

Abstract
Cardiac-specific Na+–Ca2+ exchanger (NCX) knockout (KO) mice surprisingly survive into adulthood without compensatory changes in protein expression levels. To determine how cardiac function is maintained in the absence of NCX, we investigated membrane currents, intracellular Ca2+, and action potentials (APs) in whole cell patch-clamped myocytes from wild-type (WT) and NCX knockout mice. There was no difference in resting Ca2+ or sarcoplasmic reticular Ca2+ load between KO and WT. During prolonged caffeine exposure, the decrease of the Ca2+ transient was drastically slowed in KO versus WT myocytes, indicating that no alternative Ca2+-extrusion mechanism is upregulated to compensate for the absence of NCX. Peak L-type Ca2+ current (ICa) was reduced by 62% in KO myocytes compared with WT. Nevertheless, the corresponding Ca2+ transients were similar, implying an increase in the gain of excitation–contraction coupling in KO cells. APs recorded from KO cells repolarized more rapidly than in WT. In WT myocytes, applying a KO AP waveform voltage clamp reduced Ca2+ influx via ICa by 59% compared with WT AP waveform clamps. Again, the corresponding Ca2+ transients remained similar. Our findings indicate that NCX KO myocytes limit Ca2+ influx to ≈20% of that in WT by reducing ICa and by abbreviating the AP. Contractility is maintained by an increase in the gain of excitation–contraction coupling resulting from both a more rapid repolarization of the AP and an as yet unidentified AP-independent mechanism.
Keywords: excitation—contraction coupling, Na+—Ca2+ exchanger, L-type Ca2+ channel, mouse, heart, action potential
Na+–Ca2+ exchange (NCX) maintains intracellular Ca2+ homeostasis in cardiac myocytes by removing Ca2+ after each contraction.1–3 Not surprisingly, global knockout (KO) of NCX causes prenatal death in a murine model.4–6 We recently produced a viable cardiac-specific NCX KO mouse7 in which we were able to completely delete NCX from 80% to 90% of ventricular cardiomyocytes. These animals live into adulthood and exhibit only modest cardiac dysfunction. Immunoblot and microarray analyses showed no adaptations in the expression levels of other proteins involved in Ca2+ regulation, such as the plasma membrane Ca2+ ATPase (PMCA), the L-type Ca2+ channel (DHPR), the sarcoendoplasmic reticular (SR) Ca2+ ATPase, or the ryanodine receptor (RyR). How these animals survive without myocardial NCX is unclear.
These observations bring into question the physiological significance of NCX. Maintained cardiac function in the absence of NCX is inconsistent with current models of excitation–contraction (EC) coupling. In the absence of a robust Ca2+-extrusion mechanism, we expect the key features of cardiac EC coupling, especially transsarcolemmal Ca2+ fluxes, to be distinctly altered. Unrecognized autoregulatory mechanisms of fundamental importance for cardiac physiology may be involved.
The purpose of the present study was to determine whether NCX KO myocytes activate an alternative mechanism to remove Ca2+ and, if not, to further test the hypothesis that NCX KO mice adapt by reducing Ca2+ influx, obviating the need for an alternative Ca2+ efflux mechanism.7
We find that the rate of cellular Ca2+ efflux from NCX KO myocytes is drastically reduced, indicating the absence of an upregulated alternative Ca2+ removal mechanism. To maintain Ca2+ homeostasis, the myocyte compensates by severely limiting Ca2+ influx to balance efflux. KO myocytes limit Ca2+ influx by decreasing peak ICa and by shortening action potential (AP) duration. This produces an extraordinary situation in which overall transsarcolemmal Ca2+ fluxes are reduced to ≈20% of those in WT myocytes. Strikingly, KO myocytes exhibit increased gain of EC coupling compared with WT, in part attributable to more rapid repolarization of the KO AP.
Materials and Methods
Generation of Transgenic Mice
We generated NCX cardiac-specific KO mice using Cre/loxP technology as described in detail previously.7 As reported, this results in total deletion of NCX from 80% to 90% of the cardiomyocytes. The animals used in this study were between 7 and 12 weeks of age, did not display any gross pathology, had good cardiac function, and showed no evidence of heart failure.
Isolation of Ventricular Myocytes From Adult Mouse Hearts
Isolation of ventricular myocytes by collagenase/protease digestion was performed as recently reported7 and based on the methods of Mitra and Morad.8 All procedures were in accordance with the guidelines of the University of California at Los Angeles Office for Protection of Research Subjects. Following isolation, we stored the dissociated cells for up to 6 hours at room temperature in modified Tyrode solution, containing (in mmol/L): 136 NaCl, 5.4 KCl, 10 HEPES, 1.0 MgCl2, 0.33 NaH2PO4, 1.0 CaCl2, 10 glucose (pH 7.4) with NaOH. This solution was also used, with modifications described below, as the standard bath for electrophysiological recordings.
Electrophysiology
To record whole-cell membrane currents, we placed the cells in an experimental chamber (0.5 mL) mounted on the stage of a Nikon Diaphot inverted microscope. A heated bath solution (26°C) continuously perfused the chamber. Patch electrodes were pulled from borosilicate glass (TW150F-3; World Precision Instruments, Sarasota, Fla) on a Sutter P-97 horizontal puller (Sutter Instruments, Novato, Calif). The fire-polished electrodes had a tip diameter of 2 to 3 μm and a resistance of 1 to 2 MΩ when filled with the patch electrode solution, which contained (in mmol/L): 110 CsCl, 30 TEA-Cl, 10 NaCl, 10 HEPES, 5 MgATP, 0.1 cAMP, 0.1 K5fura-2 (pH 7.2) with CsOH. We recorded whole-cell membrane current or voltage using an Axopatch 200 patch clamp amplifier (Axon Instruments, Union City, Calif) in voltage-clamp or current-clamp mode and a Digidata 1322A (Axon Instruments) data acquisition system controlled by pCLAMP 9 software (Axon Instruments). We applied series resistance compensation to all recordings.
