

Abstract
A global scan of transcription factor usage in the sea urchin embryo was carried out in the context of the S.purpuratus genome sequencing project, and results from six individual studies are here considered. Transcript prevalence data were obtained for over 280 regulatory genes encoding sequence-specific transcription factors of every known family, but excluding genes encoding zinc finger proteins. This is a statistically inclusive proxy for the total “regulome” of the sea urchin genome. Close to 80% of the regulome is expressed at significant levels by the late gastrula stage. Most regulatory genes must be used repeatedly for different functions as development progresses. An evolutionary implication is that animal complexity at the stage when the regulome first evolved was far simpler than even the last common bilaterian ancestor, and is thus of deep antiquity.
Keywords: Regulome, Transcription factor usage, Indirect development
Concepts of the evolutionary origins of bilaterian animals have been transformed by the results of genome sequencing. A most important result is that all bilaterian animals share a common qualitative repertoire of genes encoding sequence-specific transcription factors and signaling proteins, the “bilaterian regulatory toolkit”. These genes are the essential constituents of the developmental gene regulatory networks that underlie development of the body plan. The concept of a “regulatory toolkit” is now firmly established (Davidson, 2006; Erwin and Davidson, 2002), and the evidence from the new sea urchin genome sequence provides much further support (The Sea Urchin Sequencing Consortium, 2006). Every developmentally utilized signaling system, and with only a few exceptions, every subfamily of every class of transcription factor found in vertebrates and ecdysozoans is also represented in this non-chordate deuterostome genome as well. The regulatory toolkits of different bilaterians genomes differ mainly in the number of members of given gene subfamilies. Cnidarians as well share at least a large fraction of this same toolkit (Martindale et al., 2004; Seipel and Schmid, 2005). Cnidarians are also complex animals, however, which are more similar to bilaterians than once thought, and in geologic time they may have diverged from the bilaterian stem lineage not long before the bilaterians themselves diversified (Peterson et al., 2004). The existence of a shared bilaterian regulatory gene toolkit brings into focus the following question: did the regulatory toolkit, the “regulome,” evolve concomitantly with the complex adult body plans of bilaterians (or of cnidarians/bilaterians)? This would allow the hypothesis that the evolutionary assembly of the toolkit repertoire per se might have been causal with respect to the appearance of animals of the bilaterian grade of morphological complexity. Or, did the regulome predate complex animal forms? This allows the alternative hypothesis that bilaterian evolution followed from increasingly elegant modes of toolkit utilization, rather than invention and qualitative diversification of the toolkit itself. In mechanistic terms these alternatives at root amount to evolution of animal complexity driven mainly by the appearance of new genes, vs. evolution of animal complexity driven mainly by appearance of new regulatory linkages among pre-existing genes.
The sea urchin genome sequence provides a unique opportunity to address this issue. This is the only genome so far sequenced from an organism that utilizes maximum indirect development (Peterson et al., 1997). Here the primary role of the embryo is to produce a larva, which provides a life support system for the postembryonic development of the adult body plan. The body parts of the adult form later develop within the larva, from cell populations that had been set aside from embryological specification and differentiation process. In direct development, on the other hand, the primary object of embryogenesis is construction of the adult plan as immediately as possible. The embryo/larva of indirectly developing forms may possess very little similarity to the adult body plan, and are typically far simpler in structure and complexity than any adult bilaterian body plan. Morphological simplicity is an obvious character of the S. purpuratus embryo (Fig. 1). Early in cleavage the embryo blastomeres begin to express distinct sets of genes signifying the process of regulatory specification. Territories of gene expression which are also territories of prospective cell fate are color-coded in Figure 1. But in the end the embryo remains a relatively simple structure, consisting of only 10-15 cell types. In contrast to all adult bilaterian forms and all directly developing bilaterian embryos, the sea urchin embryo consists exclusively of single cell thick epithelial layers, and individual mesenchymal cells (Fig.1). It has no mesodermal tissue layers, nor organs, nor body parts formed from mesoderm plus ectoderm or endoderm.
Fig. 1.

Specification in the sea urchin embryo. Color coded tracings from from photomicrographs of the embryo of Strongylocentrotus purpuratus are shown (A. Ransick and E. Davidson; reproduced from Davidson, 2006). Veg1 and veg2 are rings of 8 cells each, arising from their parental cells at the horizontal 6th cleavage. From veg1 derives ectoderm plus (mainly) hindgut endoderm; and from veg2 nonskeletogenic (secondary) mesenchyme (mesodermal cell types) plus gut endoderm. Skeletogenic mesenchyme lineage, red; endoderm, blue; secondary mesenchyme, violet; oral ectoderm, yellow; apical oral ectoderm, hatched yellow; aboral ectoderm, green; unspecified cells, white. 6 and 10 hr, cleavage stages; 15 hr, blastula stage; 20 and 24 hr, mesenchyme blastula; 30, 33, 38 hr, gastrula stages; 55 hr, late gastrula or “prism” stage.
