

Abstract
Tagetitoxin (Tgt) inhibits plastid-encoded, bacterial and some eukaryotic RNA polymerases (RNAPs) by an unknown mechanism. A 2.4Å-resolution structure of the Thermus thermophilus RNAP/Tgt complex revealed that Tgt-binding site within the RNAP secondary channel overlaps with that of the stringent control effector ppGpp, which partially protects RNAP from Tgt inhibition. Tgt binding is mediated exclusively through polar interactions with the β and β′ residues whose substitutions confer resistance to Tgt in vitro. Importantly, a Tgt phosphate, together with two active site acidic residues, coordinates the third Mg2+ ion distinct from the two catalytic metal ions. We show that Tgt inhibits all RNAP catalytic reactions and propose a mechanism in which the Tgt-bound Mg2+ ion plays a key role in stabilization of an inactive transcription intermediate. This and other recent studies suggest that Mg-mediated remodeling of the active site could be a common theme in the regulation of catalysis by nucleic acid enzymes.
RNAP is a central enzyme of gene expression targeted by a multitude of regulatory proteins and small molecule effectors. The principal enzymatic reaction of the RNAP is the nucleotide addition, the transfer of a nucleotidyl moiety from the incoming NTP substrate to the 3′-OH of the nascent RNA, followed by the release of pyrophosphate (PPi) and enzyme translocation by one nt. Polymerization reaction is reversible: in the presence of PPi, RNAP progressively degrades the nascent RNA1, releasing NTPs from the 3′ end of the transcript (pyrophosphorolysis). RNAP also mediates two types of hydrolysis reactions. The exonucleolytic cleavage leads to the release of 3′ NMPs and is dramatically enhanced in the presence of non-cognate substrates2. The endonucleolytic cleavage occurs in backtracked transcription complexes, in which the nascent RNA is threaded through the active site into the secondary channel3; this reaction leads to the release of the 3′ extruded RNA segment and is greatly facilitated by cleavage factors, such as GreA or GreB4. All these reactions are thought to occur in a single active site and conform to the universal two-metal mediated mechanism5, in which two catalytic Mg2+ ions are coordinated by invariant Asp residues: β′ 460, 462, 464 and β 814 in E. coli enzyme (ecRNAP)6-9.
Tgt, a phytotoxin produced by Pseudomonas syringae pv. tagetis10, causes apical chlorosis in infected plants. This effect has been attributed to inhibition of transcription by the bacterial type multi-subunit RNAPs in chloroplasts11. In vitro, Tgt has been shown to efficiently inhibit only a subset of RNAPs: ecRNAP and RNA polymerase III from yeast, insects, and vertebrates are inhibited at micromolar levels of Tgt in vitro, whereas the single-subunit phage RNAPs and nuclear RNAPs I and II are resistant to inhibition11,12. However, the exact mechanism of Tgt action remains obscure, and its target site on RNAP is not known. Since the inhibitor apparently cannot be efficiently transported into bacterial cells, a “classical” genetic screen for resistant mutations has so far proved impractical. Better understanding of Tgt mechanism of inhibition can provide new insights into transcription regulation and may have practical implications for drug design.
We have determined a high-resolution structure of the inhibitor in complex with a bacterial RNAP from T. thermophilus (ttRNAP). In this structure, the binding site for Tgt is located at the base of the RNAP secondary channel, adjacent to the enzyme's active site and overlapping that of ppGpp8; consistently, Tgt inhibited all catalytic activities of RNAP and apparently competed with ppGpp for the effect on (and likely binding to) RNAP. Based on the structure, we constructed RNAP substitutions designed to perturb the inhibitor-binding site and showed that these substitutions confer resistance to Tgt in vitro. We propose that Tgt inhibits transcription by “freezing” the RNAP catalytic center in an inactive state.
RESULTS
Structure determination and overall structure
The physiological targets of Tgt, plastid-encoded RNAPs of higher plants, are architecturally similar to bacterial enzymes consisting of a five-subunit core of (α2ββ′ω); however, plastid-encoded RNAPs are missing the smallest ω subunit and contain splits in the ββ′ subunits. The broad in vitro specificity of Tgt, which inhibits RNAPs from bacteria and chloroplasts as well as yeast RNAP III11,13, argues that ttRNAP, for which the high-resolution structure is available6,8, can be used as a model system for the structural analysis of Tgt binding to bacterial-type RNAPs. Consistently, our biochemical analysis shows that Tgt inhibits transcription by the ecRNAP and ttRNAP similarly (Supplementary Fig. 1).
To elucidate structural basis for the mechanism of Tgt action we have obtained the crystals of the ttRNAP/Tgt complex diffracting to a high resolution. The RNAP/Tgt complex structure was refined at 2.4Å resolution (Table 1). The difference electron density (ED) map built using |FTgt – Fnati| coefficients (where FTgt and Fnati are the structure factor amplitudes for the RNAP/Tgt complex and apo-holoenzyme, respectively), and the phases are from the refined apo-holoenzyme structure14,15 revealed the clear ED for Tgt (Fig. 1a) that allowed an unambiguous fit of the inhibitor in the complex. Tgt binds nearly identically to both independent RNAP molecules in the asymmetric unit of the crystal and the binding site is located in the secondary channel near the RNAP active center (Fig. 1b). Except for the orientations of some side chains, the binding of Tgt did not induce significant conformational changes in RNAP.
Table 1.
Data collection and refinement statistics
| ttRNAP/Tgt | |
|---|---|
| Data collection | |
| Space group | P65 |
| Cell dimensions | |
| a, b, c (Å) | 239.5, 239.5, 253.1 |
| α, β, γ (°) | 90.0, 90.0, 120.0 |
| Resolution (Å) | 25.0 – 2.4(2.49-2.4)* |
| R merge | 0.105(0.425) |
| I / σI | 10.2(2.3) |
| Completeness (%) | 95.1(93.4) |
| Redundancy | 2.9(2.5) |
| Refinement | |
| Space group | P32# |
| Resolution (Å) | 25.0 - 2.4 |
| No. reflections | 604295 |
| Rwork / Rfree | 23.8/27.3 |
| No. atoms | |
| Protein | 53574 |
| Tgt | 52 |
| Mg2+ ions | 4 |
| Zn2+ ions | 4 |
| Water | 8187 |
| B-factors | |
| Protein | 55.7 |
| Tgt | 45.0 |
| Mg2+ ions | 11.5 |
| Zn2+ ions | 46.1 |
| Water | 34.6 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.015 |
| Bond angles (°) | 1.890 |
Highest resolution shell is shown in parenthesis.
