

Abstract
We have found that mutant acetylcholine receptor channels (AChRs) that cause slow-channel congenital myasthenic syndromes are activated by serum and that the high frequency of openings in serum is reduced by treatment with choline oxidase. Thus, slow-channel congenital myasthenic syndrome AChRs at the neuromuscular junction are likely to be activated both by steady exposure to serum choline and by transient exposure to synaptically released transmitter. Single-channel kinetic analyses indicate that the increased response to choline is caused by a reduced intrinsic stability of the closed channel. The results suggest that a mutation that destabilizes the inactive conformation of the AChR, together with the sustained exposure of endplates to serum choline, results in continuous channel activity that contributes to the pathophysiology of the disease.
Allosteric proteins in signal transduction or metabolic pathways must respond promptly on binding effector molecule(s) yet remain silent in the absence of the appropriate stimulus. Mutations that inhibit the activation process and mutations that cause the protein to be spontaneously active are both pathogenic. Examples of mutated proteins that are spontaneously active and associated with a disease include superoxide dismutase (amyotrophic lateral sclerosis; ref. 1), rhodopsin (autosomal dominant retinitis pigmentosa; ref, 2), tyrosine kinase (multiple endocrine neoplasia type 2; ref. 3), and Ca2+ receptors (familial hypoparathyroidism; ref. 4). Along similar lines, the lurcher and weaver mouse neurological phenotypes have been shown to arise from gain-of-function mutations in the δ2 glutamate receptor subunit gene (5) and a potassium channel (GIRK2) gene (6), respectively.
Nicotinic acetylcholine (ACh) receptors (AChRs) are pentameric ligand-gated ion channels that mediate excitatory synaptic transmission at the vertebrate motor endplate. Normally, they show an extremely low level of spontaneous activity (7) and activate rapidly [<20 μs (8, 9)] and with a high probability when exposed to ACh released from the presynaptic terminal. Several AChR gain-of-function mutations have been identified that result in increased spontaneous activity (10) or that cause ligands that are normally inert to become activators of the channel (11).
The slow-channel congenital myasthenic syndromes (SCCMS) (12) are dominantly inherited disorders of neuromuscular transmission characterized by muscle weakness and fatigability (13, 14). Although mutations that cause SCCMS occur in different domains and different subunits of the AChR, all SCCMS mutations produce prolonged endplate currents owing to the altered transmitter binding and/or channel-gating properties of the receptor (15–19). One consequence of these primary defects of AChR is a degeneration of the junctional folds (13, 20).
Here we present evidence that in addition to having a prolonged synaptic response, SCCMS mutant AChR exhibit increased levels of spontaneous activity and responsiveness to an ordinary serum metabolite, choline. The results suggest that the steady, inappropriate activation of AChR by serum choline is likely to contribute to the syndrome.
MATERIALS METHODS
Expression and Electrophysiology.
The details of the methodology are given in ref. 21. Human embryonic kidney cells (HEK 293) were transiently transfected by calcium phosphate precipitation either with human or mouse α-, β-, δ-, and ɛ-subunits. The human β- and ɛ-subunit cDNAs were provided by A.G.E. The human α- and δ-subunits cDNAs were provided by J. Lindstrom (Univ. of Pennsylvania, Philadelphia). The mouse subunits were provided by S. Sine (Mayo Clinic and Foundation); the α subunit had a background mutation at position 433 (21).
Forty-eight hours after transfection, single-channel currents were recorded from cell-attached patches (22). The bath solution was Dulbecco’s PBS (137 mM NaCl/0.9 mM CaCl2/2.7 mM KCl/1.5 mM KH2PO4/0.5 mM MgCl2/6.6 mM Na2HPO4, pH 7.4). The pipette solution was either 142 mM KCl, 5.4 mM NaCl, 1.8 mM CaCl2, 1.7 mM MgCl2, and 10 mM Hepes (pH 7.4) or mixed human serum (from four individuals; obtained from the Buffalo Veterans Administration Hospital, Buffalo, NY). The temperature was 23°C, and the membrane potential was estimated to be −70 mV.
To facilitate seal formation in the serum experiments, the pipette tip was filled with PBS and the shank was back-filled with serum. In these experiments, currents were recorded only >10 min after seal formation to allow the serum components to diffuse to the pipette tip (23). Currents were amplified (Axopatch-1B; Axon Instruments, Foster City, CA), digitized at 94 kHz, and recorded on tape (VR10b; Instrutech, Great Neck, NY; −3 dB at 22 kHz), and transferred directly to a PC (VR-111; Instrutech).
Although the HEK cells were always transfected with all four AChR subunits, channels with nonstandard subunit assemblies might have been expressed (24–27). To test this possibility, HEK cells were transfected with only three wild-type subunits (αδɛ, αβɛ, or αβδ). We were unable to record either un- or diliganded activity with the β- or δ-less receptors (three transfection trials), but ɛ-less AChR exhibited both spontaneous openings and clusters of openings in response to ACh. The kinetic and dose–response properties of ACh-activated, ɛ-less AChR are distinctive (27) and were not observed in cells transfected with ɛT264P (28). This result suggests that for this mutant, the expressed AChRs had the full complement of subunits and that the parameters shown in Table 1 do not reflect ɛ-less AChR.