Fluorescence Measurements
We simultaneously recorded fura-2 signals and membrane current during voltage clamp using a custom-designed photometric epifluorescence detection system, described in detail previously.9 Ca2+ concentration was calculated from the ratio (R) of the fluorescence intensities at the 2 excitation wavelengths (ratios at 600 Hz) using the method of Grynkiewicz et al10 according to the equation:
Rmax, Rmin, and β were determined by measuring the fluorescence intensity of drops of internal solution containing fura-2 and either high or low Ca2+; Kd-fura=224 nmol/L.
Determination of Cellular Phenotype
Ten to twenty percent of the myocytes isolated from KO hearts have a WT phenotype.7 Like cardiomyocytes isolated from WT hearts, these cells show NCX inward current (INCX) on rapid exposure to caffeine when patch clamped and held at a constant voltage of −40 mV. Thus, subsequent to each experimental protocol, every myocyte isolated from KO mice was rapidly exposed to 1 sec of caffeine (for details see below) to ensure KO phenotype. Those cells with a detectable inward current were excluded from the KO group.
Solution Exchange
Miniature solenoid valves (The Lee Co, Westbrook, Conn) controlled by PCLAMP digital outputs controlled the bath solution flow through a micromanifold (ALA Scientific Instruments, Westbury, NY). This enabled precise timing of solution exchanges in relation to the voltage clamp protocol. The solution surrounding a patched cell exchanged with a half-time of less than 100 ms.
Statistical Analysis
Data are expressed as means±SEM Student unpaired t test was used for direct comparisons of WT versus KO. In experiments where a range of voltages was tested in each group, we used 2-way ANOVA with Tukey–Fisher LSD post hoc testing (JMP 5.01a, SAS Institute, Cary, NC).
Results
INCX, SR Ca2+ Load, and Resting Ca2+ Concentration in WT Versus KO Myocytes
We recently reported that the SR Ca2+ load of NCX KO myocytes appeared similar to that of WT myocytes.7 To further quantify SR Ca2+ and to assess resting Ca2+ levels in KO and WT myocytes, we recorded INCX and Ca2+ transients during a 1-sec application of 5 mmol/L caffeine using a rapid solution changer. As a negative control, these experiments were also performed in the presence of the NCX inhibitor Ni2+. We administered a train of five 100-ms prepulses from −40 to 0 mV at 1 Hz immediately before caffeine application to ensure a steady-state SR Ca2+ load. We maintained a constant holding potential of −40 mV during exposure to caffeine. Figure 1A shows [Ca2+]i and INCX during caffeine application for representative WT and KO myocytes. No inward exchange current was detectable in 8 of 9 KO myocytes, consistent with the absence of NCX in 80% to 90% of the KO myocytes.7 We found no difference in the magnitude of the caffeine-induced Ca2+ transient between WT and KO myocytes (WT: 473±58 nmol/L, n=8; KO: 503±38 nmol/L, n=8; P>0.05), consistent with similar SR Ca2+ loads. Furthermore, there was no difference in resting [Ca2+]i after conditioning pulses (WT: 109±21 nmol/L, n=8; KO: 81±13 nmol/L, n=8; P=0.11). Addition of 10 mmol/L Ni2+ to block NCX during caffeine administration in WT cells abolished INCX but had no effect on the peak [Ca2+]i transient (507±71 nmol/L; n=9). The results indicate that KO of NCX does not significantly alter cellular Ca2+ stores or resting Ca2+ concentration.
Figure 1.
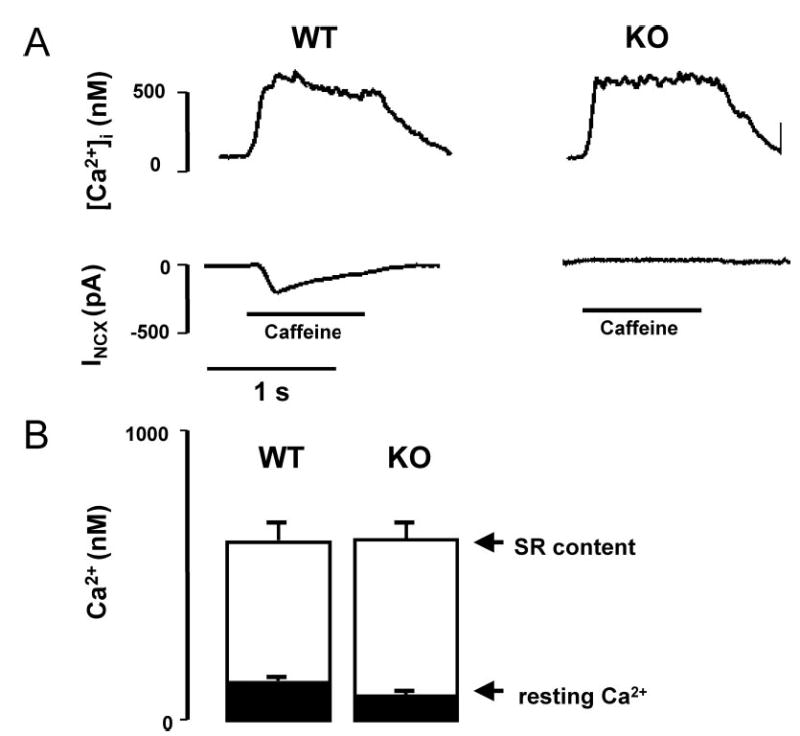
Comparison of caffeine-induced Ca2+ transients and forward NCX currents in WT vs KO myocytes. A, [Ca2+]i transients and membrane current (INCX) recordings during caffeine application in patch clamped WT (n=8) and KO (n=8) myocytes loaded with fura-2 via the patch pipette. Cells were held at −40 mV and exposed to 5 mmol/L caffeine for 1 sec, using a rapid solution exchanger, to release SR Ca2+ stores. In WT, the Ca2+ release elicited an inward NCX current, which was absent in 8 of 9 KO myocytes. B, Summary graph showing that resting [Ca2+]i and the peak of the [Ca2+]i transient, and therefore SR Ca2+ load, were similar in both groups.