The crystals belong to P32 space group with a perfect (50%) merohedral twinning mimicking P65 space group. The data were therefore processed in P65 space group and were expanded to P32 space group for refinement.
Figure 1.
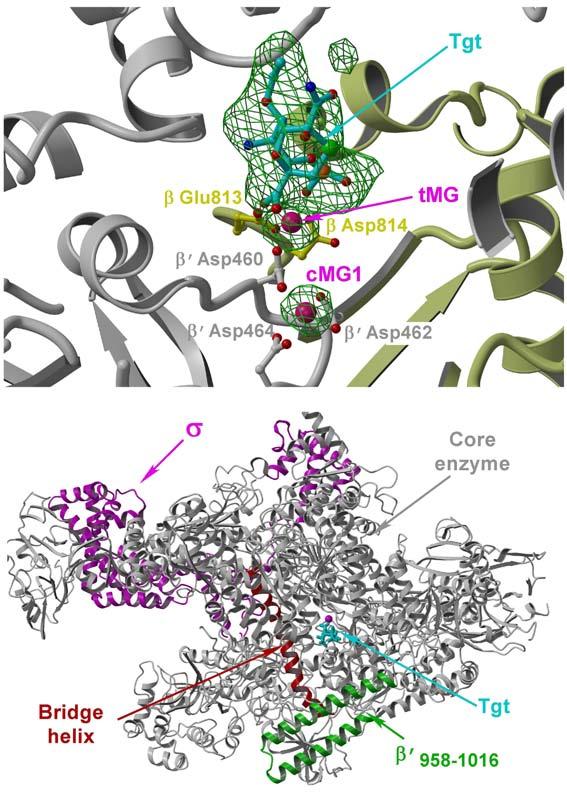
RNAP/Tgt structure. (a) Experimental 3.5Å resolution and |FTgt - Fnati| (3.0σ level both) omit ED map (green) superimposed on the RNAP1/Tgt structure. The residues (balls-and-sticks) and protein backbone (ribbon diagram) of the β and β′ subunits are shown in yellow and gray, respectively. In this and following figures, cMG1, cMG2, and tMG designate high- and low-affinity catalytic and Tgt-bound Mg2+ ions, respectively. (b) Overall view of the RNAP/Tgt complex structure showing that Tgt binds within the secondary channel in close vicinity to the active site (marked by cMG1, magenta sphere).
The Tgt-binding site
Tgt binding to RNAP is mediated exclusively by polar interactions: nine of the eleven Tgt oxygens form 18 hydrogen bonds with the adjacent protein side chains (Figs. 2a,b). A set of basic and acidic side chains that constitute the Tgt-binding site form an extensive network not only with the inhibitor but also with each other. Such a concerted mode of recognition could be essential for binding of Tgt and highly sensitive to subtle alterations in conformation/position of even a single residue. Tgt specifically interacts with three highly conserved basic residues (β Arg678, β Arg1106 and β′ Arg731; the E. coli numbering is used throughout) and β′ Asn458 likely involved in substrate recognition16; however, the Tgt and the NTP-binding sites do not directly overlap implying that competition with the substrate is not the major component of the Tgt action.
Figure 2.
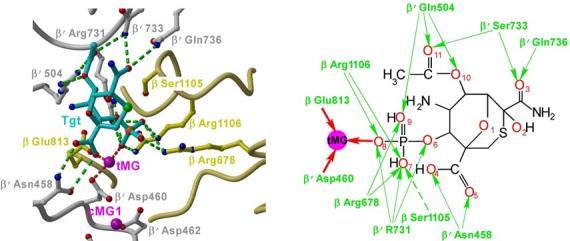

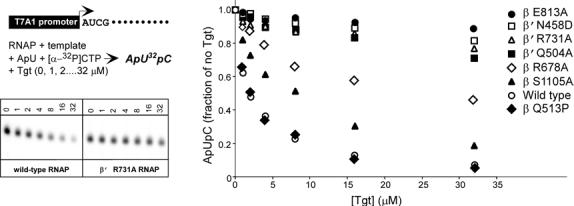
The Tgt-binding site. (a,b) A model (a) and schematic drawing (b) of the Tgt-binding site on RNAP. Residues that are not identical between E. coli and T. thermophilus are marked by their numbers only. Polar interactions are in red (tMG coordination bonds) or green (hydrogen bonds) dashed lines (a) and arrows (b). Weak contacts of β Ser1105 with Tgt are represented by a dashed arrow (b). (c) Sequence alignment of the β and β′ subunits from bacterial (E. coli, Eco; T. thermophilus, Tth), chloroplast (Arabidopsis thaliana, Ath), and yeast Saccharomyces cerevisiae (pol II, Sc II; and pol III, Sc III) enzymes in regions flanking the Tgt contact sites using DNAStar MegAlign Module. The Tgt structural determinants are highlighted by green boxes; residues substituted for the in vitro analysis are indicated by black arrows. (d) Inhibition of abortive transcription on the T7A1 promoter by Tgt. Formation of the radiolabeled ApUpC RNAs was followed as a function of Tgt concentration (from 0 to 32 μM) with wild-type or altered ecRNAP. The key is shown in the figure.
Another important structural feature of the RNAP/Tgt complex is a well-fixed Mg2+ ion (tMG) that mediates RNAP interactions with Tgt. This Mg2+ ion is coordinated by the Tgt phosphate and by two active site residues, β′ Asp460 and β Glu813. Compared to the apo-holoenzyme, the side chain of β′ Asp460 is better fixed in the complex by bridging the two Mg2+ ions (cMG1 and tMG) – this may favor coordination and subsequently strengthen binding of the catalytic cMG1; consistently, as revealed by the initial difference ED map (Fig. 1a), Tgt indeed increases RNAP affinity for the major catalytic Mg2+ ion, cMG1. At the same time, tMG binding to β′ Asp460 would compromise binding of the second catalytic Mg2+ ion (cMG2) required for catalysis5,8,17.