Table 1.
Activation parameters for unliganded and choline-occupied AChRs
| AChR | Kd, μM | nβ0, s−1 | α0, s−1 × 104 | θ0 (β0/α0) × 10−6 | β2, s−1 | α2, s−1 | θ2 (β2/α2) |
Kd/Jd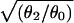
|
|---|---|---|---|---|---|---|---|---|
| Wild-type | — | 1.2, 1.3 | 1.4, 1.7 | 3.8 | 53 ± 18 (5) | 3600 ± 1100 (6) | 0.015 | 63 |
| αG153S | 540 ± 110 | 65, 18 | 0.5, 0.7 | 350 | 960 ± 230 (16) | 2700 ± 220 (3) | 0.35 | 32 |
| αN217K | 2600 ± 400 | 131 ± 36 (33) | 1.1 ± 0.1 (3) | 600 | 2100 ± 610 (15) | 1700 ± 160 (3) | 1.2 | 45 |
| αS269I | 1900 ± 370 | 3.3, 9.7 | 2.2, 1.0 | 20 | 1500 ± 1000 (5) | 1400 ± 510 (3) | 1.0 | 220 |
| βV266M | 2900 ± 800 | 164, 179 | 2.1, 1.8 | 450 | 90 ± 35 (4) | 810 ± 130 (3) | 0.11 | 16 |
| ɛT264P | 3000 ± 400 | 470 ± 120 (3) | 2.3 ± 0.7 (3) | 1020 | 800 ± 200 (7) | 450 ± 47 (3) | 1.8 | 42 |
| ɛL269F | 720 ± 700 | 113, 57 | 1.2, 1.9 | 270 | 530 ± 30 (4) | 720 ± 270 (3) | 0.74 | 52 |
All AChR were mouse, except βV266M and ɛL269F, which were human. Kd is the dissociation equilibrium constant of the closed AChR; Jd is the dissociation equilibrium constant of the open AChR, θ0 is the unliganded gating equilibrium constant, and θ2 is the diliganded gating equilibrium constant. β2 is the diliganded opening rate constant, α2 is the diliganded closing rate constant, β0 is the unliganded opening rate constant, n is the number of channels in the patch (assumed to be 20 in the calculation of θ0), and α0 is the unliganded closing rate constant. Values are mean ± SD (number of patches in parentheses).
Choline Oxidase Treatment.
Choline oxidase (oxygen 1-oxidoreductase, EC 1.1.3.17) was purchased from Sigma (catalog no. C5896). One milliliter of serum was mixed with 0.5 mg of the enzyme (≈5 units) and incubated at 37°C for 15 min. As a control, serum was incubated without adding choline oxidase.
Single-Channel Analysis.
Kinetic modeling of diliganded AChR was carried out on single-channel openings elicited by using ACh or choline in the patch pipette. Currents were idealized either by using a program that tracked the baseline and detected events via a half-amplitude algorithm (iproc; ref. 29), or a program that detected events via a hidden Markov algorithm (skm; www.qub.buffalo.edu). At high agonist concentrations, openings occur in clusters that reflect the activity of a single AChR (30). The cluster-based analysis method is described in detail elsewhere (21). Briefly, clusters were defined as a series of openings separated by closed intervals longer than some critical duration. This duration was ≈10 times greater than the longest component of intracluster closed time that scaled with the agonist concentration. For each patch, the open and closed intervals from >20 clusters were analyzed.
In adult wild-type mouse AChR, the two agonist-binding sites have similar equilibrium dissociation constants (21), and the following kinetic scheme was used to interpret the single-channel currents:
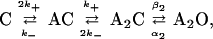 |
Scheme 1
where A is the agonist concentration, C is a closed AChR, O is an open AChR, k+ is the association rate constant, k− is the dissociation rate constant, β2 is the diliganded AChR channel-opening rate constant, and α2 is the diliganded AChR channel-closing rate constant. The rate constants were estimated by using the program mil, with a correction for missed events (31, 32) (www.qub.buffalo.edu). The dead time was 25 μs.
When the currents were clustered, the single-channel open probability (po) was estimated directly as the cluster-open probability. The rate and equilibrium constants (±SD) for diliganded AChR shown in Table 1 were estimated as follows. β2 was estimated from the saturation limit of the effective opening rate dose–response curve (fitted by the Hill equation), except for the wild-type value, which was the average effective opening rate at 20 mM choline. α2 was estimated from the inverse of the main component of the open-channel lifetime at low choline concentrations. The diliganded AChR gating equilibrium constant, θ2, is simply the ratio β2/α2. The dissociation equilibrium constant of a single transmitter binding site from a closed AChR (Kd = k−/k+) was estimated from fitting the following equation, derived from Scheme 1:
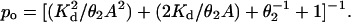 |
1 |
When the currents were not clustered, as in the absence of agonists, po could not be estimated directly. However, the probability of being open for a patch (Po) was computed by fitting the baseline component of the patch all-point amplitude histogram by a single Gaussian function. The number of samples in this component, divided by the total number of samples in the record, is the probability of being closed for the patch (Pc), and Po = 1 − Pc.
