

Abstract
The SAGE (serial analysis of gene expression) method is sensitive at detecting the lower abundance transcripts. More than a third of human SAGE tags identified are novel representing the low abundance unknown transcripts. Using the GLGI method (generation of longer 3′ EST from SAGE tag for gene identification), we converted 1,009 low-copy, human X chromosome-specific SAGE tags into the 3′ ESTs. We identified 3,418 unique 3′ ESTs, 46% of which are novel and originated from the lower abundance transcripts. However, nearly all 3′ ESTs were mapped to various regions across the genome but not X Chromosome. Detailed analysis indicates that those 3′ ESTs were isolated by SAGE tag mis-priming to the non-parent transcripts. Replacing SAGE tags with non-transcribed genomic DNA tags resulted in poor amplification, indicating that the sequence similarity between different transcripts contributed to the amplification. Our study shows the prevalence of novel low abundance transcripts that can be isolated efficiently through SAGE tags mis-priming.
Keywords: transcript, low abundance, SAGE tag, 3′ EST
1. Introduction
Transcripts are the functional carriers of genes. Transcript isolation is essential for gene identification and for functional study of genes. The abundance of different transcripts can vary over a million-fold [1-4]. The higher abundance transcripts tend to be from a limited number of genes with housekeeping functions, while the lower abundance transcripts tend to be from most of the genes with specialized functions. Isolation of full sets of transcripts expressed from a given genome, regardless of abundance, is an ultimate goal in transcriptome studies.
Transcript isolation has been highly successful during the last decade. For example, the large-scale human EST collection has isolated over 7 million ESTs from the human genome [5-7. http://www.ncbi.nlm.nih.gov/dbEST/dbEST_summary.html). However, recent evidence shows that novel, low-abundance transcripts are widely present in yeast, fly, mouse, rat, Arabidopsis, rice, and human [8-17], indicating that the transcriptome is far more complex than thought [18], and transcript identification is far from complete, even in these extensively characterized model genomes.
The approaches used for large-scale transcript identification include the EST approach that detects transcripts with several hundred bases [5-7] and the SAGE approach that detects transcripts with 10-20 bases [19-20]. Over 7 million copies of human ESTs and 20 million copies of human SAGE tags have been isolated (http://www.ncbi.nlm.nih.gov/projects/SAGE/). While novel transcript identification by the EST approach has decreased dramatically [14], the SAGE approach continues to detect more low abundant transcripts due to its high sensitivity [21]. However, SAGE provides only short sequence information for the detected transcripts. Identification of the original transcripts with longer sequence information will be essential for annotation and functional studies. In this study, we used the GLGI method to convert over a thousand human X chromosome-specific SAGE tags into the 3′ ESTs with the primary aim of identifying the novel transcripts from X chromosome. While nearly half of the isolated 3′ ESTs are novel, most of the 3′ ESTs represent the transcripts from non-X chromosomes. Further analysis reveals that those 3′ ESTs were isolated by SAGE tags through mis-priming mechanism. Here we report the details of the study.
2. Materials and methods
2.1. SAGE data analysis
Experimental 10-base SAGE tags and 17-base long SAGE tags were downloaded from NCBI GEO (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=gpl4, http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=gpl1485). The reference 10-base and 17-base long SAGE tags were downloaded from “SAGEmap-full” of the SAGEmap database (ftp://ftp.ncbi.nlm.nih.gov/pub/sage/map/Hs/). Human genome sequences (NCBI 34) were used for extracting 10-base and 17-base genomic tags at the CATG sites. For sense strand, tags were extracted immediately after each CATG site; for anti-sense strand, tags were extracted before each CATG site with reverse/complementary sequences. Human ESTs were downloaded from NCBI dbEST (ftp://ftp.ncbi.nih.gov/repository/dbEST/) and 10-base and 17-base tags were extracted after the last CATG sites in the 3′ ESTs. Different computational programs written in Perl were used for the comparison between the experimental SAGE tags, reference SAGE tags, and the genomic tags.
2.2. Selection of X chromosome-specific novel SAGE tags
The 315 human SAGE libraries containing 18,966,751 SAGE tags were downloaded (http://www.ncbi.nlm.nih.gov/geo/, December 9, 2005). The SAGE tags were matched to the human SAGEmap reference database (SAGEmap full, release 172, October 25, 2005) to identify the non-matched SAGE tags as novel SAGE tag candidates. A chromosome-based human genomic SAGE tag reference database was constructed by extracting 10-bases and 17-bases after all CATG and before CATG (reverse complementary) using the human genome sequences (NCBI 34). All novel SAGE tags were mapped to the genomic tag reference database to identify those specifically mapped to the human X chromosome.
2.3. Conversion of SAGE tags into 3′ ESTs
Each X chromosome-specific novel SAGE tag was used to design a primer with sequences CAGGGACATGxxxxxxxxxx, where CAGGGA is used to increase the length of the primer, CATG in the SAGE tag is the NlaIII restriction site used for releasing SAGE tags from cDNA templates, and xxxxxxxxxx is the 10-bp SAGE tag sequence. RNA samples from brain and placenta tissues (Stratagene, La Jolla, CA) were treated by using DNase I digestion to eliminate potential genomic DNA contamination, and the purity of digested RNA samples was tested by PCR amplification of the beta-actin gene (GenBank ID NM_001101) using sense primerGGACTTCGAGCAAGAGATGG and antisense primer AGCACTGTGTTGGCGTACAG that span the intron 4 of the beta-actin gene. The amplified genomic DNA will be 329 bp, and the amplified mRNA will be 234 bp.