Disruption of RNAP contacts with Tgt confers resistance in vitro
Generation and analysis of Tgt-resistant mutants would be complicated not only due to the low permeability of bacterial cells to this compound but also due to close proximity of its binding site to the RNAP catalytic center. Substitution of the ecRNAP residues in contact with Tgt have been shown to interfere with RNA extension and exonuclease cleavage2, ribodiscrimination16 and substrate binding (VS, DGV, and IA, in preparation); most of these mutants are inviable. Interpretation of the resistant phenotypes is further complicated by their allosteric and pleiotropic effects away from the actual binding site15,18,19. Even more intriguingly, certain RNAP variants are not merely resistant to, but critically dependent upon antibiotics Mcc and CBR for their viability20,21, indicating that some resistant substitutions exploit rather than ameliorate the effects of antibiotic binding. Therefore, we decided to assess the functional importance of the Tgt/RNAP interactions observed in the co-crystal directly, using a highly purified in vitro transcription system. Since tt and ecRNAPs respond to Tgt similarly (see above), we engineered Ala substitutions of the ecRNAP residues to disrupt the observed polar contacts with Tgt.
Residues from six non-contiguous regions of RNAP come together to form a binding site for Tgt in the holoenzyme (Fig. 2c). These regions are highly conserved in all RNAPs from chloroplasts to humans; however, eukaryotic RNAPs display differential response to Tgt11 (see Supplementary Fig. 2 for discussion of Tgt effects on nuclear enzymes). We selected four conserved residues that, according to the co-crystal structure, should be crucial for the inhibitor binding: β′ Gln504, β Arg678 and β′ Arg731, all making direct hydrogen bonds with the inhibitor, and β Glu813 implicated in Mg2+-mediated interactions with the Tgt phosphate (Fig. 2c, shown by arrows). We also picked β Ser1105, which makes only weak polar or van der Waals interactions with Tgt and thus could be partially dispensable for binding.
We introduced mutations into the E. coli rpoB and rpoC genes by site-directed mutagenesis, purified the altered RNAPs, and tested their response to Tgt using a steady-state transcription assay in vitro (Fig. 2d). With the wild-type ecRNAP, an increase in Tgt concentration to 32 μM led to an over 90% decline in the synthesis of the ApUpC transcript. In contrast, β R678A, β E813A, β′ Q504A and β′ R731A RNAPs retained 45-95% activity at 32 μM of the inhibitor. While the resistance of the β′ Q504A, β R678A and β′ R731A variants is likely caused by the loss of contacts essential for binding, the β E813A substitution probably results in the loss of tMG, which might be critical for the function rather than for the binding of Tgt. In excellent agreement with the structural data, the level of resistance demonstrated by β S1105A RNAP was considerably lower. We also tested β′ N458D RNAP, which was insensitive to inhibition by ppGpp8. Since β′ Asn458 interacts with the carboxyl group of Tgt (Fig. 2c,d), introduction of the negative charge on its side chain would likely result in repulsive forces destabilizing Tgt binding. As expected, the β′ N458D substitution conferred strong resistance to Tgt (Fig. 2d). In contrast, neither the distant rifampicin-resistant substitution β Q513P nor the substitution of the residue adjacent to the Tgt contact site, β N677A, conferred resistance to Tgt (Fig. 2d and data not shown). These results support the importance of the Tgt/RNAP contacts observed in the co-crystal and indicate that the structural data obtained with the holoenzyme/Tgt complex correlate with the biochemical data collected with the transcriptionally active complexes.
Tgt and ppGpp bind to overlapping sites on RNAP
Tgt-binding site is located at the base of the secondary channel, the focal point of regulation by various protein factors and small effectors8,22-26. Notably, superposition of the RNAP/Tgt complex structure with that of the ttRNAP bound to the “magic spot” (ppGpp), the regulator of stringent response,8 revealed that the Tgt- and ppGpp-binding sites overlap (Fig. 3a) and the Tgt-binding mode resembles that of ppGpp, particularly in the 3′ orientation. The Tgt phosphate superposes well on the 3′ ppGpp β-phosphate and makes similar contacts with β Arg1106 and β′ Arg731, whereas the O10 oxygen and attached acetate group make hydrogen bonds with β′ Gln504 (Fig. 3b), reminiscent of the ppGpp 5′ phosphate in the 3′ orientation (Fig. 3c). In addition, the Tgt carboxyl group forms hydrogen bonds with β′ Asn458, mimicking interactions with the ppGpp base that are crucial for the ppGpp function8. Finally, tMG and pMG1 in the 3′ (but not in the 5′) orientation induce similar active site alterations: they are coordinated by β′ Asp460, thereby likely compromising binding of the catalytic cMG2 ion (Fig. 3b).
Figure 3.
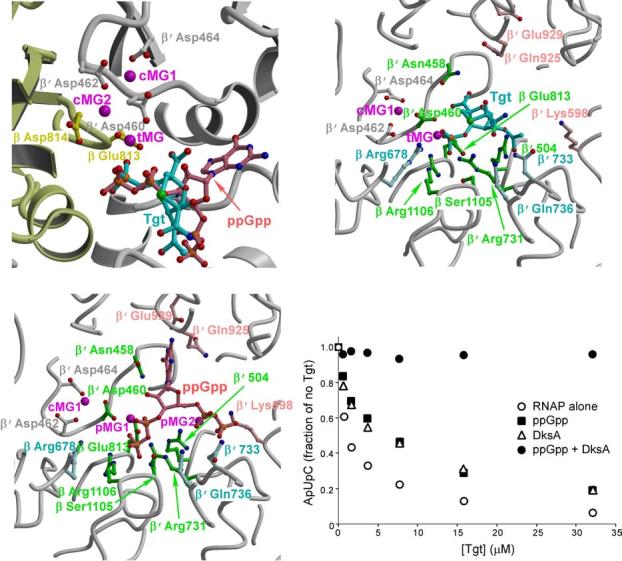
Tgt and ppGpp bind to overlapping sites on RNAP. (a) Superposition of the ttRNAP/Tgt and ttRNAP/ppGpp complexes. The color scheme is the same as in Figures 1a and 2a. (b,c) Structural determinants likely crucial for the Tgt and ppGpp binding. The RNAP/Tgt (b) and RNAP/ppGpp (c) complexes are shown in the same orientation for better comparison. RNAP residues (balls-and-sticks) that interact with both Tgt and ppGpp are shown in green, whereas those specific for Tgt and ppGpp are shown in light cyan and light pink, respectively. (d) ppGpp and DksA compete with Tgt for the inhibition of abortive transcription by the wild-type ecRNAP from the T7A1 promoter. The assay was performed as in Figure 2d. ppGpp was added to 0.5 mM, DksA – to 500 nM.
