

Abstract
Mutations of proto-oncogenes are common events in the pathogenesis of cancers, as shown in a wide range of studies during the 30 years since the discovery of these genes. The benefits of novel therapies that target the products of mutant alleles in human cancers, and the demonstrated dependence of cancers in mouse models on continued expression of initiating oncogenes, are especially promising signs that revolutionary improvements in cancer care are possible. Full realization of the promise of targeted therapies, however, will require better definitions of the genotypes of human cancers, new approaches to interrupt the biochemical consequences of oncogenic mutations, and a greater understanding of drug resistance and tumor progression. In this paper, we summarize recent efforts toward these goals in our laboratory and others.
It is now about 30 years since the cellular progenitors of retroviral oncogenes, the first proto-oncogenes, were identified in vertebrate genomes (Stehelin et al. 1976; Rosenberg and Jolicoeur 1997), and over 20 years since mutant versions of such genes were first discovered in human cancers. In the past two decades, the list of mutant proto-oncogenes in human tumors has grown dramatically; many genes belonging to at least two other categories—tumor suppressors and governors of genomic integrity—have been implicated in carcinogenesis, and the proteins encoded by such genes have been extensively characterized. These developments have set the stage for more rational approaches to the detection, diagnosis, classification, treatment, and prevention of human cancers. Yet the most common and most lethal of these diseases are still inadequately controlled with traditional methods (chemotherapy and radiation), which do not take advantage of our new understanding of cancer at the molecular level.
In this survey of recent work from our laboratory and many others, we emphasize evidence that encourages the belief that therapies targeted against the specific genetic damage present in each cancer, especially therapies affecting proteins encoded by mutant proto-oncogenes, are likely to have increasingly prominent roles in future efforts to control cancer. Reaching this objective will require a fuller description of cancer genotypes through a nationally coordinated effort; a better understanding of signaling pathways altered by oncogenic mutations; a deeper picture of interactions among the multiple cancer genes in a single tumor; more drugs and antibodies that counter the effects of such mutations, in part to prevent the emergence of drug resistance; and a more refined description of how tumors progress as a consequence of changes within cancer cells and the microenvironment.
We address these issues by considering mutant proto-oncogenes in several mouse models of human cancers and, in at least one case, a human disease, adenocarcinoma of the lung. One overriding notion that we stress is the idea that oncogenes are not required simply to initiate and maintain tumor growth; in several contexts, continued expression of mutant oncogenes is required to maintain the viability of the cancer cell. Such “oncogene dependence” provides an important vulnerability that some drugs already in clinical use—most obviously imatinib (Gleevec) and other inhibitors of protein-tyrosine kinases—effectively exploit. Since different types of tumors can be dependent on the same or similar oncogenes, and, conversely, histologically indistinguishable tumors from the same organ often depend on different oncogenes, it is essential that tumor genotypes be precisely and fully determined. We contend that much of contemporary cancer research should now be directed toward defining the molecular targets and therapeutic agents that show promise of producing “imatinib equivalents” for all forms of human cancer.
ONCOGENE DEPENDENCE IN CANCER
Viral Mutants
The idea that cancers are dependent on continued production of an oncogenic protein has its most explicit origin in classic studies of a temperature-sensitive mutant of the src gene of Rous sarcoma virus (RSV) (Martin 1970). These experiments not only clearly separated the oncogenic from the replicative functions (genes) of RSV; they also showed that the viral oncogene (v-src) was required to maintain as well as to initiate the transformed state.
Transgenic Mice with Inducible Oncogenes
More recent studies with transgenic mice that express oncogenes under the control of regulated promoters have made a dramatic point: After the oncogenes are turned on and tumors emerge, extinction of expression often leads to rapid disappearance of the tumor, as a result of apoptosis, differentiation of the cancer cells, or disappearance of vascular endothelial cells (Table 1). For example, about 5 years ago, our group constructed mice in which a mutant transgenic form of the Kirsten Ras gene (K-RasG12D) is regulated by a tetracycline-dependent transcription factor encoded by a second lung-specific transgene (Fisher et al. 2001). Using this system K-RasG12D is expressed in the lung at levels similar to the endogenous normal K-Ras gene, when a tetracycline analog, doxycycline, is provided in the diet. Lungs from such animals appear normal, and the mutant transgene is silent in the absence of the antibiotic. If the mice are maintained on doxycycline, foci of hyperplastic cells appear throughout the lung fields after about a month; by 2–3 months of doxycycline adminstration, adenomas and adenocarcinomas appear at multiple sites. If, however, doxycycline is then withdrawn, levels of mutant K-Ras RNA (and presumably protein) fall precipitously, and the tumor cells display signs of programmed cell death; as a result, the tumors disappear within 3–7 days, as judged by magnetic resonance imaging or by histopathology, and do not recur in the absence of drug (Fig. 1A).