Regulome utilization in embryogenesis
In the course of the S. purpuratus genome project all genes encoding recognizable transcription factors were identified and annotated, and their expression during embryonic development was measured quantitatively. Here we have tabulated these gene expression data and reduced them to a common format for analysis. Included are the forkhead genes (Tu et al., 2006), the ets genes (Rizzo et al., 2006), the hox and parahox genes (Arnone et al., 2006), all other homeobox genes (Howard-Ashby et al., 2006b), the nuclear hormone receptor genes, bhlh, smad, tbox, basic zipper, and sox transcription factor genes, as well as members of other smaller regulatory gene families (Howard-Ashby et al., 2006a). In addition, prior knowledge was incorporated, particularly the large number of regulatory genes encompassed in the endomesoderm gene regulatory network for S. purpuratus.(Davidson, 2006; Levine and Davidson, 2005). This network indicates the genomically encoded regulatory logic underlying the specification of the mesoderm, endoderm, and skeletogenic domain of the embryo, that is, those parts colored red, blue, and purple in Fig.1. A recent version of the network is shown in Fig.2 (the network is available in continuously updated form at http://sugp.caltech.edu/endomes/). Given the genome-wide gene prediction analysis (The Sea Urchin Sequencing Consortium, 2006), the concordance of an entirely independent search for regulatory genes (Howard-Ashby et al., 2006b), and of a whole genome tiling array analysis of the embryo transcriptome (Samanta et al., 2006), most DNA-binding transcription factors of known families have evidently been identified. At the very least, the 283 genes included here represent a very large, unbiased sampling of genes encoding transcription factors in the S. purpuratus genome.
Fig.2.

Gene regulatory network (GRN) for endomesoderm specification in sea urchin embryos. GRN for period from initiation of zygotic regulatory control shortly after fertilization to just before gastrulation (∼6-30 h). The short horizontal lines represent relevant cis-regulatory modules of indicated genes on which the color coded inputs impinge. The sources of these inputs are other genes of the GRN, as indicated by the thin colored lines. Small open and closed circles represent protein-protein interactions that occur off the DNA and are not included explicitly in the GRN, the objective of which is to display the predicted genomic regulatory organization responsible for spatial and temporal expression of the genes it includes. For symbolism, explanations, and access to the BioTapestry software by which the GRN is built and maintained see (http://sugp.caltech.edu/endomes/webstart/bioTapestry.jnlp), where current version of GRN is posted.
Zinc finger genes were specifically excluded from the compilation considered here because it is difficult at present to generate a comparable high confidence gene set from this class of genomic sequences. Zinc finger motifs have proven difficult to group into subfamilies and to analyze phylogenetically (Knight and Shimeld, 2001). For most genes that encode C2H2 Zn finger domains it is impossible to identify clear orthologues known to function as regulatory genes in other species, or even to know whether all such domains identified in the genome have been correctly included in gene models. It is often unclear whether given domains represent splice variants, distinct genes, or assembly errors. Another difficulty is that not all C2H2 zinc finger proteins are transcription factors, as proteins including these domains have been demonstrated to function in RNA binding and in protein-protein interactions (Laity et al., 2001; Lu et al., 2003). Illustrating this uncertainty, of the approximately 380 C2H2 Zn finger genes identified in S. purpuratus, nearly 40 have only one zinc finger domain (Materna et al., 2006), but at least two such domains are required for DNA binding specificity. A comprehensive set of true and unique zinc finger regulatory genes cannot be defined on the basis of genomic sequence and expression data alone. In contrast, identification of most other classes of DNA binding domain in the regulome is unequivocal, given their high conservation and clear orthology across the Bilateria. We therefore took genes encoding all DNA sequence specific transcription factors other than zinc finger factors to be representative of the total regulome, and considered their deployment in embryonic development.
Quantitative PCR (QPCR) was used to determine the expression profile of each of the 283 regulatory genes, from fertilization to 48 h post-fertilization (Howard-Ashby et al., 2006a, b; Rizzo et al., 2006; Tu et al., 2006). This is an extremely sensitive and accurate method which enables the detection of <1 molecule of mRNA per cell. In addition the spatial patterns of expression were determined for all genes expressed sufficiently to permit in situ hybridization (>5-10 copies per cell). The number of regulatory genes in each transcription factor family expressed only maternally; expressed maternally and zygotically at constant levels; activated zygotically during embryogenesis; or remaining silent or expressed at extremely low, insignificant levels by 48 h is collated in Table 1. The threshold of biologically significant expression was set, conservatively, at 150-350 molecules of mRNA per embryo, as follows: From late cleavage onward in the sea urchin embryo the populations expressing given regulatory states are all at least 16 cells, and by gastrula stage the largest territories are 60-200 cells. Thus at 350 mRNAs per embryo there would be 2-20 mRNAs per cell for territorially specific messages. In these embryos the rate of translation is two molecules of protein/mRNA-min (Davidson, 1986), and so within a few hours these threshold mRNA concentrations could suffice for production of the several hundred to few thousand molecules of transcription factor per cell required for significant target site occupancy (Bolouri and Davidson, 2003; Calzone et al., 1988). Factual observations support these arguments. Thus studies on expression of functional genes in the endomesoderm network of Fig.2 show that functionally essential regulatory gene transcript concentrations range indeed from a few to only about 40 molecules of mRNA per cell. The 150-350 molecule per embryo threshold thus represents a functional level of expression, though close to a minimal one. In any case, however, the great majority of the mRNAs with which we are here concerned are present either at >1,000 molecules or zero-10 molecules per embryo.