The durations of unliganded openings of both wild-type and SCCMS mutant AChR were distributed as the sum of one to three exponential components (ref. 33; C. Grosman and A.A., unpublished data). The gating rate constants of unliganded AChR were estimated by using a kinetic model that had a single closed state and multiple open states (one for each component) connected only to the closed state. In Table 1, the spontaneous opening rate (nβ0, where n is the number of channels in the patch) and closing rate constant (α0) pertain to the shortest-lived open state, which was also the most prevalent.
Because we studied cell-attached patches, the value of n was unknown. To estimate the single-channel spontaneous opening equilibrium constant (θ0 = β0/α0), we assumed that n = 20 (34). The Kd/Jd ratios shown in Table 1 (where Jd is the dissociation equilibrium constant of a single transmitter binding site from an open AChR) were calculated from the estimates of θ2 and θ0, by using Eq. 2. The estimates of θ0 and Kd/Jd totally depend on the estimated number of channels in the patch and therefore should be considered to be approximations.
Endplate Current Simulations.
Endplate currents were simulated by using mcell (Version 1.21; ref. 9). The geometry of the synaptic cleft was defined as an open box, with the AChR distributed on a 5 × 5 μm area that was 0.05 μm from the presynaptic membrane. The AChR density was 7,250/μm2, and the acetylcholine esterase (AChE) density was 2,000/μm2. A packet of 7,000 agonists molecules was released at time zero, and each molecule was followed with a time resolution of 1 μs. The diffusion constant of ligand in the cleft was 2.1 × 10−6 cm2/s.
The action of AChE was modeled by the following scheme: AChE + ACh → AChE–ACh. The association rate constant rate was 1.5 × 108 M−1⋅s−1, and the formation of AChE–ACh resulted in the disappearance of one ACh at a rate constant of 3,750 s−1 (9).
Scheme 1 was used to describe AChR activation; channel block and desensitization were not taken into account. The rate constants for wild-type mouse AChR exposed to ACh were from ref. 21: k+ = 1.35 × 108 M−1⋅s−1, k− = 33,000 s−1, β2 = 48,000 s−1, and α2 = 1,250 s−1. The rate constants for αG153S AChR exposed to ACh were (from ref. 16): k+1 = 7.88 × 108 M−1⋅s−1, k+2 = 9.5 × 107 M−1⋅s−1, k−1 = 1,300 s−1, k−2 = 970 s−1, β2 = 45,000 s−1, and α2 = 883 s−1.
To simulate the maximum possible contribution of choline to the endplate current, a packet of 7,000 choline molecules was released at time zero, and the effect of AChE was turned off. For αG153S AChR, the activation rate constants were derived from single-channel kinetic analysis (Fig. 4): k+ = 8.5 × 108 M−1⋅s−1, k− = 5,300 s−1, β2 = 800 s−1, and α2 = 3,020 s−1.
Figure 4.

Kinetic analysis and miniature endplate current simulations of mouse αG153S AChR. (A) Kinetic modeling of αG153S single-channel currents. (Top) Optimized rate constants (Scheme 1, assuming equivalent binding sites). (Bottom) Example αG153S AChR clusters elicited by choline (0.5, 1, and 2 mM in saline; top to bottom). To the right are the corresponding interval duration histograms. The solid lines are drawn according to the optimized rate constants. (B) Simulated miniature endplate currents. The rate constants and other parameters used in the simulations are given in Materials and Methods. The wild-type mepc has been scaled to have the same peak amplitude as the αG153S mepc. At time = 0, 7,000 molecules of ACh or choline were released into the synaptic gap. With the release of ACh, the simulated mepc decay time constant for αG153S AChR is much longer than for the wild-type, which agrees quantitatively with in vitro results. With the release of choline, the maximum endplate current is ≈3% of that generated by the release of ACh. This result suggests that choline arising from the hydrolysis of ACh does not contribute significantly to the αG153S endplate current.
RESULTS
Fig. 1 shows the activity of wild-type and a SCCMS mutant AChR (ɛT264P) recorded from cell-attached patches containing either pure saline or serum. In these experiments, there was no exogenous agonist added to the pipette solution. For wild-type AChR, Po in pure saline was extremely low and was only slightly higher in serum. For ɛT264P, Po in saline was low but was higher than for the wild type. However, in serum, Po for ɛT264P AChRs was >40 times higher than for the wild type (10 patches).
Figure 1.