GLGI reactions were performed to convert SAGE tags into 3′ cDNAs following the procedures described previously [22-23]. Briefly, mRNAs were purified from total RNA samples using oligo (dT)25 magnetic beads (Dynal, Brown Deer, WI). Double strand poly dA/dT (-) cDNAs were synthesized using M-MLV Reverse Transcriptase (Invitrogen, Carlsbad, CA) and the 5′ biotinylated, 3′ anchored oligo (dT) primers (5′ biotin-ATCTAGAGCGGCCGC-T16-A/G/CA/CG/CC), and digested by NlaIII. The 3′ cDNAs after the last CATG were then isolated using streptavidin magnetic beads (Dynal). GLGI reactions were performed in 96-well plates including Hotstart Tag polymerase (Denville, Metuchen, NJ), each sense primer, universal antisense primer ACTATCTAGAGCGGCCGCTT and 3′ cDNAs. The amplified products of each reaction were cloned into the pGEM-T vector (Promega, Madison, WI), transformed into E.coli TOP10 (Invitrogen) and plated in a single well of the 48-well Qtrays (Genetix, Hampshire, UK). Twelve clones from each transformation were selected for sequencing collection. Plasmids were purified by using the Montage Plasmid Miniprep96 Kit (Millipore, Billerica, MA). DNA sequencing reactions were performed using the Big-Dye Terminator v3.1 Cycle Sequencing Kit (ABI, Foster City, CA), and sequences were collected in an ABI 3730XL DNA sequencer using Phred20 as the cut-off. Only the sequences containing the SAGE tag-based sense primer in the 5′ end and the polyA tail at the 3′ end were considered as the qualified 3′ ESTs. The same sequences originated from the same sense primer were combined as a unique sequence. Poly A signals were identified in each sequence by searching the poly A signal sequences AATAAA, ATTAAA AATTAA, AATAAC, AATAAT, AATACA, ACTAAA, AGTAAA, CATAAA, GATAAA, and TATAAA upstream 100 bps from the 3′ end of each sequence [24].
2.4. Confirmation of 3′ ESTs by using RT-PCR
Sense primer and antisense primer were designed based on each selected 3′ EST sequence to generate the amplicons between 100 to 300 bases. One hundred ng of brain or placenta total RNA were used as the templates for cDNA synthesis. For antisense confirmation, cDNA was synthesized by using each antisense primer, and PCR was followed by adding each sense primer. Conditions for PCR amplification were 35 cycles of 94°C for 30 sec, 60°C for 30 sec (50°C was set for DR978181), and 72°C for 1 min, and then extended at 72°C for 7 min. PCR products were checked on 1% agarose gels.
2.5. Estimation of the abundance of the identified transcripts by using real-time PCR
The same sequences and primers used for RT-PCR confirmation were used for real-time PCR. The reactions were performed following the manufacturer′s protocol (Stratagene, La Jolla, CA). Briefly, first-strand cDNA was synthesized by using oligo dT12-18 primer and 1 ug of DNase I treated total RNA in a total volume of 20 ul. Two ul of each primer set and two ul of the synthesized cDNA were added to Full Velocity SYBR® Green QPCR master mix (Stratagene) in a total volume of 25 ul. The mixtures were placed in an Mx3000P instrument (Stratagene) and the PCR program was run at 1 cycle at 95°C for 5 minutes, 45 cycles at 95°C for 30 seconds, 60°C for 60 seconds and 72°C for 60 seconds. Beta-actin transcripts were used as a control representing the high abundance transcripts. The SAGE tag copies of beta-actin in brain and placenta tissues were identified from brain SAGE library GSM763 and placenta SAGE library GSM 14750 (http://www.ncbi.nlm.nih.gov/projects/geo/).
2.6. Control experiments using genomic tag primers and random primers
Genomic DNA sequences of 14-bps were extracted from the non-repetitive genomic regions in the human X chromosome (NCBI 34) that are free from known genes, mRNAs, and ESTs. The 14-bp sequences were filtered through the SAGE tag databases (GPL4 and SAGEmap 187) to exclude any that match existing SAGE tags. Those without matches were used as the sense primer. Six bases (CAGGGA), as used in the SAGE tag-based primers, were added to the 5′ end of each primer to increase the length to 20 bp. The same universal primer, placenta and brain 3′ cDNAs used in the GLGI were used for the reaction. For random priming PCR, the random primers CAGGGACATGnnnnnnnnnn were designed so that CAGGGACATG used in the SAGE tag-based primers was added to the 5′ end to reach the length of 20. The same placenta and brain 3′ cDNAs used for the GLGI reactions were used as the templates for the reaction.
2.7. Match 3′-ESTs to the known human transcripts
Each 3′-EST was searched in known human mRNAs in RefSeq database (ftp://ftp.ncbi.nih.gov/refseq/H_sapiens/mRNA_Prot/) and ESTs (ftp://ftp.ncbi.nih.gov/repository/dbEST/). If the 3′-EST could align with any mRNA or EST with over 95% identity with 90% coverage, the 3′-EST was annotated as “known”. Otherwise, it was defined as “novel”. Each 3′ EST was also matched to the trEST database that contains the predicted human transcript contigs based on UniGene and the EMBL databases [25-26]. Evalue 1.0e-15 was used as the cut-off to divide the matched ones from the unmatched ones [27]. When we compared 3′-EST with trEST and trGEN, we separated 3′-ESTs into five classes: a) known transcript, which the 3′-end of 3′-EST matches less than 10 bp upstream of 3′-end of transcript in database, or the 3′-end of transcript in database matches less than 10 bp upstream of 3′-end of 3′-EST; b) 3′-end shortened transcript, which the 3′-end of 3′-EST matches more than 10 bp upstream of 3′-end of transcript; c) 3′-end extended transcript, which the 3′-end of transcript in the database matches more than 10 bp upstream of 3′-end of 3′-EST; d) other alternative transcript, which are those 3′-ESTs that match transcript in the database with e-value lower than 1.0e-15 but don′t belong to any of the above classes; e) unmatched 3′-ESTs.“.
2.8. Map 3′-ESTs to the human genome
Each 3′-EST was mapped to the human genome by using BLAT with the default parameter settings (NCBI 34). The 3′ ESTs that could not be mapped by BLAT were mapped by BLASTN with the e-value cutoff as 1.0e-1. For fine genomic mapping, we extracted the mapped genomic DNA sequence, and ran the global alignment program ALIGN (from FASTA package) to align the 3′-EST with the corresponding genomic DNA sequences to calculate the global sequence identity. A mapping was determined if more than 90% of a 3′-EST mapped to the genome, and the identity between genomic DNA and 3′-EST was at least 90%. In case of multiple mapped regions, only those with a BLAT or BLAST score no more than 0.5% lower than the score of the best-mapped region were kept, which is the cut-off value used in the human genome browser (http://genome.ucsc.edu/). The 3′-ESTs with more than five mapped loci in the genome were excluded for further analysis. The immediate downstream region of the mapped genomic region was checked to identify potential genomic poly-A sequences. A 3′-EST was marked as ”internal poly A priming“ if more than eight continuous As were located within the 20bp downstream flanking region [28]. Using the annotated RefSeqs and mRNAs in the mapped regions, we classified each mapped 3′ EST into the ”Intergenic“ or ”Intragenic“ group. The ”Intragenic“ group was further classified into “sense” and “antisense” subsets. The “sense” includes “known”, “intronic” (completely mapped in a single intron), “extended 5′ end” (extended more than 10 bp upstream of the annotated 5′-end), “extended 3′ end” (extended more than 10 bp downstream of the annotated 3′ end), “shortened 3′ end” (the 3′-end of a 3′-EST maps more than 10 bp upstream of the annotated 3′ end), “cross-conjunction” (the 3′-EST overlaps the exon-intron conjunction), and “antisense” (the 3′-EST maps to the reversed orientation of annotated gene). Genes mapped to the “intergenic” region were classified into the “known” subset (with EST information) and “novel” subset (with no known transcript information).