Table 1.
Examples of Tumor Maintenance in Mouse Models
| Tumor type | Oncogene | Mechanism of regression | Reference |
|---|---|---|---|
| Melanoma | H-ras | Ink4a/Arf and VEGF independent; apoptosis in tumor cells and host-derived endothelial cells; decreased proliferation; not immune-related | Chin et al. (1999) |
| T-cell lymphoma/acute myeloid leukemia | Myc | differentiation, proliferative arrest, and apoptosis in tumor cells | Felsher and Bishop (1999) |
| Osteosarcoma | Myc | differentiation in tumor cells | Jain et al. (2002) |
| Mammary | Myc | unknown | D’Cruz et al. (2001) |
| Mammary | Neu | apoptosis and decreased proliferation in tumor cells | Moody et al. (2002) |
| Mammary | Wnt | unknown; p53 independent | Gunther et al. (2003) |
| Pancreatic beta-cell | Myc | apoptosis, redifferentiation, decreased proliferation in tumor cells and vascular degeneration; occurs despite presence of Bcl-xL; p19Arf and p53 independent | Pelengaris et al. (2002); G.I. Evans (unpubl.) |
| Non-small-cell lung cancer | K-ras | apoptosis (Ink4A/Arf and p53 independent) and decreased proliferation in tumor cells | Fisher et al. (2001) |
| Non-small-cell lung cancer | EGFR | unknown | K. Politi et al. (in prep.) |
| Sarcoma | K-ras | apoptosis and decreased proliferation in tumor cells; p53 independent; T-cell independent | Pao et al. (2003) |
| Leukemia | Bcr-abl | apoptosis in tumor cells | Huettner et al. (2000) |
| Skin | Myc | growth arrest and irreversible differentiation | Flores et al. (2004) |
| Hepatocellular carcinoma | Myc | differentiation | Shachaf et al. (2004) |
| Hepatocellular carcinoma | Met | apoptosis and decreased proliferation in tumor cells | Wang et al. (2001) |
Figure 1.
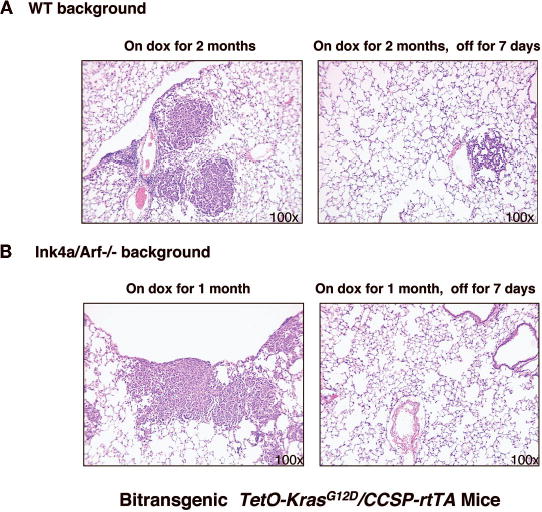
Oncogenic K-ras is required for lung tumor maintenance with or without loss of tumor suppressor genes. Samples of lung tissue, stained with hematoxylin and eosin, after exposure to doxycycline for 1 or 2 months and after withdrawal for 7 days are shown (Fisher et al. 2001). Withdrawal of doxycycline leads to rapid tumor regression in bi-transgenic CCSP-rtTA/TetOKrasG12D in a wild-type (WT) background (A) and in an Ink4A/Arf-deficient background (B). Similar results were obtained in a p53-deficient background (Fisher et al. 2001). Magnification, 100x.