Serum choline activates SCCMS mutant AChRs. Example currents (each trace is 1 sec; traces are continuous and are separated by 10 pA; inward current is down) and all-point amplitude histograms (logarithmic scale) from human AChR exposed to different solutions that did not have any exogenous agonist. Wild-type AChR rarely opens when exposed either to saline (A) or to human serum (B). The SCCMS mutant ɛT264P opens with a significant probability when exposed to serum (B). Treatment of the serum with choline oxidase eliminates this activation (C), indicating that choline is the serum component that is causing the activity.
In cell-attached patches from transiently transfected HEK cells, the level of activity of ɛT264P AChR exposed to serum remained high for >2 h, i.e., the response did not diminish over time. This result indicates that exposure to serum causes cells expressing ɛT264P AChR to have a continuous, high probability of being open.
Po in serum and in saline was examined for seven different human SCCMS AChRs. The results are summarized in Fig. 2. For all seven mutants, Po in serum was significantly higher than for the wild type. In addition, all of the SCCMS mutants showed an increased spontaneous activity in saline. The rate and equilibrium constants for unliganded gating (i.e., in pure saline) are shown in Table 1.
Figure 2.

Spontaneous activity of human AChR in saline and in serum. (Left) Example currents recorded in serum, without the addition of agonist to the patch solution. (Right) The mean patch open probability (2–10 patches) for wild-type and SCCMS AChRs exposed to saline (filled bars) or to human serum (open bars). SCCMS AChR have a higher open probability in saline and are more strongly activated by serum than the wild type (note the logarithmic scale). The vertical white lines in ɛT264P and αS269I indicate the patch open probabilities in serum that was pretreated with choline oxidase. The reduction in spontaneous activity after choline oxidase treatment indicates that the component of serum that activates the mutant AChR is choline. The calibration of time is 50 ms except for αV249F, which is 750 ms.
Choline, a quaternary amine precursor in the synthesis of ACh, is present in serum at a concentration of 10–20 μM (35). Choline is an agonist of the α7 subtype of neuronal AChRs (36–38), and therefore we considered whether this serum metabolite might be responsible for the increased activity of the mutant AChR. The bottom row of Fig. 1C shows that preincubation of serum with choline oxidase, an enzyme that converts choline into betaine, greatly reduces the ability of serum to induce openings. The effects of choline oxidase treatment for two SCCMS mutants, ɛT264P and αS269I, are shown in Fig. 2B. In both cases, pretreatment of the serum with the enzyme reduced the probability of being open in serum. These results suggest that the serum component responsible for activation of SCCMS AChR is choline and that betaine is not a significant agonist of these mutants.
Choline is an extremely weak agonist of wild-type AChRs (Fig. 3 and Table 1). The cluster po of wild-type AChR activated by all concentrations of choline was low and an estimate of the dissociation equilibrium constant (Kd) could not be obtained by fitting Eq. 1. For wild-type AChRs, the probability of being open remained low even at very high choline concentrations, indicating that choline is an extremely low-efficacy agonist. For wild-type AChR, θ2 with choline is ≈1,000 times smaller than with ACh (21) because receptors occupied by two choline molecules open ≈1,000 times more slowly than those occupied by ACh (8, 17, 39).
Figure 3.

Dose–response curves for wild-type and SCCMS mutant mouse AChRs. Each symbol represents one or more cell-attached patches containing saline plus agonist. ■ are ACh on wild type (48); all others pertain to choline (●, wild type; ▿, αG153S; ⋄, αN217K; ▵, αS269I; ○, ɛT264P, □, ɛL269F, ⊕, βV266M). (A) Single-channel probability. Choline is a very weak agonist of wild-type AChR and a much more potent agonist of the SCCMS mutants. (B) The effective channel opening rate. The value at limiting high agonist concentration is the intrinsic diliganded channel opening rate constant, which for choline is >10 times faster in the SCCMS mutants compared with the wild-type. The kinetic and equilibrium constants for choline activation of wild-type and SCCMS AChR are given in Table 1.
In contrast, choline is a significant agonist of SCCMS AChRs. For all of the mutants, the Kd for choline was 0.5–3 mM, and the θ2 with choline was 7- to 120-fold higher than the wild type. The increase in θ2 in the mutants was caused mainly by an increase in the channel-opening rate constant. Even though the mutations apparently did not significantly alter the affinity of the receptor for choline, they increased θ2 values enough to cause the SCCMS AChR to be open with a higher probability in the presence of the low levels of choline, such as are present in serum. In all of the SCCMS mutants the closing rate constant with choline was slower (by a factor of 2–8) than the wild type, and this, too, contributes to the higher open probability observed in serum.
In addition to being a component of serum, choline is generated via the hydrolysis of ACh, and with each synaptic impulse, endplate AChRs are transiently exposed to high concentrations of choline. To test whether this exposure might activate SCCMS AChRs to a significant extent, miniature endplate currents (mepcs) were simulated for one SCCMS mutant, αG153S. The results are shown in Fig. 4.