Table 1.
Regulome usage in development by gene family
| Family | Total | M | Z | C | − | % exp | Localizedc |
|---|---|---|---|---|---|---|---|
| hox cluster | 11 | 0 | 2 | 0 | 9 | 18.2 | 2 |
| homeobox | 85 | 0 | 58 | 3 | 24 | 71.8 | 26 |
| T-Box | 6 | 0 | 5 | 0 | 1 | 83.3 | 3 |
| smad | 4 | 0 | 4 | 0 | 0 | 100 | 1 |
| forkhead | 22 | 1 | 20 | 0 | 1 | 95.5 | 20 |
| Sox/HMG | 10 | 1 | 5 | 2 | 2 | 80.0 | 3 |
| bHLH | 48a | 0 | 24 | 2 | 17 | 59.5 | 5 |
| ets | 11 | 0 | 10 | 0 | 1 | 90.9 | 4 |
| bZip | 14a | 0 | 9 | 2 | 2 | 84.6 | 2 |
| nuclear receptor | 33 | 0 | 22 | 1 | 10 | 69.7 | 4 |
| other types | 45 | 1 | 37 | 4 | 3 | 93.3 | 10 |
| all genes | 283 | 3 | 196 | 14 | 70 | 75.3/ 77.6b | 76 |
No expression data is reported for five bHLH genes and one bZip gene.
Statistic is recalculated omitting the hox cluster genes.
Only genes with sufficient expression to likely be detectable were examined by in situ hybridization.
M, maternal expression; Z, zygotic expression; C, constant expression; (−), no expression at or above threshold of 150-350 molecules per whole embryo.
The majority of all regulatory genes in the sample have been activated by late gastrula stage. More than 80% of members of the forkhead, ets, bZip, smad, sox, and many other families are utilized in the embryo by 48 h post-fertilization (Table 1). The largest family, the non-hox homeobox genes, are >70% expressed by late gastrula. Only the nuclear receptor and bHLH families are expressed at somewhat lower levels, but the majority of even these have been activated by 48 h. The hox genes are a special case. As predicted (Davidson, 1990) and later experimentally demonstrated (Arenas-Mena et al., 1998), the hox cluster as such is not utilized until formation of the adult body plan in postembryonic sea urchin development (Arenas-Mena et al., 2000). Only two of the 11 hox cluster genes are expressed during embryogenesis. Since the hox cluster is utilized as a functional unit, expression of individual hox genes cannot be considered as statistically independent events. Overall, 75% of the regulome has already been used at least once by late gastrula stage, when development of this embryo is only two-thirds complete. If the hox genes are removed from the calculation, the fraction rises to 77% by 48 h. The cumulative time course of regulome use is plotted in Fig. 3 (green and blue curves).
Fig. 3.
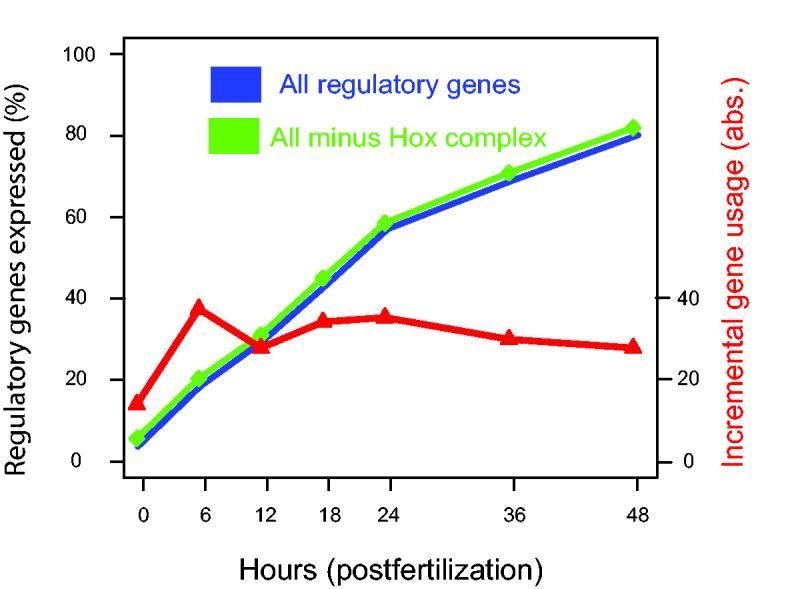
Regulatory gene usage in development. Regulome usage is plotted as a function of developmental time. Data were collated from references cited in text. A total of 283 regulatory genes is included in the analysis. The threshold for biological significance was set at 150-350 copies per embryo (see text). Genes were classified as first activated zygotically at 6, 12, 18, 24, 36, or 48 h postfertilization; or not expressed significantly by late gastrula stage. Genes expressed only maternally or at a constant level including maternal expression are included at the 0 h time point. The blue curve is the percentage of all regulatory genes which have been zygotically expressed by the given time after fertilization. The green curve is the same discounting the genes of the hox complex (see text). The red line (right ordinate) indicates the number of regulatory genes newly activated in each time interval. Transcript levels in each cDNA sample were measured by comparing the QPCR amplification of the target sequence to that of a standard of known concentration in cDNA prepared from embryos of the appropriate stage (cf. primary references for details). A fluorescent reporter dye is used to measure the increasing concentration of the unknown and standard amplicons at the end of every PCR cycle. If the copy number of the standard is known, given that each PCR cycle produces an amplification of approximately 1.9-fold, the embryonic copy number of the unknown can be calculated from the difference in cycle numbers needed to produce an arbitrary fluorescent signal between standard and unknown (Howard-Ashby et al., 2006b). Ubiquitin, which is present at the same concentration at all developmental time points, rRNA, and other constant sequences were used as the internal standards. Data from the S. purpuratus embryonic transcriptome analysis (Samanta et al., 2006) were used for external validation of whether individual genes were truly expressed. For some genes, a slightly different set of time points was used, and the expression at the above time points was extrapolated.