To ask whether this apparent oncogene dependence occurs only in the absence of other oncogenic mutations, we repeated the experiments in mice known to be deficient in the tumor suppressor genes p53 or Ink4a/Arf. Under these conditions, adenocarcinomas appeared more rapidly, usually within a month after doxycycline induction, but again quickly regressed by an apoptotic mechanism when the doxycycline was withdrawn (Fig. 1B). Thus, dependence of the mouse lung adenocarcinomas on the initiating K-Ras oncogene is not relieved by additional mutations in important tumor suppressor genes.
Our K-Ras-dependent lung cancer model and many of the other doxycycline-inducible mouse models listed in Table 1 prompt a general comment about oncogene dependence. It seems from these studies that cells that were at one time fully viable in the absence of the product of each inducible oncogene have become “imbalanced” in some way, so that sudden deprivation of the oncogene product now triggers an event—apoptosis, differentiation, or loss of angiogenic signals—that leads to tumor regression. Understanding such imbalances and their consequences could offer new ways to interfere with signaling downstream of an oncogenic activity. Particularly in the case of RAS genes, which are mutant in about a third of human tumors but for which no effective targeted inhibitor has been found, such insights might be an important step toward improving therapies of common human cancers.
Cancer Therapies in Patients
An example of dramatically improved therapy for a human cancer underscores the importance of the concept of oncogene dependence. As discussed elsewhere (see Sawyers, this volume), the use of imatinib—a potent inhibitor of at least three protein-tyrosine kinases, including ABL (and its mutant form BCR-ABL)—for the treatment of chronic myeloid leukemia (CML), a leading form of adult leukemia, rapidly reverses the hematological and symptomatic manifestations of the disease and maintains remissions for up to 5 years and more, especially when therapy is begun during early stages of the disease (for review, see Deininger et al. 2005). The appearance of second-site mutations in the kinase domain of BCR-ABL that confer imatinib resistance (Gorre et al. 2001) constitutes powerful evidence that the drug response is indeed due to the dependence of the leukemic cells on mutant ABL kinase activity. Strategies based on these mutations are now being pursued to prevent or overcome drug resistance (Shah et al. 2004; Burgess et al. 2005; Carter et al. 2005; Gumireddy et al. 2005; Sawyers, this volume).
Several other human cancers, including solid tumors, bearing changes in ABL—or mutations in the other two genes, CKIT and PDGFR-A, encoding kinases known to be inhibited by imatinib—also respond to this drug. This strengthens the argument that oncogene dependence is a general phenomenon, affecting solid and liquid cancers. Thus, therapeutic responses in human patients, not just regulated expression of transgenes in mice or temperature-sensitive mutant oncogenes in cell culture, provide important evidence for oncogene dependence.
EVIDENCE FOR EGFR ONCOGENE DEPENDENCE IN HUMAN LUNG ADENOCARCINOMA
Two small chemical inhibitors of the EGFR tyrosine kinase, gefitinib (Iressa) and erlotinib (Tarceva), were tested for efficacy in human lung cancer for several years (Fukuoka et al. 2003; Kris et al. 2003) before it was recognized that some lung cancers carry dominantly acting mutations in the EGFR gene (Lynch et al. 2004; Paez et al. 2004; Pao et al. 2004). The recent discovery of such EGFR mutations offers a striking example of how drug responses in patients can reveal dependence of cancers on a mutant oncogene.
Non-small-cell lung cancer (NSCLC) is the leading cause of cancer death worldwide (Parkin et al. 2005). In about 10% of patients in the U.S. and Europe with adenocarcinoma of the lung, the most common form of NSCLC, rapid partial remissions occur when these drugs are used, even late in the course of metastatic disease, and in parts of Asia the response rate is significantly higher (Fukuoka et al. 2003; Kris et al. 2003). Sometimes (as shown in Fig. 2A) the response is dramatic, resembling the tumor regression seen in our K-Ras-based mouse model of lung cancer when doxycycline is withdrawn (Fisher et al. 2001).
Figure 2.
