 1999;274:7165–7171. doi: 10.1074/jbc.274.11.7165. [DOI] [PubMed] [Google Scholar]
- 45.Peterson D R, Hjelle J T, Carone F A, Moore P A. Kidney Int. 1984;26:411–421. doi: 10.1038/ki.1984.190. [DOI] [PubMed] [Google Scholar]
- 46.DeLamatre J G, Sarphie T G, Archibold R C, Hornick C A. J Lipid Res. 1990;31:191–202. [PubMed] [Google Scholar]
- 47.Rodewald R. J Cell Biol. 1973;58:189–211. doi: 10.1083/jcb.58.1.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dehouck B, Fenart L, Dehouck M P, Pierce A, Torpier G, Cecchelli R. J Cell Biol. 1997;138:877–889. doi: 10.1083/jcb.138.4.877. [DOI] [PMC free article] [PubMed] [Google Scholar]