To test the utility of the simulation approach, mepcs first were simulated by using rate constants that pertain to the action of ACh. When rate constants for wild-type mouse AChR were used, the decay-time constant of the simulated mepc was 1.1 ms (Fig. 4B). This is in good agreement with the experimental mepc decay time constant recorded from adult mouse endplates, τ = 1.0 ms (40). When the rate constants for αG153S mouse AChR were used (16), the decay-time constant of the simulated mepc was 36 ms. There is no corresponding experimental decay-time constant for αG153S mouse endplates. However, αG153S mepcs from human endplates exhibit a slow component with a decay-time constant of 26 ± 3.3 ms (16). The quantitative difference in the experimental and simulated decay time constants could reflect differences between the kinetics of human vs. mouse αG153S AChR, and/or differences between the structural parameters of the simulated vs. experimental endplates. Nonetheless, the similarity between the experimental and the simulated mepcs for both wild-type and αG153S endplates suggests that the simulation paradigm was appropriate and could be used to investigate the possibility that choline might activate SCCMS AChRs during synaptic transmission.
Before attempting to simulate the effects of choline at αG153S endplates, it was first necessary to estimate the rate constants for choline association and dissociation for the mutant AChR. The results of kinetic modeling of choline-activated αG153S single-channel currents are shown in Fig. 4A. Choline associates more slowly by more than a factor of 10, and dissociates ≈5 times more rapidly, than ACh. To assess the maximum possible effect of choline during a synaptic impulse, simulations were carried out assuming that all of the released ACh was instantly hydrolyzed. The results, shown in Fig. 4B, indicate that the peak current generated by choline activation of αG153S AChR during a synaptic impulse is small and was only 3% of the current generated by ACh. In summary, the simulation results suggest that choline derived from ACh hydrolysis is not a significant route of AChR activation at αG153 endplates.
The diliganded AChR gating equilibrium constant is a function of both the increased affinity for the agonist(s) in the activated conformation and the unliganded gating equilibrium constant:
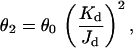 |
2 |
where Kd and Jd are the dissociation equilibrium constants of the closed- and open-channel conformations, respectively. Thus, the high values of θ2 for choline in the SCCMS mutant AChR could arise either from high values for the unliganded gating equilibrium constant, θ0, and/or from high values for the driving function, Kd/Jd. To distinguish between these possibilities, the ratio Kd/Jd was computed for several SCCMS mutants (Table 1). Although there is scatter in this ratio (some of which can be traced to uncertainty in estimation of the number of channels in each patch), the value was not dissimilar for wild-type and mutant AChRs. For both wild-type and most SCCMS AChRs, the open conformation has a ≈50-fold higher affinity for choline than the closed conformation. This result suggests that the change in affinity on opening is similar for wild-type and SCCMS AChRs and that the increase in θ2 for the mutants is caused mainly by an increase in the unliganded gating equilibrium constant, θ0.
DISCUSSION
The primary finding is that AChRs that cause the neuromuscular disease SCCMS are activated by choline, whereas wild-type AChRs are not. Thus, AChRs at patient endplates are likely to be continuously activated by the steady exposure to low levels of choline in the serum. Although gain-of-function mutations previously have been associated with genetic diseases, the SCCMS circumstance is novel because two phenomena, a mutation and contact with an ordinary metabolite, combine to generate the “constitutive” activity in a signaling pathway.
We assume that the degeneration of the endplate is a direct consequence of the functional anomalies of the mutated AChR. The three distinguishing functional properties of SCCMS mutant AChRs are the prolonged endplate current, the activation by serum choline, and the spontaneous activity in the absence of choline. We next consider the relative endplate currents from these three sources for one SCCMS mutant, ɛT264P.
The total steady current per endplate is equal to the product npoi, where n is the number of AChRs at the endplate (5 × 106; refs. 19 and 41), po is the probability of a single channel being open, and i is the single-channel current amplitude (−5.6 pA at a membrane potential of −80 mV, 22°C). From Eq. 1, at low agonist concentrations po, choline ≈ A2θ2/Kd2, which, from the dose–response parameters for ɛT264P in saline, is equal to 3 × 10−4 at 15 μM choline (the concentration in serum; ref. 35). From these values we estimate that the total current per endplate because of serum choline is 8.4 nA, which corresponds to ≈3 × 1012 cations per minute. The unliganded AChR current can be similarly calculated (po, unliganded ≈θ0), and for ɛT264P the estimated current leak is ≈1013 cations per minute per endplate. Note that this approximation depends on the assumed number of channels in a patch. The SCCMS choline-induced and unliganded currents are ≈300 times larger than the wild type (assuming Kd = 2 mM).