New transcription factors are activated steadily during development (red line in Fig. 3, essentially the experimentally measured derivative of the blue line). Every regulatory gene can be thought of as a node in the gene regulatory network which reads, processes, and transmits spatial and temporal information (Davidson, 2006). A given gene is activated when the correct set of upstream inputs is presented, and the resulting regulatory protein conveys new spatial and temporal cues when it interacts with its cis-regulatory targets in downstream genes. Thus Fig. 3 shows that new information processing nodes are being activated continuously, with concomitant increase in the regulatory complexity of the embryo, even though this is yet not apparent morphologically (Fig. 1). If the integral percent usage plot is projected forward to 72 h when embryogenesis is complete and the larva becomes capable of feeding (Fig. 1C), 95% of the regulome will have been used at least once. Measurements on the forkhead transcription factor family did extend out to 72 h (Tu et al., 2006), and indeed 95.5% of these factors are in play by then.
Why is early development so expensive in regulatory apparatus?
The complexity of the regulatory apparatus required to execute a given developmental process is a system level property, which can only be interpreted accurately by means of a system level functional analysis. The endomesoderm gene regulatory network of Fig.2 is such an analysis (Davidson, 2006; Davidson et al., 2002; Howard and Davidson, 2004; Levine and Davidson, 2005; Oliveri and Davidson, 2004). This network pertains to only part of the embryo, and to only about half of the developmental period from fertilization to late gastrula. It covers the period from about 6 h after fertilization, when spatially confined zygotic regulatory gene expression begins to dominate the developmental process, to mesenchyme blastula stage. Specific regulatory states have by then been established in all its territories (Davidson, 2006), that is, specific sets of regulatory genes are being expressed, but gastrulation has not yet taken place. The endomesoderm network includes the specification of skeletogenic and other mesodermal precursors and of gut endoderm, but it excludes the aboral and oral ectodermal territories, and also the neurogenic apical territory (Fig.1). Between mesenchyme blastula stage and late gastrula much additional development occurs, including the subdivision of the archenteron into fore-, mid- and hind-gut, and of the oral ectoderm into stomodaeal, lateral and ciliary band subdomains, and the 48 h embryo has significantly more diverse parts than it does at mesenchyme blastula stage. Furthermore, the network in Fig.2 is a “driver gene network”, i.e., it is focused on regulatory genes that are expressed in spatially and/or temporally specific ways, since these are the regulatory genes that must execute the control logic which specifies cells differentially in space and time.(Davidson, 2006; Yuh et al., 2001) However, ubiquitous regulatory factors that are also necessary for the normal operation of developmentally active cis-regulatory modules, as shown explicitly for the endo16 control system (Yuh et al., 2001; Yuh et al., 2005), and these are not systematically represented in the endomesoderm network. Despite these limitations in coverage, the endomesoderm gene regulatory network includes > 40 sequence specific regulatory genes.
Specific aspects of regulatory gene usage in the sea urchin endomesoderm network, and in other developmental gene regulatory networks (Koide et al., 2005; Loose and Patient, 2004; Stathopoulos and Levine, 2005), illuminate the need for large regulatory apparatus in embryonic development. First, if a regulatory gene is expressed, it will have a function. If its expression is blocked the expression of downstream genes will be affected and therefore the fractions of regulatory genes expressed as shown in Fig. 3 are likely to be directly meaningful. The functional interactions that are the basis of these networks show that expression of regulatory genes almost always has specific downstream effects and except for specific signal mediated effects on transcription factor activity (as specified in the networks) there is no support for the idea that a subsequent level of control at the translational level is important in these embryonic control systems (Davidson, 2006). Second, individual regulatory genes at the nodes of developmental gene regulatory networks respond to unique sets of inputs, and the outputs they send onwards have unique sets of destinations; i.e., no two nodes do the same things. Therefore the number of nodes represents the number of cis-regulatory input information processing units the network must encompass. This number is never small. Third, individual developmental jobs the network mediates are each performed by modular subcircuits not used elsewhere in that spatial and temporal stage of development, every one of which consists of several regulatory genes. Such jobs include specification of given territories, such as the prospective skeletogenic or gut territory; or operation of given differentiation gene batteries. The endomesoderm network includes many such subcircuits because there are many such jobs to be done.