Ca2+-Extrusion Capability Is Drastically Reduced in KO Versus WT Myocytes
To investigate whether an alternative Ca2+-extrusion mechanism compensates for the absence of NCX, we estimated the Ca2+-extrusion capacity of the KO myocytes by using a longer exposure to caffeine (8.5 sec; Figure 2A). In the presence of caffeine, SR Ca2+ accumulation is prevented, so that the decline of the caffeine-induced Ca2+ transient is attributable mainly to the ability of the myocyte to extrude Ca2+ from the cell. In WT, the caffeine-induced transient decayed to 50% of peak magnitude (t0.5) in 2.4±0.4 sec and was reduced by 86±3% after 8.5 sec of caffeine exposure (n=10). In KO myocytes, t0.5 was not reached during the 8.5 sec of caffeine exposure as only 18±6% of the initial transient decayed within this time (n=8). When NCX was blocked in WT cells by 10 mmol/L Ni2+, the [Ca2+]i decay rate mimicked that of KO myocytes (n=3). Figure 2B quantifies the amount of Ca2+ that is extruded during 8.5 sec of caffeine exposure in WT, KO, and WT plus Ni2+. These observations demonstrate that alternative Ca2+-extrusion mechanisms are not upregulated to compensate for the absence of NCX in KO myocytes.
Figure 2.
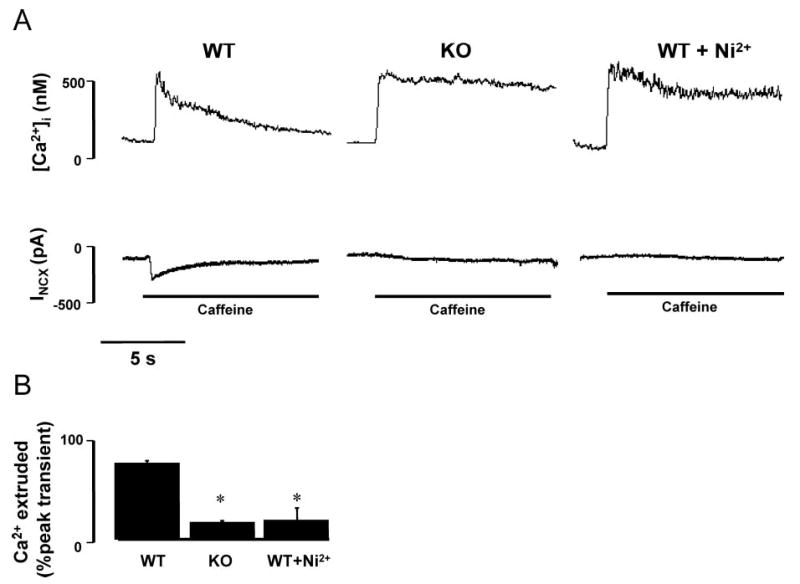
Decreased Ca2+-extrusion capability of KO vs WT myocytes. A, Ca2+ extrusion in patch clamped myocytes (VH = −40 mV) loaded with fura-2 during prolonged exposure of caffeine (8.5 sec) to disable SR Ca2+ uptake. Under these conditions, decay of the caffeine-induced Ca2+ transient is mainly attributable to the ability of the cell to extrude Ca2+ into the extracellular space. B, Bar graph showing the amount of Ca2+ that is extruded from the myocytes during 8.5 sec of caffeine exposure as a percentage of the peak caffeine-induced Ca2+ transient. *P≤0.05 vs WT group.
Decreased ICa and Increased EC Coupling Gain in KO Myocytes
We have previously reported that Ca2+ transients are unchanged in externally paced KO myocytes but that the amplitude of ICa is decreased when measured in patch-clamped KO myocytes.7 We proposed that there was an increase in the gain of EC coupling defined as the ratio of SR Ca2+ release (Δ[Ca2+]i) to trigger Ca2+ (ICa) in KO cells. To directly test this hypothesis, we simultaneously measured Ca2+ transients and ICa during voltage clamps in fura-2–loaded cells studied with the whole-cell patch-clamp technique. Cells were depolarized for 200 ms from a holding potential (VH) of −40 mV to a family of test potentials ranging from −30 to +40 mV in 10-mV steps. Each voltage clamp was preceded by a train of 5 conditioning pulses (100 ms; 1 Hz) from −40 to 0 mV. For clamps to 0 mV, the peak amplitude of ICa in KO myocytes (3.6±0.6 pA/pF; n=4) was significantly smaller than in WT (9.6±1.5 pA/pF; n=7, P<0.01). However, [Ca2+]i release was similar (Δ[Ca2+]i=502±20 nmol/L in KO versus 518±73 nmol/L in WT) (see Figure 3 for representative tracings). Similar results were observed at other potentials, as shown by the summary data in Figure 4. The gain of EC coupling was significantly increased in KO myocytes compared with WT (P≤0.01 by 2-way ANOVA, Figure 4C).
Figure 3.
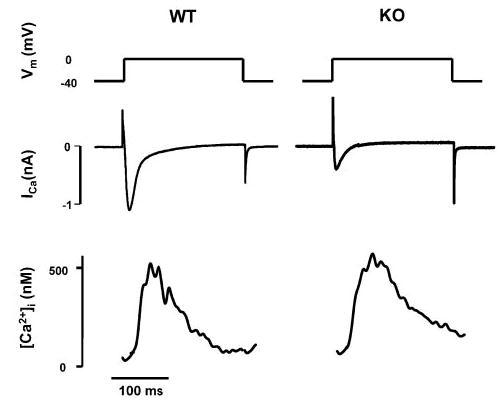
Reduced ICa but increased EC coupling gain in WT vs KO myocytes. Representative tracings showing ICa and [Ca2+]i measured simultaneously during depolarization of the myocytes from a membrane potential (Vm) of −40 mV to 0 mV.