There are, however, a number of essential differences in the modes of Tgt and ppGpp binding. First, although Tgt contacts to β′ Asn458 resembles interactions of the ppGpp base, Tgt is lacking other important specific contacts formed by the ppGpp base with β′ Glu929, β′ Gln925, and β′ Lys598. These interactions link the “catalytic” domain of the enzyme (bound to the ppGpp phosphates), which participates in the formation of binding sites for the substrate, RNA/DNA hybrid and unwound non-template DNA strand, with the β′ domain that constitutes a base for the downstream DNA-binding site. Second, Tgt binding induces a substantial alteration of the side chain orientation of β Arg678, which is stabilized by multiple interactions with the Tgt phosphate (Fig. 3b). Similar interactions are absent in the RNAP/ppGpp complex (Fig. 3c). Due to its proximity to the catalytic Asp residues, β Arg678 could be crucial for the substrate positioning in the active site. Third, despite the similarities in Mg2+-mediated interactions with RNAP, tMG ion is located substantially closer to the active site than pMG1 in the 3′ and 5′ orientations: the distances from cMG2 ion are ∼4Å, ∼5.2Å and 6.3Å, respectively.
The overlap between the ppGpp- and Tgt-binding sites implies that both inhibitors would compete for binding to RNAP. In addition, Tgt action could be interfered with by DksA, which acts synergetically with ppGpp27 and binds within the secondary channel26. To test this prediction, we used an abortive initiation assay on the T7A1 promoter template (Fig. 3d). While RNAP alone was susceptible to inhibition, addition of either ppGpp or DksA partially protected RNAP against Tgt, and together ppGpp and DksA eliminated the inhibition completely. The inability of ppGpp to block Tgt action in the absence of DksA completely is likely due to the fact that very high levels of ppGpp are required to inhibit transcription in vitro, whereas the addition of DksA reduces the effective ppGpp concentration dramatically27. Since in our hands the ratio of the inhibitory concentrations of ppGpp and Tgt is ∼100, the requirement for DksA for efficient competition is not surprising.
Together, the large number of direct polar contacts between RNAP and Tgt observed in the structure, the fact that substitutions of the residues making these contacts confer resistance to Tgt, and the apparent competition between Tgt and ppGpp/DksA provide strong evidence for the high specificity and functional relevance of the observed Tgt-binding site.
Tgt inhibits all catalytic reactions of RNAP
Proximity of the Tgt-binding site to the enzyme's active site suggests that Tgt may inhibit not only RNA synthesis but also other RNAP catalytic activities. To test this hypothesis, we used ecRNAP as a model system. First, we tested Tgt effect on pre-formed, radiolabeled A26 TECs halted on a λPR template pIA22628. Upon addition of either PPi or GreA, the A26 RNA was rapidly degraded (Fig. 4a,b, left panels). However, addition of Tgt slowed the RNA cleavage dramatically in both cases (right panels).
Figure 4.

Tgt inhibits all catalytic reactions. (a,b) Pyrophosphorolysis and GreA-enhanced transcript cleavage assays. Halted radiolabeled A26 TECs were purified from NTPs using G50 spin columns and pre-incubated with Tgt at 37 °C for 2 min. GreA (300 nM) or PPi (400 μM) were added at time 0, followed by incubation at 37 °C. (c) Exonuclease cleavage assay. 3′ radiolabeled C27 complexes were incubated with water (left panel), 2 μM Tgt (center), or 1 mM ATP (right panel) at 37 °C. Aliquots were removed at the indicated times and quenched.
Next, we tested the effect of Tgt on the exonuclease activity. We prepared unlabeled A26 TECs on the same template, removed NTPs by gel filtration, and extended the nascent RNA by one nt upon addition of [α-32P]CMP. The 3′ labeled C27 TECs were used to monitor the release of the labeled CMP that occurred upon exonucleolytic transcript cleavage (Fig. 4c). When C27 TECs were incubated at 37 °C in the presence of 10 mM Mn2+, a rapid disappearance of the C27 RNA and an accumulation of [α-32P]CMP were easily detectable after 5-min incubation. The increase in the CMP production observed at later points likely represents multiple rounds of CMP addition and release, since the amount of released CMP exceeded that of the C27 RNA. As reported previously2, the addition of a non-cognate NTP dramatically increased the exonuclease activity. In contrast, upon addition of Tgt cleavage of the C27 RNA was markedly decreased. These results demonstrate that Tgt inhibits all catalytic reactions of RNAP.
DISCUSSION
Mechanism of Tgt action
The simplest mechanism of Tgt action would be direct competition with the NTP substrate. Indeed, such a mechanism was proposed for MccJ25 that also binds within the secondary channel22,23. Two lines of data appear to rule out this possibility. First, in agreement with our structural data, Tgt acts as an uncompetitive inhibitor12,29 implying that it does not prevent the substrate binding. Second, Tgt inhibits catalytic reactions that utilize different substrates (Fig. 4).
Given that Tgt binds near the active site and interacts with RNAP residues that in turn likely interact with the incoming NTP, Tgt may alter the substrate loading or stabilize it in a non-productive state. In the recently proposed pathway of the substrate loading14, the incoming NTP first occupies an inactive, non-specific entry site (E-site)17,30-33; then rotates towards the pre-insertion site, another catalytically-inactive but specific intermediate, where it is recognized through base pairing with the acceptor template DNA base and sugar-discriminating interactions with the protein; lastly, the cognate NTP is delivered to the active, insertion site31,33 for catalysis.