In short, developmental gene regulatory networks provide a basis for comprehending the high usage of regulatory genes in development. With respect to the sea urchin embryo, the endomesoderm network by itself would predict by extrapolation to the whole embryo at 48h, a quantitative requirement for regulatory gene usage consistent with that shown in Fig. 3.
The regulome in development
It is a commonplace that genes encoding given transcription factors are utilized in multiple times and places during the development of an organism, participating in entirely independent processes. Even within the three days required for sea urchin embryogenesis, many specific regulatory genes have been found to be expressed in a succession of diverse domains where they execute distinct and unrelated functions. For example, the hnf6 gene is initially expressed ubiquitously, when it has targets in many parts of the embryo, then it becomes an oral ectoderm regulator, and later it is required specifically in ciliated band (Otim et al., 2004); the deadringer gene and the goosecoid genes are first utilized in skeletogenic cells and later in oral ectoderm (Amore et al., 2003; Angerer et al., 2001); the diverse regulatory modules of the otx gene drive expression in many different domains of the embryo (Yuh et al., 2002); the “early” and “late” modules of the blimp1/krox gene respectively control a dynamic pattern of expression in cleavage stage endomesoderm, and later contribute to a dedicated midgut/hindgut regulatory state in the invaginated archenteron (Livi and Davidson, 2006).
Here we see that repeated reutilization must indeed be the overwhelming majority pattern of regulatory gene utilization. This implication follows directly from the finding that most regulatory genes are required for development just to the late gastrula stage. The embryo itself will become significantly more complex after this stage, with the elaboration of its nervous system, the development of the stomodaeum, the ciliated band, the coelomic pouches, the tripartite gut, and so forth. But the development of the adult body plan in postembryonic development dwarfs the whole of the embryonic process in the complexity of its multilayered morphology, and its numerous new cell types. The regulome from which are constituted the many developmental gene regulatory networks required to organize adult body plan development must be the same regulome required to make the gastrula, for there is no more, save the 20-25% of regulatory genes not yet deployed by this stage. Some of the regulatory genes not used in the embryo up to gastrula stage have specific roles. For example, a cohort of these genes is expressed specifically in oogenesis (Song et al., 2006); and most of the genes of the hox complex are silent until activation in the course of formation of the adult body plan in postembryonic larval development (Arenas-Mena et al., 2000). What is perhaps unexpected is that such a small fraction of the regulome is dedicated to such “special purposes.”
The conclusions, then, are that even simple territorial specification functions require complex networks of many genes of multiple transcription factor families; and that more complex later development is driven by recursive utilization of the same regulatory genes. These same conclusions must inform consideration of early animal evolution as well.
The regulome in evolution
A “minimalist” interpretation of the last common bilaterian ancestor, based on the logic of incontrovertibly shared characters, provides an image of a creature much simpler in morphological organization than any modern bilaterian. It must have had a tripartite through gut (foregut, midgut, hindgut), bilateral anterior/ posterior nervous system organization, organ grade internal body parts perhaps including heart (Erwin and Davidson, 2002), and mesodermal layers, used both as major functional and structural components of the body and for developmental signaling interactions with endodermal and ectodermal layers. But such an organism would have been very significantly more complex than embryos of animals such as the sea urchin: these have no organ level structures nor mesodermal layers, only a few types of free-wandering mesodermal cells and some muscular sphincters in the gut. Such embryos do generate bilateral anterior/posterior organization and tripartite gut with mouth and anus. Because it had very significantly more diverse morphology, the last common bilaterian ancestor must necessarily have required for its development a more extensive and elaborated genomic regulatory apparatus, more and deeper networks of regulatory gene interactions encoded in its genome, than does the embryonic phase of modern indirect development.
The palaeontological record of bilaterian origins is famously enigmatic, though in recent years valuable clues have accumulated. Molecular phylogeny based on calibrated protein divergence rates across the Bilateria indicate that bilaterian divergence from a common ancestral lineage probably occurred after the Marinoan Glaciation (Aris-Brosou and Yang, 2003; Douzery et al., 2004; Peterson et al., 2004); the last of the world wide snowball earth episodes which ended about 630 mya, i.e., 70 million years before the beginning of the Cambrian (Peterson and Butterfield, 2005). A variegated assemblage of microfossils from S. W. China dating to about 590 mya, includes a large variety of eggs and embryos that have earmarks of bilaterian forms, such as distinctive patterns of unequal cleavage (Chen et al., 2006; Chen et al., 2000; Dornbos et al., 2005; Xiao and Knoll, 1999). Among these microfossils is a complex, unusually well preserved form that has unmistakable bilaterian structural features (Chen et al., 2004). Later on, by 10 or 15 million years before the beginning of the Cambrian at 542 mya, there appear trace fossils, bore holes in the benthic deposits that were undoubtedly made by bilaterian animals (Bottjer et al., 2000), and also the first macroscopic bilaterian body fossils, such as the complex, mollusk-like Kimberella (Fedonkin and Waggoner, 1997).