3. Results and Discussion
3.1. SAGE data analysis
Three sets of data were used for the analysis, including 1) the experimental human SAGE tags that represent the human transcripts detected by SAGE; 2) the reference human SAGE tags extracted from well-annotated mRNAs and ESTs that represent the known human transcripts detectable by SAGE [29]; and 3) the reference human genomic tags extracted from the human genome sequences that represent the possibly transcribed loci and detectable by SAGE in the human genome. The experimental human SAGE data include the standard SAGE tags of 10-bases (664,615 unique tags identified from 16,350,661 tag copies originated from 307 human SAGE libraries), and long SAGE tags of 17-bases (630,837 unique tags identified from 3,616,090 copies originated from 29 human long SAGE libraries); the reference human SAGE tags include reference 10-base standard SAGE tags and reference 17-base long SAGE tags (577,224 10-base tags and 1,291,619 17-base tags, respectively); the reference human genomic tags include standard 10-base and 17-base long SAGE genomic tags extracted from the human genome sequences adjunct to CATG sites in sense and anti-sense orientations (957,056 standard and 19,618,123 long genomic tags from 26,201,271 genomic locations). The genomic tags provide evidence for the genomic origin of SAGE tags and serve as a discriminator to eliminate artificial SAGE tags from true SAGE tags. An experimental SAGE tag that matches a reference SAGE tag and a reference genomic tag implies that this SAGE tag is originated from a known transcript expressed from the genome; an experimental SAGE tag that has no match to reference SAGE tag but maps to genomic tag implies that this SAGE tag is likely originated from a novel transcript expressed from the genome; an experimental SAGE tag that has no match to reference SAGE tag nor maps to genomic tag implies that the SAGE tag is potentially an artificial tag generated by experimental errors.
Figure 1 shows the results of the comparison. For the 664,615 experimental standard 10-base SAGE tags, 645,061 map to the reference genomic tags, 447,618 (69.4%) of which match to both the reference SAGE tags of known transcripts and reference genomic tags, and 197,443 (30.6%) have no match to the reference SAGE tags but map to the reference genomic tags; for the 630,837 long SAGE tags, 217,640 map to the reference genomic tags, 109,548 (50.3%) of which match both the reference SAGE tags of known transcripts and genomic tags, and 108,092 (49.7%) have no match to the reference SAGE tags but map to the reference genomictags. The abundance of these novel SAGE tags based on their copy numbers in the SAGE libraries shows that the novel SAGE tags have low copy numbers, implying that most of the transcripts detected by novel SAGE tags are the low-abundance transcripts.
Fig. 1.
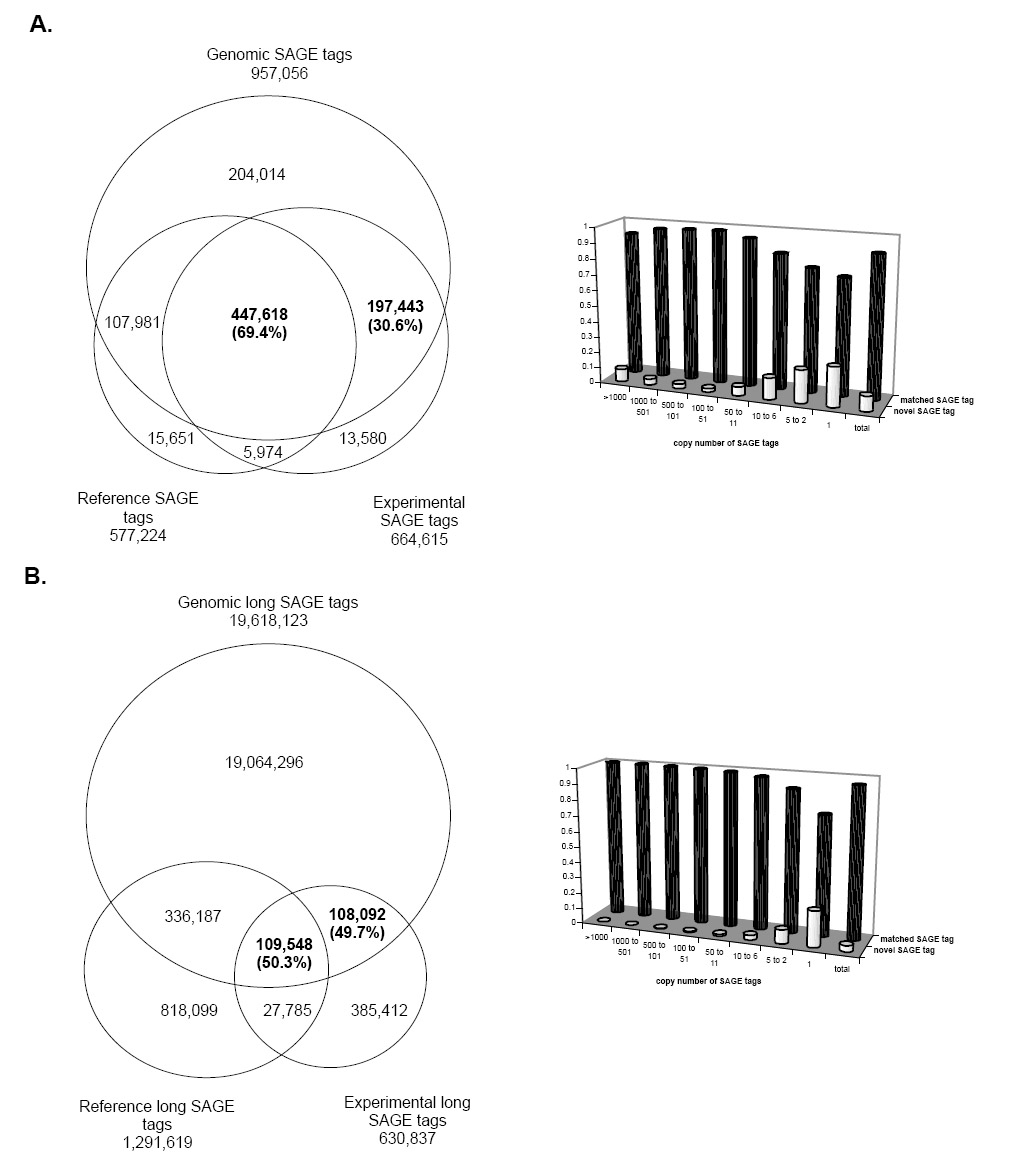
The comparison between SAGE tags, reference SAGE tags of known human transcripts and genomic tags of the human genome sequences. Figure 1A shows the analysis of the standard 10-bp tags. Figure 1B shows the analysis of the 17-bp long tags. Of the experimentally collected SAGE tags that mapped to the genome, 30.6% of the 10-base tags and 49.7% of the 17-base tags have no match to the corresponding reference SAGE tags. The histograms show the copy number distribution of the matched SAGE tags and the novel SAGE tags in their original SAGE libraries. In both types of SAGE tags, the novel SAGE tags have lower copies than the matched SAGE tags.