Structural comparison shows that all three substrate-binding sites are proximal to the Tgt-binding site and thus each of them might be a potential Tgt target. In search for deeper insight into the mechanism of Tgt, we undertook homology modeling of Tgt bound to the TECs with the substrates occupying the E-site, the pre-insertion, and the insertion sites based on a simple superposition of the bacterial and eukaryotic RNAP structures by the residues constituting the substrate-binding site. The superposition resulted in root mean square deviations of ∼1.3Å over 30 Cα positions, revealing a high structural conservation of the respective RNAP segments and therefore suggesting that the results of this analysis would be applicable to both systems.
The E-site substantially overlaps with the Tgt-binding site (Fig. 5a). As revealed by the pattern of structural contacts with the protein17, the NTP binding to the E-site is likely very weak and therefore should not contribute much to the overall substrate affinity; hence, Tgt would not be expected to act as a competitive inhibitor of transcription if it acts exclusively through competition with the substrate bound in the E-site. The major role of the E-site could be to shift equilibrium of the TEC toward the post-translocated state through competition of the NTP phosphates with the 3′ terminus of the nascent RNA in the pre-translocated TECs. Thus inability of the substrate to occupy the E-site in the presence of Tgt may result in stabilization of a non-productive pre-translocated state. This may slow down transcription, but likely will not block it completely, as the post-translocated TECs may form spontaneously (without NTP stimulation) and become stabilized by the NTP bound to the pre-insertion site bypassing the E-site14.
Figure 5.
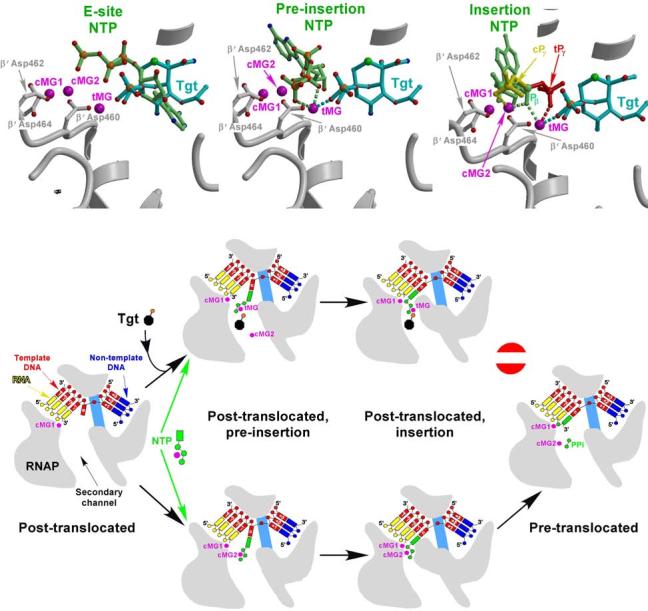
Mechanism of Tgt action. (a,b,c) Models of the substrates (light green) corresponding to the three postulated consecutive steps during NTP loading to the RNAP active site are superimposed on the RNAP/Tgt structure; the E-site (a), the pre-insertion site (b), and the insertion site (c); the PDB accession codes used in panels a, b and c were 1R9T, 1Y77, and 1R9S, respectively. The putative coordination bonds with tMG and/or cMG2 of Tgt (cyan), the NTP in the pre-insertion site and the β-phosphate (Pβ) of the NTP in the insertion site (light green) (b,c), as well as of the NTP γ-phosphates in the insertion site (c) in the “catalytic” cPγ (yellow) and “inactive”, tMG-bound tPγ (red) configurations are shown by dashed lines. (d) Stabilization of an inactive transcription intermediate by Tgt.
In the pre-insertion site, the modeled NTP triphosphate slightly clashes with Tgt (Fig. 5b); however, subtle torsion rotations of the β and γ phosphate moieties eliminate the clash, arguing against Tgt competition with the substrate. Moreover, the modeling suggests that Tgt might even additionally stabilize the cognate NTP in the pre-insertion site through the tMG-mediated interactions between the NTP and Tgt phosphates and the complementary van der Waals interactions of the NTP phosphates with the Tgt backbone.
The TEC model with the NTP loaded into the insertion site (Fig. 5c) also did not reveal serious steric hindrance between the substrate and Tgt. However, it suggested that while bound to the catalytic Mg2+ ions in a productive conformation, NTP might simultaneously coordinate tMG by its β phosphate (Fig. 5a). In a paused conformation proposed to be particularly sensitive to Tgt29, a swap of β′ Asp460 to tMG might initiate competition between tMG and the low affinity cMG2 for the γ phosphate of the substrate. If tMG would take over in this competition, the NTP might maintain base pairing with the DNA template, while its phosphates would be fixed (likely with no loss in affinity) in a non-productive conformation (Fig. 5c). An increased affinity for the major catalytic Mg2+ ion (cMG1) observed in the presence of Tgt (see above) may additionally stabilize this inactive intermediate, thereby enhancing pausing.
Altogether, in agreement with the biochemical analysis (see Supplementary Fig. 3), the structural modeling argues against the competition between Tgt and NTP substrate and suggests the mechanism in which Tgt stabilizes some inactive intermediate during the substrate loading into the active site. While the structural analysis indicates that such an intermediate may be formed at either the pre-insertion or insertion step, we favor a concerted two-step model, in which this intermediate is pre-formed in the pre-insertion site and then is finally stabilized in the insertion complex (Fig. 5d). Indeed, while binding to the pre-insertion site in the presence of Tgt, the substrate phosphates that coordinate cMG2 ion would likely switch interactions to a well-fixed tMG, with a subsequent loss of cMG2. As the substrate loading in the insertion site is likely achieved by a simple rotation without significant repositioning of the NTP phosphates, the established interactions of NTP with tMG would not be disrupted during this isomerization. The presumably more compact conformation of the active site in the insertion complex14,31 would result in a tighter binding of the tMG-bound substrate to prevent both the dissociation of the substrate and the catalytic reaction, thereby irreversibly locking RNAP in a non-productive state.