What was the nature of the Precambrian genomic landscape in which the Bilateria originated; how complex was it? In terms of cellular organization, the simplest current free living bilaterian forms, the larvae of maximally indirectly developing animals, lack distinctive features of the last common bilaterian ancestor and are much less complicated. It is here entirely irrelevant whether the gene regulatory networks directing the development of such larval forms are themselves evolutionary “simplifications” adaptively derived for the ecological conditions of larval life; or on the other hand, are plesiomorphic survivals of early evolving gene regulatory networks for generation of simple organisms. For, the evidence in Fig. 3 shows that the large majority of the shared bilaterian regulome is required for the mechanism of development of the mere gastrula of an indirectly developing animal. It follows that the development of forms much simpler than the last common bilaterian ancestor must still have required most of the current bilaterian regulome. Therefore, the bilaterian regulome considered in Fig. 3 is thus at least of Upper Neoproterozoic antiquity.
There is yet no evidence as to how deep in time evolutionary assembly of the regulome occurred, or what was the morphology of the form for the development of which it was deployed. If there was an evolutionary stage when the developmental (organismal) complexity of bilaterian ancestors was driven by the assembly of the regulatory toolkit, it was at a remote period, preceding the last common bilaterian ancestor. Ever since, the evolution of animal form has depended mainly on endless reutilization of the same regulome. This of course means endless reorganization of the genomic regulatory apparatus controlling regulatory gene use; primarily evolution of gene regulatory pathways, not evolution of new kinds of regulatory genes.
Acknowledgements
Research was supported by NIH grant HD37105; DOE grant DE-FG0203ER63584, and NASA/Ames NAG-1587.
References
- Amore G, Yavrouian RG, Peterson KJ, Ransick A, McClay DR, Davidson EH. Spdeadringer, a sea urchin embryo gene required separately in skeletogenic and oral ectoderm gene regulatory networks. Dev. Biol. 2003;261:55–81. doi: 10.1016/s0012-1606(03)00278-1. [DOI] [PubMed] [Google Scholar]
- Angerer LM, Oleksyn DW, Levine AM, Li XT, Klein WH, Angerer RC. Sea urchin goosecoid function links fate specification along the animal-vegetal and oral-aboral embryonic axes. Development. 2001;128:4393–4404. doi: 10.1242/dev.128.22.4393. [DOI] [PubMed] [Google Scholar]
- Arenas-Mena C, Cameron AR, Davidson EH. Spatial expression of Hox cluster genes in the ontogeny of a sea urchin. Development. 2000;127:4631–4643. doi: 10.1242/dev.127.21.4631. [DOI] [PubMed] [Google Scholar]
- Arenas-Mena C, Martinez P, Cameron RA, Davidson EH. Expression of the Hox gene complex in the indirect development of a sea urchin. Proc. Natl. Acad. Sci. U. S. A. 1998;95:13062–13067. doi: 10.1073/pnas.95.22.13062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aris-Brosou S, Yang ZH. Bayesian models of episodic evolution support a late Precambrian explosive diversification of the Metazoa. Mol. Biol. Evol. 2003;20:1947–1954. doi: 10.1093/molbev/msg226. [DOI] [PubMed] [Google Scholar]
- Arnone MI, Rizzo F, Annunciata R, Cameron RA, Peterson KJ. Genetic organization and embryonic expression of the ParaHox genes in the sea urchin S. purpuratus: insights into the relationship between clustering and colinearity. Dev. Biol. 2006 doi: 10.1016/j.ydbio.2006.07.037. in press. [DOI] [PubMed] [Google Scholar]
- Bolouri H, Davidson EH. Transcriptional regulatory cascades in development: Initial rates, not steady state, determine network kinetics. Proc. Natl. Acad. Sci. U. S. A. 2003;100:9371–9376. doi: 10.1073/pnas.1533293100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottjer DJ, Hagadorn JW, Dornbos SQ. The Cambrian substrate revolution. GSA Today. 2000;10:1–7. [Google Scholar]
- Calzone FJ, Theze N, Thiebaud P, Hill RL, Britten RJ, Davidson EH. Developmental Appearance of Factors That Bind Specifically to Cis-Regulatory Sequences of a gene expressed in the sea-urchin embryo. Genes Dev. 1988;2:1074–1088. doi: 10.1101/gad.2.9.1074. [DOI] [PubMed] [Google Scholar]