The actual number of novel transcripts should be higher than that of the novel SAGE tags, based upon the following considerations. Many low abundance transcripts are below the threshold of SAGE detection; a third of the standard 10-base SAGE tags are shared by different transcripts [30]; transcripts lacking the NlaIII site for tag releasing are not detected by SAGE; and certain SAGE tags excluded from this analysis might be from true novel transcripts.
3.2. Conversion of human X chromosome-specific SAGE tags into 3′ ESTs
By searching human SAGE data, we identified 1,009 novel SAGE tags that do not match known human transcripts but map solely to the genomic sequences of the human X chromosome. All these novel SAGE tags have low-copies in their original SAGE libraries. Using the GLGI method, we converted these SAGE tags into 3′ ESTs using RNA samples from brain and placenta. By sequencing 12 clones per GLGI reaction, we obtained 13,824 raw sequences. After excluding unqualified sequences and combining redundant sequences, we identified 3,418 unique 3′ ESTs, each of which contains the original SAGE tag at its 5′ end and polyA tail at its 3′ end (Table 1A). A total of 945 of the 1,009 SAGE tags (94%) contributed these final sequences (Table 1B). The 3,418 3′ ESTs have been deposited in GenBank with accession numbers from DR977574 to DR980991.
Table 1.
Summary of the resulting 3′ ESTs converted from SAGE tags
| A. 3′ ESTs isolated from X chromosome-specific SAGE tags | ||
|---|---|---|
| Items | Numbers | |
| X chromosome-specific SAGE tags | 1,009 | |
| Raw sequences generated from SAGE tags | 13,824 | |
| Sequences not qualified | 3,614 | |
| Sequences qualified | 10,210 | |
| Placenta | 6,561 | |
| Brain | 3,649 | |
| Final set of unique 3′ ESTs | 3,418 | |
| Placenta | 2,328 | |
| Brain | 1,090 | |
| Containing polyA signal | 1,374 | |
| length distribution (bp) | 30 to 688 | |
| B. Number of 3′ ESTs contributed by SAGE tags | ||
|---|---|---|
| Number of SAGE Tags (%) | Contributed 3′ ESTs | Number of 3′ ESTs (%) |
| 134 (13) | 1 | 134 (4) |
| 180 (18) | 2 | 360 (11) |
| 185 (18) | 3 | 561 (16) |
| 176 (17) | 4 | 692 (20) |
| 123 (12) | 5 | 645 (19) |
| 77 (8) | 6 | 474 (14) |
| 31 (3) | 7 | 196 (6) |
| 27 (3) | 8 | 248 (7) |
| 12 (1) | 9 | 108 (3) |
| Total 945 (100) | Total 3,418 (100) | |
3.3. Confirmation of the isolated transcripts
To verify that the isolated sequences were not from contaminated genomic DNA but rather from transcripts, we performed PCR to test the RNA samples by using the sense and anti-sense primers that span an intron of the beta-actin gene. No genomic DNA signal was detected in the RNA samples treated after RNase A. Therefore, the isolated sequences likely originated from RNA rather than genomic DNA contamination (Figure 2A). We then used RT-PCR to verify the isolated transcripts. Sense and antisense primers were designed based on each selected 3′ EST (Supplementary table 1), and RNA samples from brain and placenta were used as the template. To confirm the antisense sequences, cDNA was synthesized by using each antisense primer for PCR amplification. Of the 30 selected 3′ ESTs, 28 were detected, including all 5 antisense sequences detected in both RNA samples, 18 sense sequences detected in both RNA samples and 5 sense sequences detected in either RNA sample (Figure 2B). The positive detection reveals that most of the detected transcripts were indeed present in the RNA sample. Those two negatively detected 3′ ESTs might be related with the poor amplification efficiency of the primers.
Fig. 2.
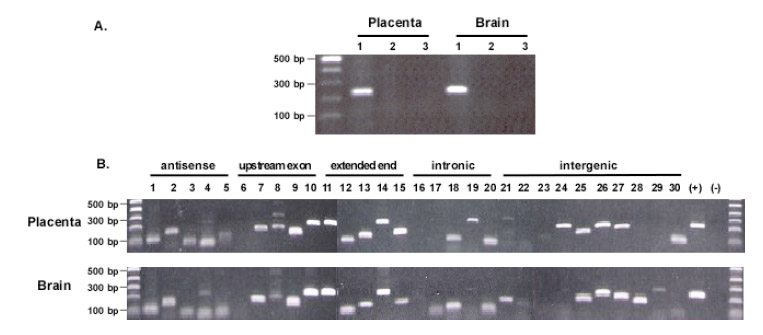
Confirmation of the origin of the isolated sequences. A. Determination of genomic DNA contamination. DNase I digested RNA samples were tested by direct PCR amplification of beta-actin genomic DNA with primers spanning an intron. The amplicons from genomic DNA will be 329 bps, whereas that from mRNA will be 234 bp. 1. RT-PCR with RNA samples. 2. PCR with RNase A digested RNA samples. 3. Negative control without RNA. B. RT-PCR confirmation of detected sequences. A total of 30 isolated sequences were selected for the confirmation. Sense primer and antisense primer were designed based on each sequence. For antisense confirmation, antisense primer was used for cDNA synthesis; for other types of confirmation, oligo dT was used for cDNA synthesis. No. 1, 2, 3, 4, and 5 are the antisense confirmation. The order of the tested sequences is the same as listed in the Supplementary table 1. (+): positive control with beta-actin transcripts; (-): negative control with RNase A digested RNA samples. Most of the sequences, except for a few cases, were detected in both RNA samples with similar size distribution.