Tgt-bound Mg2+ ion and remodeling of the RNAP active site
All nucleic acid polymerases are thought to rely on the universal two-metal mechanism5 to mediate the phosphodiester bond formation by a bimolecular nucleophilic substitution (SN2-like) mechanism. This mechanism typically requires several carboxylate residues in the enzyme's active site to coordinate two Mg2+ ions: the NTP-binding ion (cMG2), which coordinates the α, β, and γ phosphates of the incoming NTP, and the catalytic ion (cMG1), which coordinates the α phosphate of NTP and the 3′-O of the primer strand. The precise positioning of the Mg2+ ions, and hence the proper orientation of the coordinating active site side chains, are required to facilitate the chemical reaction, in-line nucleophilic attack of 3′ OH on NTPs Pα and to stabilize the pentavalent transition state. Dynamics simulations of catalysis by DNA polymerase β indicate that the two Mg ions are required to maintain the closed, catalytically-competent state of the enzyme and orchestrate slow adjustments of the active site groups to attain the “perfect” configuration34.
The structures of the RNAP/Tgt and RNAP/ppGpp complexes revealed a specific binding site for a third Mg2+ ion in vicinity of the active site. Binding of this ion closely resembles that of cMG2, as it requires external ligands (Tgt or ppGpp) for stable coordination. Since the tMG ion is coordinated by the active site residue β′ Asp460, it may compromise binding of the cMG2, as was previously suggested for ppGpp in the 3′-orientation8, or at least perturb the optimal geometry of the cMG1 and cMG2, thereby inhibiting all catalytic reactions (Fig. 4). This mechanism of inhibition is analogous to the attenuation of the RNaseH activity where at high Mg2+ concentrations an additional carboxylate was proposed to recruit a third, inhibitory metal ion35, compromising coordination of the active site Mg2+ ions. These findings underscore the importance of Mg2+ ions not only in catalysis by nucleic acid polymerases but also in modulation of the active site reactions by auxiliary regulators, which may misalign the catalytic Mg2+ ions or adventitiously recruit additional, inhibitory ions.
Implications for the differential regulatory effects of Tgt and ppGpp
Although Tgt and ppGpp bind to overlapping sites on RNAP (Fig. 3), their effects on transcription are strikingly distinct. Tgt does not destabilize the RNAP open promoter complexes, the key element of the transcription control by ppGpp and DksA27. Conversely, ppGpp does not inhibit transcript cleavage reactions and the sites at which ppGpp and Tgt induce RNAP pausing are quite distinct (IA, data not shown). In contrast to Tgt, ppGpp is not likely to affect substrate binding to the insertion site, since ppGpp-bound pMG1 ion (in both 3′ and 5′ orientations) appears too distant from the NTP in the active configuration to allow its coordination by the β phosphate, thereby excluding potential competition with the active site metal ion and subsequent formation of the inactive conformation. At the same time, Tgt and ppGpp may share some modes of inhibition of the RNA synthesis, such as interference with the cMG2 binding/positioning. Both ppGpp and Tgt likely affect mobile, functionally crucial RNAP elements, whose structural transitions might be sensitive to subtle differences in the inhibitors' shape and mode of binding as well as to the nucleic acid sequence.
The primary (substrate-independent) effect of ppGpp on the open complex stability might be explained by the fact that, unlike Tgt, binding of the ppGpp phosphates and the base moiety (missing in Tgt) bridges the two RNAP domains that do not interact with each other in the apo-holoenzyme. One of these domains is likely involved in the interactions with the downstream DNA, whereas the other might contact the unwound DNA strands in the open complex. The mobility of these structural segments, which is likely constrained by ppGpp binding, might be required during transcription initiation to form a stable open complex.
RNAP secondary channel as a broad specificity target for transcriptional control
Our findings emphasize the role of the RNAP secondary channel as a versatile target for transcriptional regulation. A number of recent studies implicated the secondary channel as an entry point for several factors that gain access to the RNAP catalytic site and modulate its activity: ppGpp, transcript cleavage factors, antibiotic microcin J25, and DksA8,22-26,36,37. Another important conclusion from our and other relevant studies is that the RNAP secondary channel is fairly structurally “insensitive,” allowing for binding of many structurally distinct molecules. At the same time, the secondary channel is extremely functionally “sensitive,” allowing mediators that are quite similar in shape and mode of binding (like Tgt and ppGpp, GreA/B and DksA) to confer dramatically different effects on transcription. These unique properties suggest that we may anticipate discovery of additional factors targeting the secondary channel.
METHODS
Proteins and reagents
All general reagents were obtained from Sigma and Fisher; NTPs, from GE Health; PCR reagents, from Eppendorf; restriction and modification enzymes, from NEB; [α–32P] NTPs, from NEN. Oligonucleotides were obtained from Integrated DNA Technologies. DNA purification kits were from Qiagen, Tgt from Epicentre Technologies. GreA was purified as described previously38. Substitutions in the E. coli RNAP subunits were constructed by site-directed mutagenesis; sequences of all plasmid constructs were verified at the OSU PMGF. Altered RNAP were purified as described previously15,16. All plasmid constructs used in this work are listed in the Supplementary Table 1.
Structure determination and refinement
The ttRNAP holoenzyme was purified and crystallized as described previously15. To obtain the complex crystals, the crystals of the apo-holoenzyme were transferred for 6 hours into the mother liquor containing 2mM Tgt. The data for apo-holoenzyme15 and the ttRNAP/Tgt complex were collected at the beam line BL5 at Photon Factory (Tsukuba, Japan) using ADSC Quantum-315 CCD detector and processed using HKL2000 data processing package39. The refinement was carried out using the CNS program40. The Tgt model was built into the initial experimental difference ED map (Fig. 1a). The rigid body refinement followed by several rounds of the B-factor, positional, simulated annealing refinements, and water “pick” and water “delete” procedures, alternating by manual model building using the O program41 yielded the final R-factor of 23.8% and R-free of 27.3% for the ttRNAP/Tgt complex (Table 1). Three-dimensional structural figures were prepared using programs Molscript42, Bobscript43, and Raster3D44.