Figure 4.
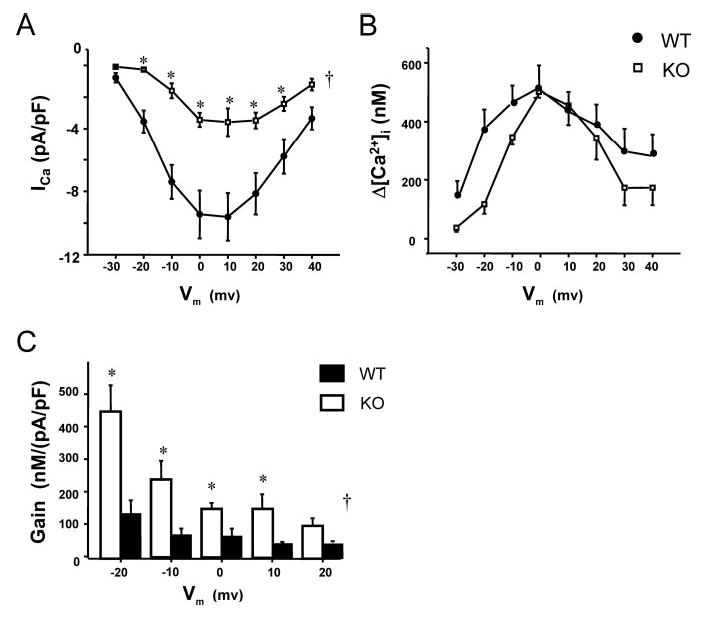
Summary data for the voltage dependence of L-type Ca2+ current, intracellular Ca2+ transients, and EC-coupling gain in WT vs KO. In KO, a clear reduction in peak ICa is observed at all voltages (A), whereas the corresponding Δ[Ca2+]i is largely unaffected (B). Bar graphs (C) show EC-coupling gain, calculated as Δ[Ca2+]i divided by ICa for KO (n=4, except for −20 to 0 mV, n=3) vs WT (n=7). Gain was higher in KO across all voltages. †P<0.05 for KO vs WT by 2-way ANOVA. *P<0.05 at indicated voltages by Fisher–Tukey post hoc testing.
Faster Repolarization of Action Potentials from KO Versus WT Mice
We recorded APs using the current clamp mode of the patch clamp amplifier. Consistent with our previous observations,7 there was no difference in resting potential between WT (n=10) and KO (n=9) (WT, −75±1 mV; KO, −74±1 mV), but the amplitude of the KO AP spike was slightly smaller than the WT AP (WT, 121±3 mV; KO, 113±3 mV; P<0.05). The AP plateau phase was absent in KO myocytes (AP duration to 90% repolarization [APD90, 8.7±1.0 ms] as compared with WT [APD90, 75.1±10.8 ms], P<0.05; Figure 5A). This is consistent with the finding that inward INCX prolongs the plateau phase of the AP.11–14 The spike of the AP was also narrower in KO myocytes (Figure 5B). Thus, both the AP duration at 0 mV (WT, 1.7±0.2 ms; KO, 1.3±0.1 ms; P<0.05) and the AP duration at 50% repolarization (APD50; WT 2.0±0.4 ms; KO 1.2±0.1 ms; P<0.05) were smaller in KO as compared with WT myocytes.
Figure 5.
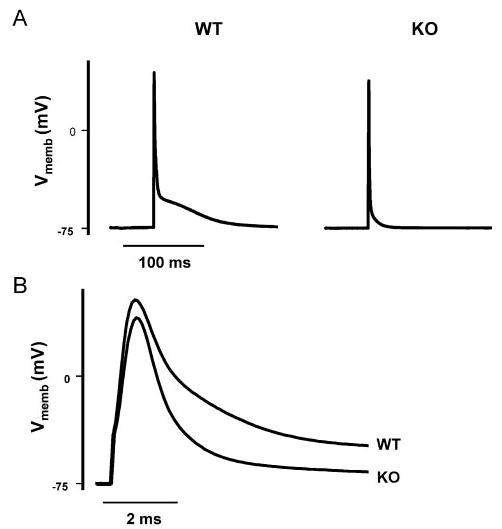
Faster AP repolarization in KO vs WT myocytes. AP tracings are representative for 9 KO and 10 WT myocytes. The pipette solution contained (in mmol/L): 110 KCl, 5 MgCl, 10 NaCl, and 20 HEPES (pH 7.2) with KOH. A, KO APs lack a plateau phase as compared with WT. B, First 10 ms of the tracings shown in A displayed on an expanded time scale. Vmemb indicates membrane potential.
Abbreviated KO AP Limits Ca2+ Influx but Maintains EC Coupling
To investigate the influence of the KO AP shape on ICa and EC coupling, we used representative KO and WT AP waveforms as voltage-clamp commands in patch-clamped KO and WT myocytes while simultaneously monitoring ICa and the Ca2+ transient (Figures 6 and 7). Cells were dialyzed with fura-2 via the patch pipette. After 5 conditioning pulses from −75 mV to 0 mV (100 ms; 1 Hz) to equilibrate the SR, the KO AP waveform (referred to as KO-clamp) was applied from a holding potential of −75 mV. Immediately thereafter, the protocol was repeated in the same myocyte, this time applying the WT AP waveform (referred to as WT-clamp). To isolate L-type Ca2+ current under these conditions, Na+ and K+ were both replaced with Cs+ in the external and pipette solution. The bath solution contained 10 μmol/L tetrodotoxin to block INa. To distinguish the resulting current from the capacitance artifact, both waveforms were again applied to the cell in the presence of Cd2+ (1.5 mmol/L) to inhibit ICa. Only the Cd2+-sensitive difference currents are shown and analyzed.
Figure 6.