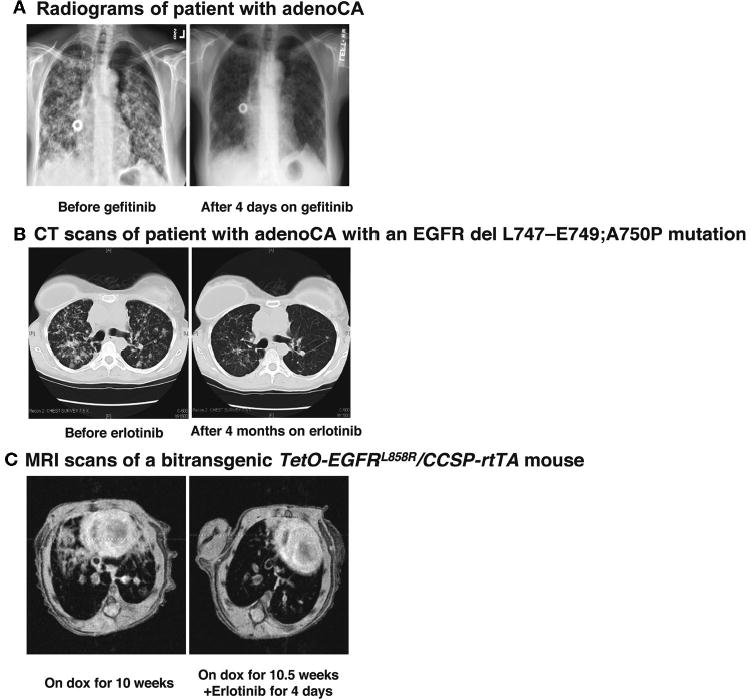
Lung tumors with EGFR mutations respond dramatically to treatment with tyrosine kinase inhibitors. Chest radiographs (A) and chest computerized tomography-generated images (B) showing a tumor response to gefitinib (A) after 4 days of treatment and erlotinib (B) after 4 months of treatment in two patients, one of which is known to have a tumor with an EGFR mutation. (C) Magnetic resonance images of lungs from a mouse expressing the EGFR L858R mutant before (left panel) and after 4 days (right panel) of erlotinib treatment.
Observations like these suggested that the two tyrosine kinase inhibitors (TKIs) might be inactivating mutant kinases, either the known target for these drugs, EGFR, or some other unsuspected protein-tyrosine kinase among the 90 encoded in the human genome (Manning et al. 2002). Sequencing of the coding exons of EGFR genes in tumors that showed radiographic responses to the TKIs revealed that sensitivity to gefitinib and erlotinib were highly associated with mutations in the EGFR kinase domain (Lynch et al. 2004; Paez et al. 2004; Pao et al. 2004; see Thomas et al.; Haber et al.; both this volume). (For an example, see the computerized tomography scans in Fig. 2B.) Surprisingly, these mutations are highly idiosyncratic: Nearly 90% are either point mutations that change leucine at position 858 to arginine (L858R) or three- to seven-codon deletions affecting the highly conserved sequence, LREA, that is positioned close to the ATP-binding site in the kinase domain (for review, see Pao and Miller 2005).
The biochemical, physiological, and structural consequences of these mutations and several other substitution mutations that have been associated with TKI responsiveness are still under study (Sordella et al. 2004; Tracy et al. 2004; Amann et al. 2005; Engelman et al. 2005; Greulich et al. 2005; Chen et al. 2006), but several observations support the idea that this subset of lung adenocarcinomas is dependent on the mutant EGFR kinase. The mutations are strongly associated with a measurable radiological response to the TKIs; such responses are rarely observed in tumors without detectable EGFR mutations, including the 20–30% of tumors with mutations in KRAS (Eberhard et al. 2005; Pao et al. 2005b), the product of which acts “downstream” of EGFR. In addition, second-site mutations in one of the exons encoding the EGFR kinase domain are observed in about half of drug-resistant tumors that resume growth during treatment with the TKIs (Kobayashi et al. 2005; Pao et al. 2005a).
Strikingly, these resistance-inducing secondary EGFR mutations all encode the same change, threonine to methionine at position 790, a change that is strictly analogous to one of the mutations observed in imatinib-resistant forms of BCR-ABL, CKIT, and PDGFR-A in various types of tumors treated with imatinib (Gorre et al. 2001; Cools et al. 2003; Tamborini et al. 2004; see Sawyers, this volume). The introduction of a bulky side chain in the methionine residue is likely to interfere with drug binding; screening of additional TKIs for their ability to bind to or inhibit the doubly mutant kinases have already identified agents that are effective in the presence of T790M and analogous mutations (Carter et al. 2005; Kwak et al. 2005); such drugs might be used after primary resistance occurs or in combination with the first TKI to prevent the emergence of resistance.