Halted complex formation
Linear DNA template generated by PCR amplification (40 nM), holo RNAP (50 nM), ApU (100 μM), and starting NTP subsets (indicated in figure legends) were mixed on ice in 50 μl of TGA buffer (20 mM Tris-acetate, 20 mM Na-acetate, 10 mM Mg-acetate, 5% (v/v) glycerol, 14 mM 2-mercaptoethanol, 0.1 mM EDTA, pH 8.0). Reactions were incubated at 37 °C (for ecRNAP) or at 55 °C (for ttRNAP) and stored on ice prior to use in elongation assays or quenched.
Transcript cleavage assays
Halted A26 elongation complexes were formed on pIA226-derived template in 50 μl of TGA buffer and purified by gel-filtration through G50 spin columns (GE Health) equilibrated in TGA buffer. For endonucleolytic assays, A26 TECs were internally labeled with [α-32P]GMP and incubated GreA or PPi at 37 °C. For exonucleolytic assays, unlabeled G50-purified A26 TECs were incubated with [α–32P]CTP (1 μM) for 3 min at 37 °C. The resulting 3′-end labeled C27 TECs were supplemented with MnCl2 (10 mM final). The reactions were incubated at 37 °C. Samples were removed at indicated times, quenched, and separated on 12% (w/v) denaturing gels.
Steady-state abortive initiation assays
Linear T7A1 promoter template (2 nM), holo RNAP (50 nM), ApU (100 μM), and various concentrations of CTP (a mixture of cold and radiolabeled CTP at a constant specific activity) were mixed on ice in 50 μl of TGA buffer. Tgt was added at concentrations indicated in the figures, reactions were transferred to 37 °C for 20 min, quenched, and analyzed on 12% (w/v) denaturing gels.
Accession codes
The coordinates and structure factors for the crystal structure of the RNAP/Tgt complex have been deposited in the Protein Data Bank under ID code XXX.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Bob Landick and Jeff Roberts for helpful comments. This work was supported by GM74252 and GM74840 (to DGV) and GM67153 (to IA) grants from National Institutes of Health, and by RIKEN (DGV).
REFERENCES
- 1.Rozovskaia T, Chenchik A, Bibilashvili R. Reaction of pyrophosphorolysis catalyzed by Escherichia coli RNA polymerase. Mol. Biol. (Mosk) 1981;15:636–652. [PubMed] [Google Scholar]
- 2.Sosunov V, et al. Unified two-metal mechanism of RNA synthesis and degradation by RNA polymerase. EMBO J. 2003;22:2234–44. doi: 10.1093/emboj/cdg193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Komissarova N, Kashlev M. Transcriptional arrest: Escherichia coli RNA polymerase translocates backward, leaving the 3' end of the RNA intact and extruded. Proc Natl Acad Sci U S A. 1997;94:1755–60. doi: 10.1073/pnas.94.5.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Orlova M, et al. Intrinsic transcript cleavage activity of RNA polymerase. Proc. Natl. Acad. Sci. USA. 1995;92:4596–4600. doi: 10.1073/pnas.92.10.4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steitz TA. A mechanism for all polymerases. Nature. 1998;391:231–2. doi: 10.1038/34542. [DOI] [PubMed] [Google Scholar]
- 6.Vassylyev DG, et al. Crystal structure of a bacterial RNA polymerase holoenzyme at 2.6 A resolution. Nature. 2002;417:712–9. doi: 10.1038/nature752. [DOI] [PubMed] [Google Scholar]
- 7.Zhang G, et al. Crystal structure of Thermus aquaticus core RNA polymerase at 3.3 A resolution. Cell. 1999;98:811–824. doi: 10.1016/s0092-8674(00)81515-9. [DOI] [PubMed] [Google Scholar]
- 8.Artsimovitch I, et al. Structural basis for transcription regulation by alarmone ppGpp. Cell. 2004;117:299–310. doi: 10.1016/s0092-8674(04)00401-5. [DOI] [PubMed] [Google Scholar]
- 9.Cramer P. Structure and function of RNA polymerase II. Adv Protein Chem. 2004;67:1–42. doi: 10.1016/S0065-3233(04)67001-X. [DOI] [PubMed] [Google Scholar]
- 10.Lukens JH, Durbin RD. Tagetitoxin affects plastid development in seedlings leaves of wheat. Planta. 1985;165:311–321. doi: 10.1007/BF00392227. [DOI] [PubMed] [Google Scholar]
- 11.Mathews DE, Durbin RD. Tagetitoxin inhibits RNA synthesis directed by RNA polymerases from chloroplasts and Escherichia coli. J Biol Chem. 1990;265:493–8. [PubMed] [Google Scholar]
- 12.Mathews DE, Durbin RD. Mechanistic aspects of tagetitoxin inhibition of RNA polymerase from Escherichia coli. Biochemistry. 1994;33:11987–92. doi: 10.1021/bi00205a038. [DOI] [PubMed] [Google Scholar]
- 13.Steinberg TH, Mathews DE, Durbin RD, Burgess RR. Tagetitoxin: a new inhibitor of eukaryotic transcription by RNA polymerase III. J Biol Chem. 1990;265:499–505. [PubMed] [Google Scholar]
- 14.Temiakov D, et al. Structural basis of transcription inhibition by antibiotic streptolydigin. Mol Cell. 2005;19:655–66. doi: 10.1016/j.molcel.2005.07.020. [DOI] [PubMed] [Google Scholar]
- 15.Artsimovitch I, et al. Allosteric modulation of the RNA polymerase catalytic reaction is an essential component of transcription control by rifamycins. Cell. 2005;122:351–63. doi: 10.1016/j.cell.2005.07.014. [DOI] [PubMed] [Google Scholar]
- 16.Svetlov V, Vassylyev DG, Artsimovitch I. Discrimination against deoxyribonucleotide substrates by bacterial RNA polymerase. J Biol Chem. 2004;279:38087–90. doi: 10.1074/jbc.C400316200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Westover KD, Bushnell DA, Kornberg RD. Structural basis of transcription: nucleotide selection by rotation in the RNA polymerase II active center. Cell. 2004;119:481–489. doi: 10.1016/j.cell.2004.10.016. [DOI] [PubMed] [Google Scholar]