With regard to the synaptic current, in ɛT264P endplates, the mepc amplitude is 2.12 nA, with a fast (τ = 1.5 ms) and a slow (τ = 16.5 ms) decay component of about equal amplitudes. The integrated current is a1τ1 + a2τ2 or 1.06 × (1.5 + 16.5) = 19.1 pC. This is equivalent to an influx of 1.1 × 108 ions per mepc. If the quantal content is 36 (19), then ≈4.1 × 109 ions enter the endplate with each synaptic impulse. The number of impulses will depend on the level of muscle activity. As a baseline example, an endplate in the diaphragm may receive 10 impulses for each inspiration (42), which occur at a rate of 12 per min. In this example, the net total ionic flux per endplate from synaptically released ACh is ≈5 × 1011 ions per min. This current is within a factor of two of the wild type, estimated from the parameters given in ref. 19 (3.92 pA × 2.32 ms = 12.7 pC; quantal content = 31).
From these calculations, we estimate that in continuously active muscles of ɛT264P SCCMS patients, the number of ions entering the endplate from activation by serum choline, from unliganded activity, and from activation by synaptically released ACh are of the same order of magnitude. The presence of extrajunctional AChRs (43) would increase the current arising from serum choline and unliganded activity, and increased muscle activity will increase the current arising from synaptic activation. The calculations suggest that in SCCMS, spontaneous activity, activation by serum choline, and the slow endplate current may all be pathogenic.
The fraction of AChR current arising from activation by serum choline and by synaptically released ACh may vary between mutants. The clustering of the currents elicited in serum in αV249F (Fig. 2) indicates that these AChR desensitize in the steady presence of choline. As a consequence, in endplates with this mutant AChR, the total ionic flux arising from serum choline is likely to be reduced compared with those with ɛT264P AChR and may even be comparable to the wild-type. By the same token, in the patient, the desensitization of αV249F AChR caused by continuous exposure to serum choline might reduce the number of available AChR at the endplate and therefore the amplitude of the endplate current. Although in vitro assays indicate that the αV249F mepc is of normal amplitude (19), desensitization by choline would only prevail under in vivo conditions.
Quinidine, a long-lived open-channel blocker of AChR mitigates the symptoms of SCCMS caused by diverse mutations (44, 45), presumably because these agents block the current arising from both the serum choline and synaptic pathways. Lowering serum choline should further reduce the ionic flux into muscle, but this may be difficult to achieve because the choline concentration of serum is not readily manipulated by diet or pharmacological agents (35, 46).
The SCCMS mutations we have studied are widely distributed in different subunits of the AChR. Some are in the α-subunit and are likely to be near the transmitter-binding sites, others are in M2 segment of the β- and ɛ-subunits and are probably far from the docked agonist. The question arises, by what mechanism(s) do such spatially diverse mutations all render the AChR sensitive to choline?
In wild-type AChR, θ2 for choline is small because there is only a relatively small increase in the affinity of the binding site for this ligand when the channel opens. The results indicate that in the SCCMS mutants, this driving function remains small. The newfound sensitivity to choline in the SCCMS mutant AChR can be mainly attributed to a decrease in the relative stability of the closed vs. the open conformation of the unliganded protein.
This conclusion is consistent with the scattered locations of the mutations. Mutations in the M2 region, where the subunits are likely to be in close apposition, might be expected to reduce the intrinsic stability of the closed AChR, either by increasing the frequency of spontaneous openings and/or by making these openings longer lived (10, 19, 47). In support of this notion, mutations (that have not been associated with SCCMS) at the 12′ position of the M2 region (28) of α-, β-, δ-, and ɛ-subunits both increase the level of spontaneous activity and make the AChR sensitive to choline (C. Grosman and A.A., unpublished data).
The inference that the increased response to choline arises from changes in the intrinsic stability of the protein demonstrates that the response of a receptor can be determined by events occurring at locations that are far removed from the ligand binding site. The ability of a molecule to activate a protein, or, inversely, the ability of the protein to remain inactive when contacted by an inappropriate ligand is determined both by the specific nature of the ligand-protein interaction, as well as by the intrinsic stability of the inactive, unliganded protein.
Choline is an essential nutrient that has long been considered to be pharmacologically inactive and have little immediate consequence to neuromuscular transmission. This is true at normal endplates because the extreme stability of unliganded-closed conformation and instability of the unliganded-open conformation prevent the channel from being open even when the transmitter-binding sites are occupied by this small, ever-present quaternary amine. It appears that a breakdown in these mechanisms caused by a mutation results in AChRs that are constitutively activated by choline in the serum and that this activation contributes to the pathogenesis of SCCMS.
This disease mechanism may be relevant to other genetic diseases of signaling, metabolic, and protein-folding pathways. Allosteric proteins such as receptors, kinases, and transcription factors reside in complex environments and are constantly bombarded by small molecules. Normally, these low-affinity contacts have little or no consequence with regard to protein activation. However, it is not unlikely that a random mutation could reduce the intrinsic stability of the inactive protein, thereby allowing ordinarily inert molecules to trigger activation. The gain-of-function induced by such a mutation might not be apparent in in vitro assays, but only in the complex intra- or extracellular milieu in which the protein naturally exists. Our findings should spur the search for other genetic diseases that are caused by constitutive activity in a signaling and/or metabolic pathway generated by molecules that normally serve only housekeeping functions.