- Chen JY, Bottjer DJ, Davidson EH, Dornbos SQ, Gao X, Yang YH, Li CW, Li G, Wang XQ, Xian DC, Wu HJ, Hwu YK, Tafforeau P. Phosphatized polar lobe-forming embryos from the Precambrian of Southwest China. Science. 2006;312:1644–1646. doi: 10.1126/science.1125964. [DOI] [PubMed] [Google Scholar]
- Chen JY, Bottjer DJ, Oliveri P, Dornbos SQ, Gao F, Ruffins S, Chi HM, Li CW, Davidson EH. Small bilaterian fossils from 40 to 55 million years before the Cambrian. Science. 2004;305:218–222. doi: 10.1126/science.1099213. [DOI] [PubMed] [Google Scholar]
- Chen JY, Oliveri P, Li CW, Zhou GQ, Gao F, Hagadorn JW, Peterson KJ, Davidson EH. Precambrian animal diversity: Putative phosphatized embryos from the Doushantuo formation of China. Proc. Natl. Acad. Sci. U. S. A. 2000;97:4457–4462. doi: 10.1073/pnas.97.9.4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson EH. Gene Activity in Early Development. 3rd ed. Academic Press; Orlando, FL: 1986. [Google Scholar]
- Davidson EH. How Embryos Work - a Comparative View of Diverse Modes of Cell Fate Specification. Development. 1990;108:365–389. doi: 10.1242/dev.108.3.365. [DOI] [PubMed] [Google Scholar]
- Davidson EH, Rast JP, Oliveri P, Ransick A, Calestani C, Yuh CH, Minokawa T, Amore G, Hinman V, Arenas-Mena C, Otim O, Brown CT, Livi CB, Lee PY, Revilla R, Rust AG, Pan ZJ, Schilstra MJ, Clarke PJC, Arnone MI, Rowen L, Cameron RA, McClay DR, Hood L, Bolouri H. A genomic regulatory network for development. Science. 2002;295:1669–1678. doi: 10.1126/science.1069883. [DOI] [PubMed] [Google Scholar]
- Dornbos SQ, Bottjer DJ, Chen JY, Oliveri P, Gao F, Li CW. Precambrian animal life: Taphonomy of phosphatized metazoan embryos from southwest China. Lethaia. 2005;38:101–109. [Google Scholar]
- Douzery EJP, Snell EA, Bapteste E, Delsuc F, Philippe H. The timing of eukaryotic evolution: Does a relaxed molecular clock reconcile proteins and fossils? Proc. Natl. Acad. Sci. U. S. A. 2004;101:15386–15391. doi: 10.1073/pnas.0403984101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erwin DH, Davidson EH. The last common bilaterian ancestor. Development. 2002;129:3021–3032. doi: 10.1242/dev.129.13.3021. [DOI] [PubMed] [Google Scholar]
- Fedonkin MA, Waggoner BM. The Late Precambrian fossil Kimberella is a mollusc-like bilaterian organism. Nature. 1997;388:868–871. [Google Scholar]
- Howard-Ashby M, Materna S, Brown CT, Chen L, Cameron AR, Davidson EH. Gene families encoding transcription factors expressed in early development of Strongylocentrotus purpuratus. Dev. Biol. 2006a doi: 10.1016/j.ydbio.2006.08.033. in press. [DOI] [PubMed] [Google Scholar]
- Howard-Ashby M, Materna S, Brown CT, Chen L, Cameron AR, Davidson EH. Identification and characterization of homeobox transcription factor genes in S. purpuratus, and their expression in embryonic development. Dev. Biol. 2006b doi: 10.1016/j.ydbio.2006.08.039. [DOI] [PubMed] [Google Scholar]
- Howard ML, Davidson EH. cis-Regulatory control circuits in development. Dev. Biol. 2004;271:109–118. doi: 10.1016/j.ydbio.2004.03.031. [DOI] [PubMed] [Google Scholar]
- Knight RD, Shimeld SM. Identification of conserved C2H2 zinc-finger gene families in the Bilateria. Genome Biol. 2001;2:0016.1–0016.8. doi: 10.1186/gb-2001-2-5-research0016. research. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koide T, Hayata T, Cho KWY. Xenopus as a model system to study transcriptional regulatory networks. Proc. Natl. Acad. Sci. U. S. A. 2005;102:4943–4948. doi: 10.1073/pnas.0408125102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laity JH, Lee BM, Wright PE. Zinc finger proteins: new insights into structural and functional diversity. Curr. Opin. Struct. Biol. 2001;11:39–46. doi: 10.1016/s0959-440x(00)00167-6. [DOI] [PubMed] [Google Scholar]
- Levine M, Davidson EH. Gene regulatory networks for development. Proc. Natl. Acad. Sci. U. S. A. 2005;102:4936–4942. doi: 10.1073/pnas.0408031102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livi CB, Davidson EH. Expression and function of blimp1/krox, an alternatively transcribed regulatory gene of the sea urchin endomesoderm network. Dev. Biol. 2006;293:513–525. doi: 10.1016/j.ydbio.2006.02.021. [DOI] [PubMed] [Google Scholar]
- Loose M, Patient R. A genetic regulatory network for Xenopus mesendoderm formation. Dev. Biol. 2004;271:467–478. doi: 10.1016/j.ydbio.2004.04.014. [DOI] [PubMed] [Google Scholar]