- 18.Zhou YN, Jin DJ. The rpoB mutants destabilizing initiation complexes at stringently controlled promoters behave like “stringent” RNA polymerases in Escherichia coli. Proc Natl Acad Sci U S A. 1998;95:2908–13. doi: 10.1073/pnas.95.6.2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jin DJ, Gross C. Mapping and sequencing of mutations in the Escherichia colirpoB gene that lead to rifampicin resistance. J. Mol. Biol. 1988;202:45–58. doi: 10.1016/0022-2836(88)90517-7. [DOI] [PubMed] [Google Scholar]
- 20.Yuzenkova J, et al. Mutations of bacterial RNA polymerase leading to resistance to microcin J25. J Biol Chem. 2002;277:50867–75. doi: 10.1074/jbc.M209425200. [DOI] [PubMed] [Google Scholar]
- 21.Artsimovitch I, Chu C, Lynch AS, Landick R. A new class of bacterial RNA polymerase inhibitor affects nucleotide addition. Science. 2003;302:650–4. doi: 10.1126/science.1087526. [DOI] [PubMed] [Google Scholar]
- 22.Adelman K, et al. Molecular mechanism of transcription inhibition by peptide antibiotic microcin J25. Mol Cell. 2004;14:753–62. doi: 10.1016/j.molcel.2004.05.017. [DOI] [PubMed] [Google Scholar]
- 23.Mukhopadhyay J, et al. Antibacterial peptide microcin J25 inhibits transcription by binding within and obstructing the RNA polymerase secondary channel. Mol Cell. 2004;14:739–51. doi: 10.1016/j.molcel.2004.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Opalka N, et al. Structure and function of the transcription elongation factor GreB bound to bacterial RNA polymerase. Cell. 2003;114:335–45. doi: 10.1016/s0092-8674(03)00600-7. [DOI] [PubMed] [Google Scholar]
- 25.Laptenko O, Lee J, Lomakin I, Borukhov S. Transcript cleavage factors GreA and GreB act as transient catalytic components of RNA polymerase. EMBO J. 2003;22:6322–34. doi: 10.1093/emboj/cdg610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perederina A, et al. Regulation through the secondary channel--structural framework for ppGpp-DksA synergism during transcription. Cell. 2004;118:297–309. doi: 10.1016/j.cell.2004.06.030. [DOI] [PubMed] [Google Scholar]
- 27.Paul BJ, et al. DksA: a critical component of the transcription initiation machinery that potentiates the regulation of rRNA promoters by ppGpp and the initiating NTP. Cell. 2004;118:311–22. doi: 10.1016/j.cell.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 28.Artsimovitch I, Landick R. The transcriptional regulator RfaH stimulates RNA chain synthesis after recruitment to elongation complexes by the exposed nontemplate DNA strand. Cell. 2002;109:193–203. doi: 10.1016/s0092-8674(02)00724-9. [DOI] [PubMed] [Google Scholar]
- 29.Steinberg TH, Burgess RR. Tagetitoxin inhibition of RNA polymerase III transcription results from enhanced pausing at discrete sites and is template-dependent. J Biol Chem. 1992;267:20204–11. [PubMed] [Google Scholar]
- 30.Kettenberger H, Armache KJ, Cramer P. Complete RNA polymerase II elongation complex structure and its interactions with NTP and TFIIS. Mol Cell. 2004;16:955–65. doi: 10.1016/j.molcel.2004.11.040. [DOI] [PubMed] [Google Scholar]
- 31.Armache KJ, Kettenberger H, Cramer P. The dynamic machinery of mRNA elongation. Mol Cell. 2005;15:197–203. doi: 10.1016/j.sbi.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 32.Temiakov D, et al. Structural basis for substrate selection by T7 RNA polymerase. Cell. 2004;116:381–91. doi: 10.1016/s0092-8674(04)00059-5. [DOI] [PubMed] [Google Scholar]
- 33.Yin YW, Steitz TA. The structural mechanism of translocation and helicase activity in T7 RNA polymerase. Cell. 2004;116:393–404. doi: 10.1016/s0092-8674(04)00120-5. [DOI] [PubMed] [Google Scholar]
- 34.Yang L, et al. Critical role of magnesium ions in DNA polymerase beta's closing and active site assembly. J Am Chem Soc. 2004;126:8441–53. doi: 10.1021/ja049412o. [DOI] [PubMed] [Google Scholar]
- 35.Nowotny M, Gaidamakov SA, Crouch RJ, Yang W. Crystal structures of RNase H bound to an RNA/DNA hybrid: substrate specificity and metal-dependent catalysis. Cell. 2005;121:1005–16. doi: 10.1016/j.cell.2005.04.024. [DOI] [PubMed] [Google Scholar]
- 36.Kettenberger H, Armache KJ, Cramer P. Architecture of the RNA polymerase II-TFIIS complex and implications for mRNA cleavage. Cell. 2003;114:347–57. doi: 10.1016/s0092-8674(03)00598-1. [DOI] [PubMed] [Google Scholar]
- 37.Sosunova E, et al. Donation of catalytic residues to RNA polymerase active center by transcription factor Gre. Proc Natl Acad Sci U S A. 2003;100:15469–74. doi: 10.1073/pnas.2536698100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Artsimovitch I, et al. RNA polymerases from Bacillus subtilis and Escherichia coli differ in recognition of regulatory signals in vitro. J Bacteriol. 2000;182:6027–35. doi: 10.1128/jb.182.21.6027-6035.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Otwinowski Z, Minor W. Processing X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 40.Brunger AT, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallographica. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 41.Jones TA, Zou JY, Cowan SW, Kjeldgaard Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 42.Kraulis PJ. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 1991;24:946–950. [Google Scholar]
- 43.Esnouf RM. Further additions to MolScript version 1.4, including reading and contouring of electron-density maps. Acta Crystallogr. 1999;D55:938–940. doi: 10.1107/s0907444998017363. [DOI] [PubMed] [Google Scholar]
- 44.Merrit EA, Bacon DJ. Raster3D: photorealistic molecular graphics. Methods Enzymol. 1997;277:505–524. doi: 10.1016/s0076-6879(97)77028-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.