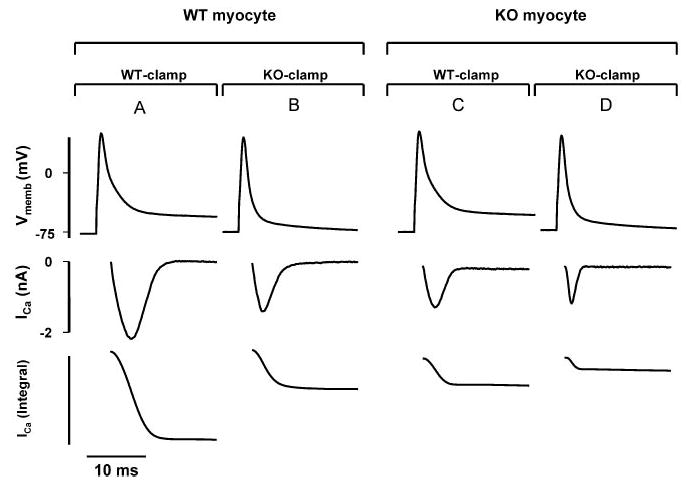
The shape of the KO AP modulates ICa. AP tracings from WT and KO myocytes were translated into voltage-clamp command waveforms and then applied to patch-clamped WT and KO myocytes. Top, Voltage-clamp commands mimicking a WT AP (WT-clamp) or a KO AP (KO-clamp). Middle, ICa measured during the respective clamp command. Bottom, Corresponding integral of ICa (∫ICa) as a measure of Ca2+ influx in WT and KO myocytes. A, ICa and ∫ICa in a WT myocyte induced by a WT-clamp. B, Stimulation of the same WT myocyte with a KO-clamp. C and D, Experiment was repeated using a KO myocyte. Experiments shown are representative for 9 WT and 7 KO myocytes. Integral is given in arbitrary units. For quantification of data, see Table.
Figure 7.
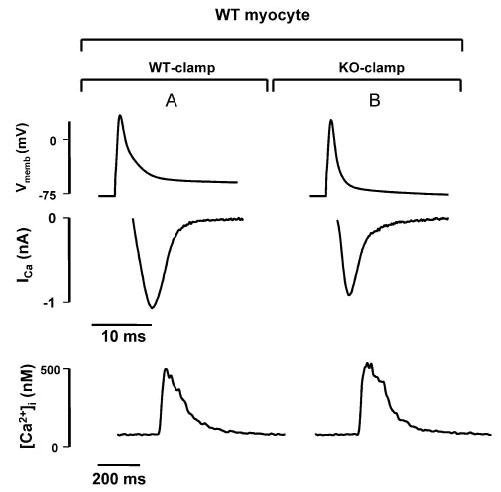
WT and KO AP voltage-clamp commands are equally effective at triggering SR Ca2+ release. Representative tracings for ICa (middle) and [Ca2+]i (bottom) during application of voltage clamps with WT and KO AP waveforms (top) to the same WT myocyte. Although ∫ICa and, therefore, the amount of trigger Ca2+ entering the cell is clearly reduced during KO-clamp (B) as compared with WT-clamp (A), the corresponding Δ[Ca2+]i is similar. Similar observations were made in 7 WT and 3 KO myocytes.
Figure 6 shows KO and WT AP waveform commands (top) and the resulting ICa (middle) in both WT and KO myocytes. The corresponding integral of ICa (∫ICa, bottom) is a measure of the amount of Ca2+ entering the myocyte during the waveform command. First, we compared ICa and ∫ICa in response to the 2 different waveforms in WT myocytes (Figure 6A and 6B; n=9). ICa generated by KO-clamp (Figure 6B) was decreased in amplitude and duration as compared with ICa generated by WT-clamp in the same myocyte (Figure 6A). This led to a reduction of ∫ICa to 41±5% during KO-clamps.
The experiment was repeated using KO myocytes (Figure 6C and 6D; n=7). We have already demonstrated that ICa is reduced in KO myocytes during voltage clamps (see Figures 3 and 4). Consistent with this finding, we observed smaller ICa during WT-clamps in KO myocytes (Figure 6C) compared with WT-clamps in WT cells (Figure 6A). Application of a KO-clamp further reduced ICa amplitude and duration in KO myocytes (Figure 6D).
The summary data in the Table show that in both WT and KO myocytes, the effect of the different command waveforms on the amplitude of ICa was modest. However, there was a significant decrease in time to peak ICa as well as time to 50% inactivation (t1/2) of ICa during KO-clamp. The net result is a substantial reduction of ∫ICa. During KO-clamps applied to KO myocytes (Figure 6D; n=7), ∫ICa was 19±4% of ∫ICa recorded in WT myocytes during stimulation with WT-clamps (Figure 6A; n=9). Thus, the shape and duration of the KO-AP reduces Ca2+ influx.
To assess the effect of the altered AP of KO myocytes on SR Ca2+ release, we also monitored [Ca2+]i during the WT- and KO-clamps (Figure 7). Although the KO-clamp results in less Ca2+ influx via ICa, triggered SR Ca2+ release was similar to the release obtained when the same cell was stimulated with a WT-clamp (Δ[Ca2+]i in WT cells during KO-clamp: 96±11% of Δ [Ca2+]i during WT-clamp; n=7). Similar results were obtained in KO myocytes stimulated with KO- and WT-clamps (n=3).
Discussion
NCX is the dominant Ca2+ efflux mechanism in cardiac myocytes. Yet, intracellular Ca2+ transients and resting Ca2+ levels are normal in ventricular myocytes from adult cardiac-specific NCX KO mice as shown previously7 and in this study. Three possible explanations are (1) an alternative Ca2+ efflux mechanism is upregulated and compensates for loss of the exchanger, (2) Ca2+ influx is reduced and obviates the need for robust Ca2+ extrusion, or (3) a combination of these mechanisms.
If an upregulated alternative Ca2+-extrusion mechanism compensates for the absence of NCX, one would expect the Ca2+-removal rate of KO myocytes to be similar to that of WT myocytes. Instead, we find a dramatic slowing of Ca2+ extrusion from the KO myocytes into the extracellular space (Figure 2). Thus, Ca2+ homeostasis in NCX KO myocytes is not maintained by the upregulation of an alternative Ca2+ efflux mechanism. Ca2+-extrusion capacity in KO myocytes is severely compromised.