3.4. Estimation of the abundance of isolated transcripts
We used real-time RT-PCR to estimate the abundance of the same 30 isolated transcripts used for RT-PCR confirmation (Supplementary Table 1). The beta-actin transcript was used as the reference that was expressed at high abundance levels in both brain and placenta tissues (721 out of the total 63,208 SAGE tag copies in the brain SAGE library and 1,141 out of the total 118,083 SAGE tag copies in the placenta SAGE library). Of the 30 selected sequences, 29 (except DR978181) were detected in both placenta and brain RNA samples. All 29 in the placenta and 28 in the brain (except DR979502) were detected after the beta-actin signal (Figure 3A). The abundance of these transcripts covered several orders of magnitude and most were lower than that of beta-actin; DF979332 had the lowest abundance (Figure 3B). Transcripts not visible in gel by regular RT-PCR at 35 PCR cycles (Figure 2B) were detected by real-time PCR at 45 cycles, suggesting very low abundance levels for no. 6, 16, 17, 22, 23 in both placenta and brain, 28 in placenta, 19, 24 and 30 in brain.
Fig. 3.
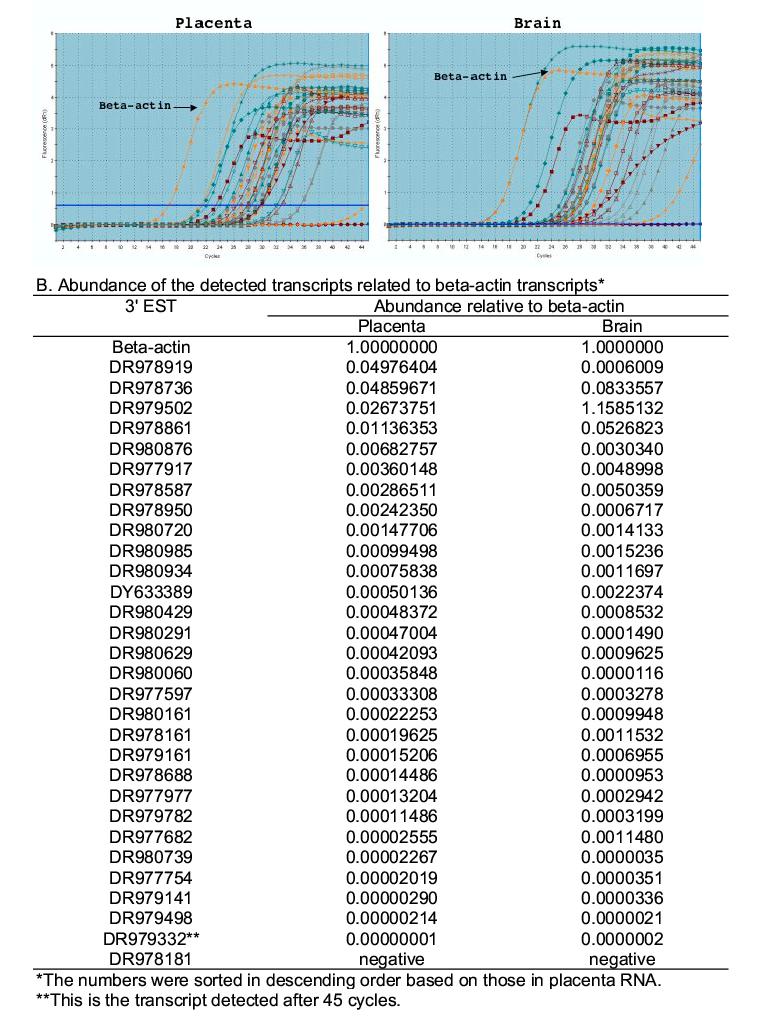
Comparison between SAGE tag-based primer, genomic sequence-based primer and random primer for transcript detection. Reactions were performed by using 30 SAGE tag-based primers (Supplementary table 1), 30 genomic sequence-based primers (Supplementary table 2), and random primers. Placenta and brain samples were used as the templates. The last lanes of the two gels marked by “SAGE tag” were the amplicons from random primers, the last lanes of the two gels marked by “Genomic tag” were the positive control using the SAGE tag primer (no. 16 in supplementary table 1). Note that in the genomic sequence-based reaction, only No. 22 and 28 in placenta RNA and 22 in brain RNA show positive amplification.
3.5. Annotation of the isolated transcripts
We compared the 3′ ESTs to the known human transcripts. The results show that 46% of the 3,418 unique 3′ ESTs represent novel transcripts not identified so far (Table 2). This rate is significantly higher than the less than 5% novelty in large-scale EST collection [14]. We mapped the 3′ ESTs to the human genome to determine their genomic origins (Table 3). Of the 3,418 3′ ESTs, 2,503 are directly mapped to 2,630 loci in the genome, including 2,408 mapped to non-X chromosomes, 89 mapped to X-chromosome and 6 mapped to both X and non-X chromosomes. Of the mapped loci, 28% are at the intergenic regions without annotated genes. Although 72% map to the intragenic regions, 24% are in the antisense strand, 7% of which are novel antisense transcripts. For the 47% mapped to the sense strand of the intragenic regions, 21% map precisely to the annotated 3′ end of the known genes, while the rest map as “intronic,” and different spliced variations. In total, 60% of the 3′ ESTs provide various degrees of novel transcriptional information for these mapped loci. For the 915 3′ ESTs that do not map to the genome, some may represent the transcripts after complex splicing processes.
Table 2.
Comparison of 3’ ESTs with known transcripts
| Class | Number | |
|---|---|---|
| Match to known transcripts | 1,857 (54) | |
| Known mRNA* | 1,175 | |
| EST** | 1,857 | |
| No match | 1,561 (46) | |
| Total | 3,418 (100) |
RefSeq (ftp://ftp.ncbi.nih.gov/refseq/H_sapiens/mRNA_Prot/)
dbEST (ftp://ftp.ncbi.nih.gov/repository/dbEST/).
Table 3.