- Lu D, Searles MA, Klug A. Crystal structure of a zinc-finger - RNA complex reveals two modes of molecular recognition. Nature. 2003;426:96–100. doi: 10.1038/nature02088. [DOI] [PubMed] [Google Scholar]
- Martindale MQ, Pang K, Finnerty JR. Investigating the origins of triploblasty: ‘mesodermal’ gene expression in a diploblastic animal, the sea anemone Nematostella vectensis (phylum, Cnidaria; class, Anthozoa) Development. 2004;131:2463–2474. doi: 10.1242/dev.01119. [DOI] [PubMed] [Google Scholar]
- Materna S, Ashby-Howard M, Gray R, Davidson EH. The C2H2 zinc finger genes of Strongylocentrotus purpuratus and their expression in embryonic development. Dev. Biol. 2006 doi: 10.1016/j.ydbio.2006.08.032. in press. [DOI] [PubMed] [Google Scholar]
- Oliveri P, Davidson EH. Gene regulatory network controlling embryonic specification in the sea urchin. Curr. Opin. Genet. Dev. 2004;14:351–360. doi: 10.1016/j.gde.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Otim O, Amore G, Minokawa T, McClay DR, Davidson EH. SpHnf6, a transcription factor that executes multiple functions in sea urchin embryogenesis. Dev. Biol. 2004;273:226–243. doi: 10.1016/j.ydbio.2004.05.033. [DOI] [PubMed] [Google Scholar]
- Peterson KJ, Butterfield NJ. Origin of the Eumetazoa: Testing ecological predictions of molecular clocks against the Proterozoic fossil record. Proc. Natl. Acad. Sci. U. S. A. 2005;102:9547–9552. doi: 10.1073/pnas.0503660102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson KJ, Cameron RA, Davidson EH. Set-aside cells in maximal indirect development: Evolutionary and developmental significance. BioEssays. 1997;19:623–631. doi: 10.1002/bies.950190713. [DOI] [PubMed] [Google Scholar]
- Peterson KJ, Lyons JB, Nowak KS, Takacs CM, Wargo MJ, McPeek MA. Estimating metazoan divergence times with a molecular clock. Proc. Natl. Acad. Sci. U. S. A. 2004;101:6536–6541. doi: 10.1073/pnas.0401670101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzo F, Fernandez-Serra M, Squarzoni P, Archimandritis A, Arnone MI. Identification and developmental expression of the ets gene family in the sea urchin (Strongylocentrotus purpuratus) Dev. Biol. 2006 doi: 10.1016/j.ydbio.2006.08.012. in press. [DOI] [PubMed] [Google Scholar]
- Samanta MP, Tongprasit W, Istrail S, Cameron AR, Tu Q, Davidson EH, Stolc V. High resolution transcriptome map of the sea urchin embryo. Science. 2006 doi: 10.1126/science.1131898. in press. [DOI] [PubMed] [Google Scholar]
- Seipel K, Schmid V. Evolution of striated muscle: Jellyfish and the origin of triploblasty. Dev. Biol. 2005;282:14–26. doi: 10.1016/j.ydbio.2005.03.032. [DOI] [PubMed] [Google Scholar]
- Song JL, Wong JL, Wessel G. Oogenesis: Single cell development and differentiation. Dev. Biol. 2006 doi: 10.1016/j.ydbio.2006.07.041. in press. [DOI] [PubMed] [Google Scholar]
- Stathopoulos A, Levine M. Genomic regulatory networks and animal development. Developmental Cell. 2005;9:449–462. doi: 10.1016/j.devcel.2005.09.005. [DOI] [PubMed] [Google Scholar]
- The Sea Urchin Sequencing Consortium The genome sequence of the purple sea urchin, Strongylocentrotus purpuratus. Science. 2006 in press. [Google Scholar]
- Tu Q, Brown CT, Davidson EH, Oliveri P. Sea urchin forkhead gene family: phylogeny and embryonic expression. Dev. Biol. 2006 doi: 10.1016/j.ydbio.2006.09.031. in press. [DOI] [PubMed] [Google Scholar]
- Xiao SH, Knoll AH. Fossil preservation in the Neoproterozoic Doushantuo phosphorite Lagerstatte, South China. Lethaia. 1999;32:219–240. doi: 10.1111/j.1502-3931.1999.tb00541.x. [DOI] [PubMed] [Google Scholar]
- Yuh CH, Bolouri H, Davidson EH. Cis-regulatory logic in the endo16 gene: switching from a specification to a differentiation mode of control. Development. 2001;128:617–629. doi: 10.1242/dev.128.5.617. [DOI] [PubMed] [Google Scholar]
- Yuh CH, Brown CT, Livi CB, Rowen L, Clarke PJC, Davidson EH. Patchy interspecific sequence similarities efficiently identify positive cis-regulatory elements in the sea urchin. Dev. Biol. 2002;246:148–161. doi: 10.1006/dbio.2002.0618. [DOI] [PubMed] [Google Scholar]
- Yuh CH, Dorman ER, Davidson EH. Brn1/2/4, the predicted midgut regulator of the endo16 gene of the sea urchin embryo. Dev. Biol. 2005;281:286–298. doi: 10.1016/j.ydbio.2005.02.034. [DOI] [PubMed] [Google Scholar]