Our AP clamp experiments suggest that Ca2+ influx through L-type Ca2+ channels is reduced by 80% in KO myocytes (Figure 6 and the Table). Thus, Ca2+ homeostasis can be maintained by the plasma membrane Ca2+ pump, a low-capacity Ca2+ efflux mechanism. However, reduced Ca2+ entry through L-type Ca2+ channels should trigger less Ca2+-induced Ca2+ release and result in a reduced Ca2+ transient, which is not the case.7 The possible mechanisms underlying the reduced Ca2+ influx and the maintained Ca2+ transient are discussed below.
Characteristics of ICa Under AP Waveform Command Conditions
| WT Myocytes | KO Myocytes | |||
|---|---|---|---|---|
| AP waveform command | WT-Clamp | KO-Clamp | WT-Clamp | KO-Clamp |
| Amplitude, pA/pF | 8.7±1.5 | 7.1±1.8 | 4.4±1.2 | 4.0±1.2 |
| Time to peak, ms | 3.6±0.5 | 1.7±0.2* | 2.6±0.4 | 1.3±0.4* |
| t1/2, ms | 6.3±0.7 | 3.7±0.4* | 4.8±0.7 | 2.4±0.8* |
| ∫I Ca, % | 100 | 40.7±4.8* | 32.8±9.8 | 19.0±4.5*† |
Amplitude, time to peak, time to 50% decrease (t0.5), and integral (∫ICa) of ICa during application of voltage clamp command waveforms mimicking APs in WT (n=9) and KO (n=7) myocytes. ICa amplitude and ∫ICa are normalized to cell capacitance. ∫ICa is expressed as a percentage of ∫ICa from WT myocytes stimulated with a WT-clamp.
P<0.05 vs WT-clamp on the same myocytes,
P<0.05 vs WT-clamp in WT myocytes.
Mechanism of Decreased Ca2+ Entry Into KO Myocytes
Two mechanisms contribute to the reduced ICa of KO myocytes. First, ICa is reduced in KO myocytes by up to 60% as compared with WT myocytes (Figure 4). The mechanism underlying this reduction is unknown. DHPR protein is not reduced in KO myocytes,7 and thus regulatory changes in ICa must be responsible. An attractive possibility is that the absence of NCX results in a tonic increase in diadic cleft Ca2+, resulting in a steady-state Ca2+-dependent inactivation of the L-type Ca2+ channels. We have reported that acute blockade of NCX in rat ventricular myocytes results in increased Ca2+ spark activity,15 which would elevate diadic cleft Ca2+.16,17 We have also shown that NCX activity can acutely regulate ICa in mice that are homozygous for overexpression of NCX.17
Second, the abbreviated spike of the KO-AP further limits Ca2+ influx through Ca2+ channels as shown in Figure 6. Ca2+ entry during KO AP waveform voltage clamps is initially very rapid, probably because of the increased driving force for Ca2+ influx through Ca2+ channels during the more rapid repolarization (Table). This rapid initial phase of influx has important implications for EC coupling, as discussed below. The mechanism underlying the altered KO-AP shape is uncertain. An obvious candidate is loss of inward NCX current, which ordinarily prolongs APD.11–14,18 However, inward NCX current influences the plateau phase of the AP rather than the initial spike. Loss of an initial outward NCX current would tend to broaden, rather than narrow the AP spike. Reduced ICa could also contribute to the shorter APD in KO myocytes, but, again, ICa is more likely to influence the plateau phase. Transient outward current (Ito) is primarily responsible for early AP repolarization,19–23 and thus upregulation of Ito may account for the more rapid initial repolarization of the AP in NCX KO myocytes. There is evidence that the expression of K+-channel subunits that generate Ito is dependent on cytosolic Ca2+.24 Perhaps Ito is increased in KO myocytes secondary to altered Ca2+ handling. Further study is required to investigate the mechanism of changes in AP shape in KO myocytes.
Normal Ca2+ Transients and Increased EC Coupling Gain in the Absence of NCX
How can the amplitude of the Ca2+ transient be unaltered in KO myocytes when the trigger for SR Ca2+ release is severely reduced? We observed no increase in SR Ca2+ load in KO cells (Figure 2) that might compensate for the reduced trigger. Recently, it has been shown that APs trigger Ca2+ sparks, the elementary events of EC coupling, with a probability approaching 100%.15 This high probability of triggering suggests that each couplon must contain multiple L-type Ca2+ channels. Loss of some Ca2+ channels does not necessarily result in reduction of spark activation if a minimum number of channels remains to ensure triggering. Thus, reduced availability of channels, manifested as a decrease in whole cell current, could be tolerated without compromising the Ca2+ transient. The net effect is an increase in macroscopic gain in KO myocytes.
Alternatively, increased diadic cleft Ca2+ caused by the absence of the exchanger may facilitate opening of RyRs, leading directly to an increased gain of EC coupling. That is, an increase in diadic cleft Ca2+ may bring RyRs closer to the threshold for opening when additional Ca2+ enters through L-type Ca2+ channels.15 This increased sensitivity of RyRs to Ca2+ influx would be expected to reduce spark latency and variance on depolarization because early openings of Ca2+ channels will be more likely to trigger associated RyRs in a couplon.25
A further possibility is that increased gain in KO myocytes may be attributable to a spatial rearrangement of key proteins involved in EC coupling. The absence of NCX proteins from the diadic cleft could reduce the average distance between L-type Ca2+ channels and RyRs, possibly enhancing the interaction between the 2 proteins. Although this is highly speculative, a similar argument has been invoked in models of heart failure, where a decrease in gain has been attributed to a hypothesized increase in the distance between L-type Ca2+ channels and RyRs.26,27
The kinetics of Ca2+ entry during APs also increases the efficiency of Ca2+-induced Ca2+ release and, therefore, gain. As noted above, Ca2+ entry during KO AP waveform voltage clamps is initially more rapid, most likely attributable to the high-temporal synchronization of Ca2+ influx through open Ca2+ channels and the greater driving force during more rapid early repolarization.28 These results are consistent with studies showing that increased rate of repolarization can improve the efficiency of triggering of SR Ca2+ release.28,29
Finally, we cannot exclude the possibility that the altered ICa and EC-coupling gain in KO myocytes are caused by the up- or downregulation of specific regulatory pathways that could alter the phosphorylation state of the DHPR or modify RyR gating. However, at present we have no evidence to support or refute these hypotheses.