Mapping 3’ ESTs to the human genome (NCBI 34)
| Classification | No. of mapped 3’ ESTs (%) | No. of mapped loci*(%) |
|---|---|---|
| Intergenic region | 691 (27) | 743 (28) |
| Intragenic region | 1,855 (73) | 1,887 (71) |
| Sense | 1,234 (49) | 1,248 (47) |
| Known | 555 (22) | 565 (21) |
| Intronic | 382 (15) | 383 (14) |
| Shortened 3’-end | 250 (10) | 253 (9) |
| Cross conjunction | 40 (2) | 40 (1) |
| Extended 5’-end | 4 (0) | 4 (0) |
| Extended 3’-end | 3 (0) | 3 (0) |
| Antisense | 621 (25) | 639 (24) |
| Known | 390 (16) | 404 (15) |
| Novel | 231 (9) | 235 (8) |
| Total | 2,546 (100) | 2,630 (100) |
The number of subset may not equal to the sum, due to multiple mapped loci for some 3’-EST.
We also compared 915 3′ ESTs which cannot be matched to genome with the predicted transcript contigs in the trEST database and whole genome transcript prediction in trGEN database [25-26]. The results show that 449 3′-ESTs have matches in trEST or trGEN under E-value cutoff of 1.0e-15, and 466 3′-ESTs still cannot find significant matches in these databases. Based on the definition described in method (section 2.7), we found 173 out of those 449 matched 3′-ESTs belong to known transcripts, while 224, 21 and 81 3′-ESTs belong to “3′-end shortened transcript”, “3′-end extended transcript”, and “other alternative transcript” respectively. Of those 915 genome-unmapped 3′-ESTs, only 173 were related to the predicted known transcripts while 742 (742/915=81%) were unrelated as “novel”.
We also analyzed those filtered sequences. We set a higher than 95% of the maximum score as a tolerable cut-off for the analysis. There are a total of 1,455 loci mapped by 157 3′-ESTs with the scores between 95% and 99.5%. Using 99.5% of maximum match score as cutoff, we found only 231 loci in the genome for those 157 3′-ESTs. The classification for those mapped loci shows that 933 mapped to the intergenic region, and 522 mapped to the intragenic region of which 319 mapped to antisense orientation. For the 203 mapped to the sense orientation, only 25 mapped to known exon, and the rest mapped to intronic, and various regions in the mapped genes. Those mapping results show that there are more novel transcribed loci detected by those sequences although their precise loci cannot be assigned precisely due to the multiple mapping.
3.6. Identification of the mismatched bases between the SAGE tag part of 3′ ESTs and the mapped genomic sequences
All SAGE tags used for the experiment map only to the X chromosome. However, the majority of isolated 3′ ESTs are the transcripts originated from non-X chromosomes. Using the 2,417 3′ ESTs that map to single loci in the genome, we compared the 14-bp SAGE tag sequences of these 3′ ESTs and their mapped genomic sequences. Interestingly, we observed widely spread mismatches/gaps between these 14-bp counterpart sequences (Table 4). Overall, 98% of the mappings contain mismatches/gaps; mismatches account for 76% and gaps account for 24%. The numbers of mismatched/gapped bases are predominated by the 1- and 2-bases that are located in the middle of the SAGE tag sequences. Because the 14-bp SAGE tag sequences are from the synthesized sense primer, the mismatches/gaps between the 14-bp of the isolated 3′ ESTs and the genomic sequences cannot be related to the issue of fidelity for the PCRamplification and DNA sequencing reaction. Therefore, these transcripts must have been isolated by the SAGE tags through mis-priming.
Table 4.
Mis-matched bases between the SAGE tag part of 3' ESTs and their mapped genomic regions*
| No. mismatched bases | Location of mismatches/gaps in SAGE tag part of 3’ ESTs |
Total No. 3’ ESTs (%) | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C | A | T | G | x | x | x | x | x | x | x | x | x | x | ||
| 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 139 (6) |
| 1 | 8 | 2 | 6 | 1 | 156 | 229 | 137 | 114 | 75 | 87 | 49 | 39 | 17 | 3 | 923 (38) |
| 2 | 74 | 33 | 27 | 6 | 238 | 236 | 180 | 160 | 95 | 72 | 31 | 25 | 12 | 5 | 597 (25) |
| 3 | 135 | 67 | 44 | 24 | 92 | 100 | 79 | 54 | 40 | 26 | 47 | 14 | 3 | 4 | 243 (10) |
| 4 | 149 | 134 | 118 | 80 | 61 | 46 | 39 | 32 | 24 | 25 | 23 | 15 | 10 | 4 | 190 (8) |
| 5 | 125 | 138 | 140 | 119 | 85 | 69 | 28 | 30 | 25 | 18 | 14 | 6 | 7 | 6 | 162 (7) |
| 6 | 77 | 82 | 75 | 68 | 63 | 59 | 31 | 17 | 16 | 18 | 6 | 5 | 8 | 3 | 88 (4) |
| 7 | 19 | 20 | 22 | 19 | 20 | 17 | 19 | 11 | 7 | 4 | 5 | 5 | 4 | 3 | 25 (1) |
| 8 | 9 | 9 | 6 | 8 | 7 | 9 | 8 | 4 | 7 | 9 | 4 | 6 | 5 | 5 | 12 (0) |
| 9 | 19 | 17 | 11 | 16 | 18 | 13 | 17 | 12 | 10 | 11 | 13 | 14 | 10 | 8 | 21 (1) |
| 10 | 8 | 8 | 8 | 8 | 7 | 10 | 7 | 8 | 4 | 6 | 6 | 4 | 8 | 8 | 10 (0) |
| 11 | 5 | 3 | 3 | 4 | 5 | 4 | 5 | 4 | 4 | 5 | 5 | 3 | 3 | 2 | 5 (0) |
| 12 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 (0) |
| 13 | 2 | 2 | 1 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 (0) |
| 14 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 (0) |
| Total | 630 | 515 | 461 | 355 | 753 | 794 | 552 | 448 | 309 | 283 | 205 | 138 | 89 | 53 | 2,417 (100) |
Those include 92 3’ ESTs mapped to X chrosome of which 23 mapped to the locations expected by the original SAGE tags.