Acknowledgments
We thank Claudio Grosman for enlightening discussions and Frank Salamone for comments on the manuscript. This work is supported by Grant NS23153 from National Institutes of Health to A.A. and NS6277 from U.S. Public Health Services and a Research Grant from the Muscular Dystrophy Association to A.G.E.
ABBREVIATIONS
- ACh
acetylcholine
- AChR
acetylcholine receptor
- SCCMS
slow channel congenital myasthenic syndromes
- mepc
miniature endplate current
- Po
patch open probability
- po
single-channel open probability
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1.Lyons T J, Liu H, Goto J J, Nersissian A, Roe J A, Graden J A, Cafe C, Ellerby L M, Bredesen D E, Gralla E B, et al. Proc Natl Acad Sci USA. 1996;93:12240–12244. doi: 10.1073/pnas.93.22.12240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.al-Maghtheh M, Gregory C, Inglehearn C, Hardcastle A, Bhattacharya S. Hum Mutat. 1993;2:249–255. doi: 10.1002/humu.1380020403. [DOI] [PubMed] [Google Scholar]
- 3.Edery P, Eng C, Munnich A, Lyonnet S. BioEssays. 1997;19:389–395. doi: 10.1002/bies.950190506. [DOI] [PubMed] [Google Scholar]
- 4.Watanabe T, Bai M, Lane C R, Matsumoto S, Minamitani K, Minagawa M, Niimi H, Brown E M, Yasuda T. J Clin Endocrinol Metab. 1998;83:2497–2502. doi: 10.1210/jcem.83.7.4920. [DOI] [PubMed] [Google Scholar]
- 5.Zuo J, De Jager P L, Takahashi K A, Jiang W, Linden D J, Heintz N. Nature (London) 1997;388:769–773. doi: 10.1038/42009. [DOI] [PubMed] [Google Scholar]
- 6.Kofuji P, Hofer M, Millen K J, Millonig J H, Davidson N, Lester H A, Hatten M E. Neuron. 1996;16:941–952. doi: 10.1016/s0896-6273(00)80117-8. [DOI] [PubMed] [Google Scholar]
- 7.Jackson M B. Proc Natl Acad Sci USA. 1989;86:2199–2203. doi: 10.1073/pnas.86.7.2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maconochie D J, Steinbach J H. J Physiol (London) 1998;506:53–72. doi: 10.1111/j.1469-7793.1998.053bx.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stiles J R, Van Helden D, Bartol T M J, Salpeter E E, Salpeter M M. Proc Natl Acad Sci USA. 1996;93:5747–5752. doi: 10.1073/pnas.93.12.5747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Auerbach A, Sigurdson W, Chen J, Akk G. J Physiol (London) 1996;494:155–170. doi: 10.1113/jphysiol.1996.sp021482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Palma E, Mileo A M, Eusebi F, Miledi R. Proc Natl Acad Sci USA. 1996;93:11231–11235. doi: 10.1073/pnas.93.20.11231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ohno K, Hutchinson D O, Milone M, Brengman J M, Bouzat C, Sine S M, Engel A G. Proc Natl Acad Sci USA. 1995;92:758–762. doi: 10.1073/pnas.92.3.758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Engel A G. In: Myology: Basic and Clinical. Engel A G, Franzini-Armstrong C, editors. New York: McGraw–Hill; 1994. pp. 1798–1835. [Google Scholar]
- 14.Engel A G, Ohno K, Wang H L, Milone M, Sine S. Neuroscientist. 1998;4:185–194. [Google Scholar]
- 15.Engel A G, Ohno K, Milone M, Wang H L, Nakano S, Bouzat, Pruitt J N, Hutchinson D O, Brengman J M, Bren N, et al. Hum Mol Genet. 1996;5:1217–1227. doi: 10.1093/hmg/5.9.1217. [DOI] [PubMed] [Google Scholar]
- 16.Sine S M, Ohno K, Bouzat C, Auerbach A, Milone M, Pruitt J N, Engel A G. Neuron. 1995;15:229–239. doi: 10.1016/0896-6273(95)90080-2. [DOI] [PubMed] [Google Scholar]
- 17.Wang H L, Auerbach A, Bren N, Ohno K, Engel A G, Sine S M. J Gen Physiol. 1997;109:757–766. doi: 10.1085/jgp.109.6.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Croxen R, Newland C, Beeson D, Oosterhuis H, Chauplannaz G, Vincent A, Newsom-Davis J. Hum Mol Genet. 1997;6:767–774. doi: 10.1093/hmg/6.5.767. [DOI] [PubMed] [Google Scholar]
- 19.Milone M, Wang H L, Ohno K, Fukudome T, Pruitt J N, Bren N, Sine S M, Engel A G. J Neurosci. 1997;17:5651–5665. doi: 10.1523/JNEUROSCI.17-15-05651.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Engel A G. Neurol Clin. 1994;12:401–437. [PubMed] [Google Scholar]