Comparison With Homozygous NCX Overexpressing Mice
We have previously reported an increase in ICa and a decrease in EC-coupling gain in homozygous NCX-overexpressing mice.17 These mice have essentially normal cardiac function despite the almost 3-fold increase in NCX activity. It is noteworthy that Ca2+ influx in these animals is substantially increased, thereby compensating for increased Ca2+ extrusion by the exchanger. Essentially, this is the mirror image to the alterations in fluxes we observe in NCX KO mice, which respond to a large reduction in Ca2+-efflux capacity with an 80% reduction in Ca2+ influx.
Conclusions
Altogether, EC coupling in adult NCX KO myocytes is characterized by (1) a decreased Ca2+ entry along with an increase in EC coupling gain and (2) a faster repolarizing AP that increases the efficiency of SR triggering. These mechanisms independently, but synergistically, limit Ca2+ influx and at the same time maintain effective Ca2+-induced Ca2+ release from the SR. An extraordinary situation is created in which transsarcolemmal Ca2+ fluxes in KO myocytes are reduced to 20% of those in WT myocytes. This provides an explanation for maintenance of Ca2+ homeostasis in the absence of NCX. Depending on species, the PMCA has been estimated to provide 10% to 25% of myocyte Ca2+ efflux.30–32 Thus, a Ca2+ influx reduced to 20% as observed in NCX KO myocytes is within the capacity of the PMCA. Our findings demonstrate a remarkable adaptive plasticity of cardiac EC coupling. Details of the mechanisms underlying these adaptations are of fundamental importance for understanding the regulation of cardiac EC coupling.
Acknowledgments
This research was supported by Köln Fortune and the German Research Foundation (DFG PO 1004\1) (to C.P.), NIH grants HL48509 (to K.D.P.) and HL70828 (to J.I.G.), and the Laubisch Foundation. We appreciate the technical assistance of M. Yip and N. Huyhn and helpful discussions with Drs R. Olcese and L. Xie.
References
- 1.Bridge JH, Smolley JR, Spitzer KW. The relationship between charge movements associated with ICa and INa-Ca in cardiac myocytes. Science. 1990;248:376–378. doi: 10.1126/science.2158147. [DOI] [PubMed] [Google Scholar]
- 2.Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 3.Philipson KD, Nicoll DA. Sodium-calcium exchange: a molecular perspective. Annu Rev Physiol. 2000;62:111–133. doi: 10.1146/annurev.physiol.62.1.111. [DOI] [PubMed] [Google Scholar]
- 4.Conway SJ, Kruzynska-Frejtag A, Wang J, Rogers R, Kneer PL, Chen H, Creazzo T, Menick DR, Koushik SV. Role of sodium-calcium exchanger (ncx1) in embryonic heart development: a transgenic rescue? Ann N Y Acad Sci. 2002;976:268–281. doi: 10.1111/j.1749-6632.2002.tb04749.x. [DOI] [PubMed] [Google Scholar]
- 5.Pott C, Goldhaber JI, Philipson KD. Genetic manipulation of cardiac Na+/Ca2+ exchange expression. Biochem Biophys Res Commun. 2004;322:1336–1340. doi: 10.1016/j.bbrc.2004.08.038. [DOI] [PubMed] [Google Scholar]
- 6.Reuter H, Henderson SA, Han T, Mottino GA, Frank JS, Ross RS, Goldhaber JI, Philipson KD. Cardiac excitation-contraction coupling in the absence of Na+-Ca2+ exchange. Cell Calcium. 2003;34:19–26. doi: 10.1016/s0143-4160(03)00018-6. [DOI] [PubMed] [Google Scholar]
- 7.Henderson SA, Goldhaber JI, So JM, Han T, Motter C, Ngo A, Chantawansri C, Ritter MR, Friedlander M, Nicoll DA, Frank JS, Jordan MC, Roos KP, Ross RS, Philipson KD. Functional adult myocardium in the absence of Na+-Ca2+ exchange: cardiac-specific knockout of ncx1. Circ Res. 2004;95:604–611. doi: 10.1161/01.RES.0000142316.08250.68. [DOI] [PubMed] [Google Scholar]
- 8.Mitra R, Morad M. A uniform enzymatic method for dissociation of myocytes from hearts and stomachs of vertebrates. Am J Physiol. 1985;249:H1056–H1060. doi: 10.1152/ajpheart.1985.249.5.H1056. [DOI] [PubMed] [Google Scholar]
- 9.Goldhaber JI, Parker JM, Weiss JN. Mechanisms of excitation-contraction coupling failure during metabolic inhibition in guinea-pig ventricular myocytes. J Physiol. 1991;443:371–386. doi: 10.1113/jphysiol.1991.sp018838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 11.Noble D. Influence of Na/Ca exchange stoichiometry on model cardiac action potentials. Ann N Y Acad Sci. 2002;976:133–136. doi: 10.1111/j.1749-6632.2002.tb04731.x. [DOI] [PubMed] [Google Scholar]
- 12.Spencer CI, Sham JS. Effects of Na+/Ca2+ exchange induced by SR Ca2+ release on action potentials and afterdepolarizations in guinea pig ventricular myocytes. Am J Physiol Heart Circ Physiol. 2003;285:H2552–H2562. doi: 10.1152/ajpheart.00274.2003. [DOI] [PubMed] [Google Scholar]
- 13.Schouten VJ, ter Keurs HE, Quaegebeur JM. Influence of electrogenic Na/Ca exchange on the action potential in human heart muscle. Cardiovasc Res. 1990;24:758–767. doi: 10.1093/cvr/24.9.758. [DOI] [PubMed] [Google Scholar]