As described elsewhere (see Hillan et al., this volume), long-term outcomes of gefitinib or erlotinib treatment of EGFR-mutant tumors, as measured by survival or by time to tumor progression, are not as good as those observed in the treatment of CML with imatinib. Although a full explanation of these disappointing results is not yet at hand, the emergence of drug resistance, generally within a year of commencing therapy, is likely to be part of the problem and may be alleviated by the use of additional TKIs. The relatively modest effects of TKIs on survival may also reflect the use of these drugs relatively late in the course of the disease, a phase more akin to the blast crisis than the early stages of CML, so that other oncogenic mutations may dampen the therapeutic efficacy. Consistent with this idea, partial rather than complete radiographic responses to the TKIs are commonly seen in patients with metastatic NSCLC. From this perspective, treatment of early-stage disease (e.g., adjuvant therapy with TKIs at the time of surgery) or first-line treatment of advanced disease with TKIs might have more potent effects on survival.
Some tumors with increased copy numbers of wild-type EGFR, as determined by fluorescent in situ hybridization, appear to be sensitive to treatment with gefitinib, since at least one group has reported a stronger correlation of the outcome, especially survival, with EGFR gene amplification than with EGFR mutation (Cappuzzo et al. 2005). Similar results have been described with erlotinib (Tsao et al. 2005). It will be important to confirm these findings in additional, larger studies and, if they prove valid, to understand how TKIs produce benefit in this situation.
MOUSE MODELS OF EGFR-DEPENDENT LUNG CANCER
Building mouse models of lung cancer is a useful means to characterize the dependence of lung adenocarcinomas on mutant EGFR, to study the mechanism of tumor induction and regression, to test candidate therapies, and to investigate mechanisms of drug resistance. To those ends, we have produced a series of transgenic mice that encode wild-type EGFR, L858R EGFR, and an LREA deletion form of EGFR under the control of a doxycycline-responsive regulatory system (K. Politi et al., in prep.). Induction of the L858R mutant form of EGFR leads to development of diffuse lung tumors comprising cells expressing markers of type II pneumocytes and highly reminiscent of human bronchioalveolar carcinoma, followed by the appearance of multifocal adenocarcinomas. The tumors rapidly regress, as observed by magnetic resonance imaging or histopathology, when the animals are deprived of doxycycline or when they are treated with erlotinib (Fig. 2C), demonstrating that the tumors are dependent on continued production or activity of the mutant EGFR. A detailed description of these lines and others, and their responses to induction and de-induction of EGFR and to drugs, will be published elsewhere (K. Politi et al., in prep.).
WHAT IS LIMITING THE IMPACT OF THE MOLECULAR UNDERSTANDING OF CANCER ON PATIENT CARE?
The widespread evidence for oncogene dependence in mouse models and human tumors and the results of treatment of some human cancers with TKIs, especially imatinib, provide strong grounds for optimism about controlling cancer more effectively in the future. Yet the outcomes of most efforts to treat the common cancers have not changed significantly over the past several decades. Why is this? And what is impeding more dramatic change?
Defining Cancer Genotypes
Despite the remarkable growth of knowledge about genes that have been implicated in carcinogenesis, we still have a very meager picture of the genotype of most of the 50 or more types of human cancer. For many cancer types, some proto-oncogene or tumor suppressor gene is known to be aberrant (subtly mutated, deleted, rearranged, amplified, or epigenetically modified). However, when studied intensively, most tumors appear to have multiple altered cancer genes. Furthermore, tumors that are histologically indistinguishable are often genetically different, and important elements in the clinical history, such as occupational exposures or never smoking, sometimes correlate more strongly than histology with the tumor genotype.
Consider, for example, the situation with lung adenocarcinoma, one of most common and lethal human cancers. About a quarter of such tumors have a mutant copy of KRAS, and this occurs nearly exclusively in smokers (Ahrendt et al. 2001). Similarly, we now know that about 10% have a mutant copy of EGFR, mainly in “never smokers” (Pao et al. 2004). In addition, small numbers of tumors have mutations in genes that encode other components of the growth factor signaling network: ERBB2, BRAF, and PIK3CA (see Fig. 3) (Brose et al. 2002; Naoki et al. 2002; Samuels et al. 2004; Stephens et al. 2004; Shigematsu et al. 2005). Interestingly, mutations in more than one of these genes are rarely encountered in a single tumor, implying that no further selective advantage is conferred by additional lesions affecting this signaling network. However, the combined percentage of tumors known to have even a single mutation in this network is relatively small; thus, a mutant oncogene that might provide a therapeutic target has not been identified in the majority of lung adenocarcinomas (Fig. 3). Furthermore, although loss of tumor suppressor genes, especially P53 and INK4A/ARF, is known to occur at high frequency in such tumors (for review, see Forgacs et al. 2001), the influence of tumor suppressor deficiencies on treatment outcomes in the context of specific oncogenic mutations is not known, nor is the effect of mutations in additional proto-oncogenes, as discussed further below.