- 21.Salamone F, Zhou M, Auerbach A. J Physiol (London) 1999;516:315–330. doi: 10.1111/j.1469-7793.1999.0315v.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hamill O P, Marty A, Neher E, Sakmann B, Sigworth F J. Pflugers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 23.Auerbach A. Biophys J. 1991;60:660–670. doi: 10.1016/S0006-3495(91)82095-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jackson M B, Imoto K, Mishina M, Konno T, Numa S, Sakmann B. Pflugers Arch. 1990;417:129–135. doi: 10.1007/BF00370689. [DOI] [PubMed] [Google Scholar]
- 25.Charnet P, Labarca C, Lester H A. Mol Pharmacol. 1992;41:708–717. [PubMed] [Google Scholar]
- 26.Golino M D, Hamill O P. J Membr Biol. 1992;129:297–309. doi: 10.1007/BF00232911. [DOI] [PubMed] [Google Scholar]
- 27.Zhou M, Grosman C, Auerbach A. Biophys J. 1999;76:A371–A371. [Google Scholar]
- 28.Chen J, Auerbach A. Biophys J. 1998;75:218–225. doi: 10.1016/S0006-3495(98)77508-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sachs F. In: Single-Channel Recording. Sakmann B, Neher E, editors. New York: Plenum; 1983. pp. 265–285. [Google Scholar]
- 30.Sakmann B, Patlak J, Neher E. Nature (London) 1980;286:71–73. doi: 10.1038/286071a0. [DOI] [PubMed] [Google Scholar]
- 31.Qin F, Auerbach A, Sachs F. Biophys J. 1996;70:264–280. doi: 10.1016/S0006-3495(96)79568-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qin F, Auerbach A, Sachs F. Proc R Soc London Ser B. 1997;264:375–383. doi: 10.1098/rspb.1997.0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jackson M B. Biophys J. 1986;49:663–672. doi: 10.1016/S0006-3495(86)83693-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Franke C, Parnas H, Hovav G, Dudel J. Biophys J. 1993;64:339–356. doi: 10.1016/S0006-3495(93)81374-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Savendahl L, Mar M H, Underwood L E, Zeisel S H. Am J Clin Nutr. 1997;66:622–625. doi: 10.1093/ajcn/66.3.622. [DOI] [PubMed] [Google Scholar]
- 36.Papke R L, Bencherif M, Lippiello P. Neurosci Lett. 1996;213:201–204. doi: 10.1016/0304-3940(96)12889-5. [DOI] [PubMed] [Google Scholar]
- 37.Alkondon M, Pereira E F, Cortes W S, Maelicke A, Albuquerque E X. Eur J Neurosci. 1997;9:2734–2742. doi: 10.1111/j.1460-9568.1997.tb01702.x. [DOI] [PubMed] [Google Scholar]
- 38.Alkondon M, Pereira E F, Eisenberg H M, Albuquerque E X. J Neurosci. 1999;19:2693–2705. doi: 10.1523/JNEUROSCI.19-07-02693.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parzefall F, Wilhelm R, Heckmann M, Dudel J. J Physiol (London) 1998;512:181–188. doi: 10.1111/j.1469-7793.1998.181bf.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Linder T M, Pennefather P, Quastel D M. J Gen Physiol. 1984;83:435–468. doi: 10.1085/jgp.83.3.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ohno K, Wang H L, Milone M, Bren N, Brengman J M, Nakano S, Quiram P, Pruitt J N, Sine S M, Engel A G. Neuron. 1996;17:157–170. doi: 10.1016/s0896-6273(00)80289-5. [DOI] [PubMed] [Google Scholar]
- 42.Arita H, Bishop B. J Appl Physiol. 1983;55:1203–1210. doi: 10.1152/jappl.1983.55.4.1203. [DOI] [PubMed] [Google Scholar]
- 43.Ringel S P, Bender A N, Engel W K. Arch Neurol. 1976;33:751–758. doi: 10.1001/archneur.1976.00500110019004. [DOI] [PubMed] [Google Scholar]
- 44.Fukudome T, Ohno K, Brengman J M, Engel A G. NeuroReport. 1998;9:1907–1911. doi: 10.1097/00001756-199806010-00044. [DOI] [PubMed] [Google Scholar]
- 45.Harper C M, Engel A G. Ann Neurol. 1998;43:480–484. doi: 10.1002/ana.410430411. [DOI] [PubMed] [Google Scholar]
- 46.Zeisel S H, Blusztajn J K. Annu Rev Nutr. 1994;14:269–296. doi: 10.1146/annurev.nu.14.070194.001413. [DOI] [PubMed] [Google Scholar]
- 47.Filatov G N, White M M. Mol Pharmacol. 1995;48:379–384. [PubMed] [Google Scholar]
- 48.Akk G, Auerbach A. Biophys J. 1996;70:2652–2658. doi: 10.1016/S0006-3495(96)79834-X. [DOI] [PMC free article] [PubMed] [Google Scholar]