3.7. Tags from non-transcribed genomic sequences provide poor amplification
To investigate if genomic sequence-based primers could also result in the amplification as seen in the SAGE tag-based primers, we used the non-transcribed genomic DNA sequences in the human X chromosome as the primers for the reaction (Supplementary table 2). The results show that, of the 30 reactions tested, 28 reactions in placenta and 29 in brain generated negative results (Figure 4). Those results show that genomic DNA sequences do not provide amplification as efficiently as SAGE tag sequences. The results also imply that the sequence similarity represented by the SAGE tag sequences between different transcripts might account for their highly efficient amplification.
Fig. 4.
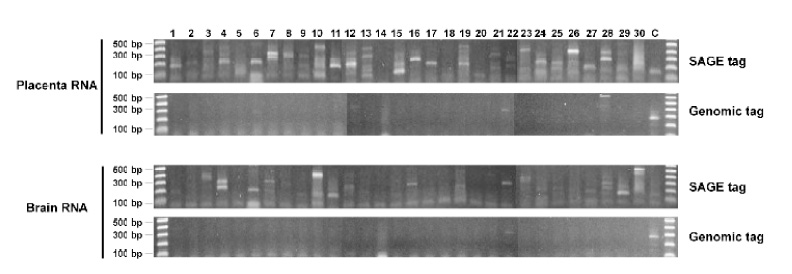
Quantitative measurement of the abundance of the detected sequences by real time PCR. The same sequences, primers and RNA samples used in Figure 2 were used for this analysis. See supplementary table 1 for detailed sequence information. (No. 2 sequence DR978181 was not used for this analysis, as its melting temperature is only 53°C, far lower than the 60°C recommended for real-time PCR). The beta-actin transcripts were used as positive control. A. Histogram of real-time PCR showing the amplification dynamics for each detected sequence. B. Relative abundance of each sequence normalized to beta-actin. Note that the weakly amplified templates by regular PCR in Figure 4 were well reflected by their lower abundance detected by real time PCR.
Our study shows that many identified transcripts are at low abundance levels from either the unannotated regions in the genome, or from the annotated genes but with complex sequence variations or from antisense of the annotated genes. The current definition of “higher” or “lower” abundance of transcripts may largely reflect our ability for transcript detection rather than biological significance of the detected genes. The quantitative range covering over six orders of magnitudes between different transcripts determines that the depth of transcript isolation depends largely upon the sensitivity of techniques used. The conventional EST approach has limited power to isolate low abundance transcripts due largely to the issue of cost-efficiency. The PCR approach is very sensitive at detecting the low abundance transcripts, but the required sequence information for two-primer design restricts its use in detecting only the known transcripts. Random primer has been applied successfully for large-scale EST isolation, but it mainly detects the middle region of the targeted transcript population without the 5′ or 3′ end sequence information [31]. In this study, we used the PCR-based GLGI technique for the detection that requires only one primer (a SAGE tag) to generate 3′ ESTs. The observed mis-priming between a SAGE tag and multiple transcripts makes it possible for pan-genome detection of low abundance transcripts.
Supplementary Material
Acknowledgments
Acknowledgements: The study was supported by National Institutes of Health (HG002600), the Daniel F. and Ada L. Rice Foundation, and Mazza Foundation.
Footnotes
Corresponding author: San Ming Wang, MD Center for Functional Genomics ENH Research Institute, Northwestern University, 1001 University Place, Evanston, IL 60201 Tel: 224-364-7491; Fax: 224-364-5003; Email: swang1@northwestern.edu
References
- [1].Bishop JO, Morton JG, Rosbach M, Richardson M. Three abundance classes in HeLa cell messenger RNA. Nature. 1974;250:199–204. doi: 10.1038/250199a0. [DOI] [PubMed] [Google Scholar]
- [2].Holland MJ. Transcript abundance in yeast varies over six orders of magnitude. Journal of Biological Chemistry. 2002;277:14363–14366. doi: 10.1074/jbc.C200101200. [DOI] [PubMed] [Google Scholar]
- [3].Czechowski T, Bari RP, Stitt M, Scheible WR, Udvardi MK. Real-time RT-PCR profiling of over 1400 Arabidopsis transcription factors: Unprecedented sensitivity reveals novel root- and shoot-specific genes. Plant Journal. 2004;38:366–379. doi: 10.1111/j.1365-313X.2004.02051.x. [DOI] [PubMed] [Google Scholar]
- [4].Carter MG, Sharov AA, VanBuren V, Dudekula DB, Carmack CE, Nelson C, Ko MS. Transcript copy number estimation using a mouse whole-genome oligonucleotide microarray. Genome Biol. 2005;6:R61. doi: 10.1186/gb-2005-6-7-r61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Adams MD, Dubnick M, Kerlavage AR, Moreno R, Kelley JM, Utterback TR, Nagle JW, Fields C, Venter JC. Sequence identification of 2,375 human brain genes. Nature. 1992;355:632–634. doi: 10.1038/355632a0. [DOI] [PubMed] [Google Scholar]
- [6].Bonaldo MF, Lennon G, Soares MB. Normalization and subtraction: Two approaches to facilitate gene discovery. Genome Research. 1996;6:791–806. doi: 10.1101/gr.6.9.791. [DOI] [PubMed] [Google Scholar]
- [7].Strausberg RL, Dahl CA, Klausner RD. New opportunities for uncovering the molecular basis of cancer. Nat Genet, Spec No. 1997;415:416. doi: 10.1038/ng0497supp-415. [DOI] [PubMed] [Google Scholar]
- [8].Wyers F, Rougemaille M, Badis G, Rousselle JC, Dufour ME, Boulay J, Regnault B, Devaux F, Namane A, Seraphin B, et al. Cryptic pol II transcripts are degraded by a nuclear quality control pathway involving a new poly (A) polymerase. Cell. 2005;121:725–737. doi: 10.1016/j.cell.2005.04.030. [DOI] [PubMed] [Google Scholar]
- [9].Lee S, Bao J, Zhou G, Shapiro J, Xu J, Shi RZ, Lu X, Clark T, Johnson D, Kim YC, et al. Detecting novel low-abundance transcripts in Drosophila. RNA. 2005;11:939–946. doi: 10.1261/rna.7239605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Okazaki Y, Furuno M, Kasukawa T, Adachi J, Bono H, Kondo S, Nikaido I, Osato N, Saito R, Suzuki H, et al. Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature. 2002;420:563–573. doi: 10.1038/nature01266. [DOI] [PubMed] [Google Scholar]