- 14.Schouten VJ, ter Keurs HE. The slow repolarization phase of the action potential in rat heart. J Physiol. 1985;360:13–25. doi: 10.1113/jphysiol.1985.sp015601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goldhaber JI, Lamp ST, Walter DO, Garfinkel A, Fukumoto GH, Weiss JN. Local regulation of the threshold for calcium sparks in rat ventricular myocytes: role of sodium-calcium exchange. J Physiol. 1999;520(pt 2):431–438. doi: 10.1111/j.1469-7793.1999.00431.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Viatchenko-Karpinski S, Gyorke S. Modulation of the Ca2+-induced Ca2+ release cascade by beta-adrenergic stimulation in rat ventricular myocytes. J Physiol. 2001;533:837–848. doi: 10.1111/j.1469-7793.2001.t01-1-00837.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reuter H, Han T, Motter C, Philipson KD, Goldhaber JI. Mice overexpressing the cardiac sodium-calcium exchanger: defects in excitation-contraction coupling. J Physiol. 2004;554:779–789. doi: 10.1113/jphysiol.2003.055046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yao A, Zu Z, Nonaka A, Lu L, Zubair I, Philipson KD, Bridge JH, Barry WH. Effects of overexpression of the Na+/Ca2+-exchanger on Ca2+ transientsin murine ventricular myocytes. Circ Res. 1998;82:657–665. doi: 10.1161/01.res.82.6.657. [DOI] [PubMed] [Google Scholar]
- 19.Xu Y, Zhang Z, Timofeyev V, Sharma D, Xu D, Tuteja D, Dong PH, Ahmmed GU, Ji Y, Shull GE, Periasamy M, Chiamvimonvat N. The effects of intracellular Ca2+ on cardiac K+ channel expression and activity: novel insights from genetically altered mice. J Physiol. 2005;562:745–758. doi: 10.1113/jphysiol.2004.076216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brouillette J, Clark RB, Giles WR, Fiset C. Functional properties of K+ currents in adult mouse ventricular myocytes. J Physiol. 2004;559:777–798. doi: 10.1113/jphysiol.2004.063446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barry DM, Nerbonne JM. Myocardial potassium channels: electrophysiological and molecular diversity. Annu Rev Physiol. 1996;58:363–394. doi: 10.1146/annurev.ph.58.030196.002051. [DOI] [PubMed] [Google Scholar]
- 22.Nuss HB, Marban E. Electrophysiological properties of neonatal mouse cardiac myocytes in primary culture. J Physiol. 1994;479:265–279. doi: 10.1113/jphysiol.1994.sp020294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oudit GY, Kassiri Z, Sah R, Ramirez RJ, Zobel C, Backx PH. The molecular physiology of the cardiac transient outward potassium current (Ito) in normal and diseased myocardium. J Mol Cell Cardiol. 2001;33:851–872. doi: 10.1006/jmcc.2001.1376. [DOI] [PubMed] [Google Scholar]
- 24.Perrier E, Perrier R, Richard S, Benitah JP. Ca2+ controls functional expression of the cardiac K+ transient outward current via the calcineurin pathway. J Biol Chem. 2004;279:40634–40639. doi: 10.1074/jbc.M407470200. [DOI] [PubMed] [Google Scholar]
- 25.Inoue M, Bridge JH. Variability in couplon size in rabbit ventricular myocytes. Biophys J. 2005;89:3102–3110. doi: 10.1529/biophysj.105.065862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gomez AM, Guatimosim S, Dilly KW, Vassort G, Lederer WJ. Heart failure after myocardial infarction: altered excitation-contraction coupling. Circulation. 2001;104:688–693. doi: 10.1161/hc3201.092285. [DOI] [PubMed] [Google Scholar]
- 27.Gomez AM, Valdivia HH, Cheng H, Lederer MR, Santana LF, Cannell MB, McCune SA, Altschuld RA, Lederer WJ. Defective excitation-contraction coupling in experimental cardiac hypertrophy and heart failure. Science. 1997;276:800–806. doi: 10.1126/science.276.5313.800. [DOI] [PubMed] [Google Scholar]
- 28.Zahradnikova A, Kubalova Z, Pavelkova J, Gyorke S, Zahradnik I. Activation of calcium release assessed by calcium release-induced inactivation of calcium current in rat cardiac myocytes. Am J Physiol Cell Physiol. 2004;286:330–341. doi: 10.1152/ajpcell.00272.2003. [DOI] [PubMed] [Google Scholar]
- 29.Sah R, Ramirez RJ, Oudit GY, Gidrewicz D, Trivieri MG, Zobel C, Backx PH. Regulation of cardiac excitation-contraction coupling by action potential repolarization: role of the transient outward potassium current (Ito) J Physiol. 2003;546:5–18. doi: 10.1113/jphysiol.2002.026468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lamont C, Eisner DA. The sarcolemmal mechanisms involved in the control of diastolic intracellular calcium in isolated rat cardiac trabeculae. Pflugers Arch. 1996;432:961–969. doi: 10.1007/s004240050223. [DOI] [PubMed] [Google Scholar]
- 31.Bassani JW, Bassani RA, Bers DM. Relaxation in rabbit and rat cardiac cells: species-dependent differences in cellular mechanisms. J Physiol. 1994;476:279–293. doi: 10.1113/jphysiol.1994.sp020130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bassani RA, Bassani JW, Bers DM. Relaxation in ferret ventricular myocytes: role of the sarcolemmal Ca ATPase. Pflugers Arch. 1995;430:573–578. doi: 10.1007/BF00373894. [DOI] [PubMed] [Google Scholar]