- [11].Scheetz TE, Laffin JJ, Berger B, Holte S, Baumes SA, Brown R, 2nd, Chang S, Coco J, Conklin J, Crouch K, et al. High-throughput gene discovery in the rat. Genome Research. 2004;14:733–741. doi: 10.1101/gr.1414204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Seki M, Narusaka M, Kamiy A, Ishida J, Satou M, Sakurai T, Nakajima M, Enju A, Akiyama K, Oono Y, et al. Functional annotation of a full-length Arabidopsis cDNA collection. Science. 2002;296:141–145. doi: 10.1126/science.1071006. [DOI] [PubMed] [Google Scholar]
- [13].Bao J, Lee S, Chen C, Zhang X, Zhang Y, Liu S, Clark T, Wang J, Cao M, Yang H, et al. Serial Analysis of Gene Expression Study of a Hybrid Rice Strain (LYP9) and Its Parental Cultivars. Plant Physiol. 2005;138:1216–1231. doi: 10.1104/pp.105.060988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wang SM, Fears SC, Zhang L, Chen JJ, Rowley JD. Screening poly (dA/dT)-cDNAs for gene identification. Proc Natl Acad Sci U S A. 2000;97:4162–4167. doi: 10.1073/pnas.97.8.4162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bertone P, Stolc V, Royce TE, Rozowsky JS, Urban AE, Zhu X, Rinn JL, Tongprasit W, Samanta M, Weissman S, et al. Global identification of human transcribed sequences with genome tiling arrays. Science. 2000;306:2242–2246. doi: 10.1126/science.1103388. [DOI] [PubMed] [Google Scholar]
- [16].Imanishi T, Itoh T, Suzuki Y, O’Donovan C, Fukuchi S, Koyanagi KO, Barrero RA, Tamura T, Yamaguchi-Kabata Y, Tanino M, et al. Integrative annotation of 21,037 human genes validated by full-length cDNA clones. PLoS Biology. 2004;2:856–875. doi: 10.1371/journal.pbio.0020162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Cheng J, Kapranov P, Drenkow J, Dike S, Brubaker S, Patel S, Long J, Stern D, Tammana H, Helt G, et al. Transcriptional Maps of 10 Human Chromosomes at 5-Nucleotide Resolution. Science. 2005;20:1149–1154. doi: 10.1126/science.1108625. [DOI] [PubMed] [Google Scholar]
- [18].Carninci P. Tagging mammalian transcription complexity. Trends in Genetics. 2006 doi: 10.1016/j.tig.2006.07.003. doi:10.1016/j.tig.2006.07.003. [DOI] [PubMed] [Google Scholar]
- [19].Velculescu VE, Zhang L, Vogelstein B, Kinzler KW. Serial analysis of gene expression. Science. 1995;270:484–487. doi: 10.1126/science.270.5235.484. [DOI] [PubMed] [Google Scholar]
- [20].Saha S, Sparks AB, Rago C, Akmaev V, Wang CJ, Vogelstein B, Kinzler KW, Velculescu VE. Using the transcriptome to annotate the genome. Nature Biotechnology. 2002;20:508–512. doi: 10.1038/nbt0502-508. [DOI] [PubMed] [Google Scholar]
- [21].Chen J, Sun M, Lee S, Zhou G, Rowley JD, Wang SM. Identifying novel transcripts and novel genes in the human genome by using novel SAGE tags. Proc. Natl. Acad. Sci. USA. 2002;99:12257–12262. doi: 10.1073/pnas.192436499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Chen JJ, Rowley JD, Wang SM. Generation of longer cDNA fragments from serial analysis of gene expression tags for gene identification. Proc. Natl. Acad. Sci. USA. 2000;97:349–453. doi: 10.1073/pnas.97.1.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Chen J, Lee S, Zhou G, Wang SM. High-throughput GLGI procedure for converting a large number of serial analysis of gene expression tag sequences into 3′ complementary DNAs. Genes, Chromosomes and Cancer. 2002;33:252–261. doi: 10.1002/gcc.10017. [DOI] [PubMed] [Google Scholar]
- [24].Caron H, van Schaik B, van der Mee M, Baas F, Riggins G, van Sluis P, Hermus MC, van Asperen R, Boon K, Voute PA, Heisterkamp S, et al. The human transcriptome map: clustering of highly expressed genes in chromosomal domains. Science. 2001;291:1289–1292. doi: 10.1126/science.1056794. [DOI] [PubMed] [Google Scholar]
- [25].Pagni M, Iseli C, Junier T, Falquet L, Jongeneel V, Bucher P. trEST, trGEN and Hits: access to databases of predicted protein sequences. Nucleic Acids Res. 2001;29:148–151. doi: 10.1093/nar/29.1.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Sperisen P, Iseli C, Pagni M, Stevenson BJ, Bucher P, Jongeneel CV. trome, trEST and trGEN: databases of predicted protein sequences. Nucleic Acids Res. 2004;32:D509–511. doi: 10.1093/nar/gkh067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Dias NE, Correa RG, Verjovski-Almeida S, Briones MR, Nagai MA, da Silva W, Jr, Zago MA, Bordin S, Costa FF, Goldman GH, et al. Shotgun sequencing of the human transcriptome with ORF expressed sequence tags. Proc. Natl Acad. Sci. USA. 2000;97:3491–3496. doi: 10.1073/pnas.97.7.3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Nam DK, Lee S, Zhou G, Cao X, Wang C, Clark T, Chen J, Rowley JD, Wang SM. Oligo(dT) primer generates a high frequency of truncated cDNAs through internal poly(A) priming during reverse transcription. Proc. Natl. Acad. Sci. USA. 2002;99:6152–6156. doi: 10.1073/pnas.092140899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lal A, Lash AE, Altschul SF, Velculescu V, Zhang L, McLendon RE, Marra MA, Prange C, Morin PJ, Polyak K, et al. A public database for gene expression in human cancers. Cancer Research. 1999;59:5403–5407. [PubMed] [Google Scholar]
- [30].Ge X, Jung YC, Wu Q, Kibbe WA, Wang SM. Annotating nonspecific SAGE tags with microarray data. Genomics. 2006;87:173–180. doi: 10.1016/j.ygeno.2005.08.014. [DOI] [PubMed] [Google Scholar]
- [31].Camargo AA, Samaia HP, Dias-Neto E, Simao DF, Migotto IA, Briones MR, Costa FF, Nagai MA, Verjovski-Almeida S, Zago MA, et al. The contribution of 700,000 ORF sequence tags to the definition of the human transcriptome. Proc. Natl. Acad. Sci. USA. 2001;98:12103–12108. doi: 10.1073/pnas.201182798. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.