Figure 3.
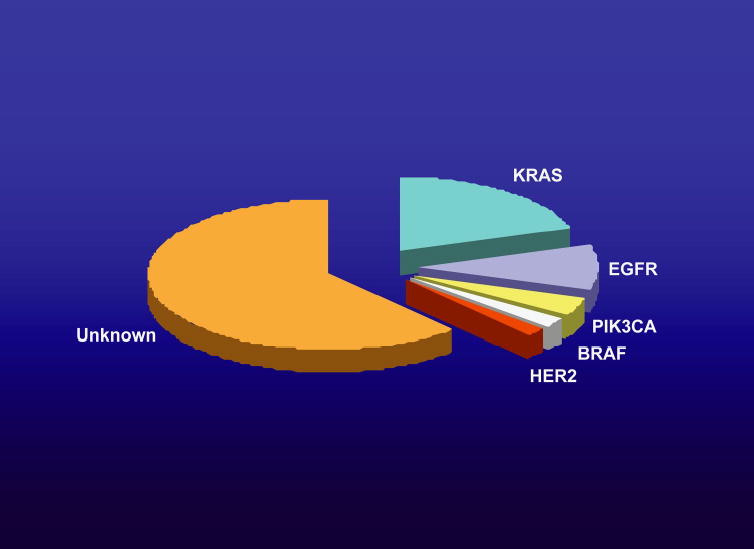
Pie chart depicting the frequency of known proto-oncogenes found to be mutated in non-small-cell lung cancers. As discussed in the text, these genes all encode components of the EGFR signaling network.
The development of high-throughput methods for assessing mutations, changes in DNA copy number, and even epigenetic changes provides an opportunity to repair, at least partially, our deficient knowledge of tumor genotypes. The National Institutes of Health is now considering a coordinated, long-term effort to find many of the genotypic changes that occur with at least 5% frequency in the 50 or so major types of human cancer (the NCAB Report can be found at http://www.genome.gov/Pages/About/NACHGR/May2005NACHGRAgenda/ReportoftheWorkingGrouponBiomedicalTechnology.pdf). Preliminary efforts at several centers to determine genotypes, mainly by re-sequencing the coding exons of genes encoding kinases, have yielded interesting results (Bardelli et al. 2003; Samuels et al. 2004; Davies et al. 2005; Parsons et al. 2005; Stephens et al. 2005; see Futreal et al., this volume), but the findings are not yet on a scale adequate to describe the molecular basis of human cancer in the way that will be required for major therapeutic advances.
Finding Novel Inhibitors of Mutant Oncogenes
A second obstacle to greater clinical impact of molecular oncology is the relative paucity of tools that affect potential therapeutic targets other than protein-tyrosine kinases (mostly inhibited by small compounds) or transmembrane proteins and their ligands (mostly blocked by monoclonal antibodies). As noted earlier, this deficiency is especially acute in relation to RAS mutants, but applies also to a wide range of oncogenic proteins. Thus far, nearly all beneficial therapies have been targeted against the products of mutant genes, but it seems reasonable to assume that nonmutated components of affected signaling pathways—for example, proteins phosphorylated by tyrosine kinases—will also prove to be vulnerable to attack.
It also seems likely to be important to understand the alterations that are responsible for the “imbalance” in cell signaling responsible for the phenomenon of oncogene dependence. For example, the apoptotic response that is observed when oncogenes are down-regulated in mouse models is presumably blocked in cancer cells expressing oncogenes by proteins induced by the oncogenes. Such proteins might be attractive therapeutic targets, especially when the mutant oncoprotein itself is refractory to inhibition by small molecules.
Minimizing Resistance to Targeted Cancer Therapies
Just as drug resistance is a problem in conventional chemotherapy and in treating infectious diseases, it is already apparent that cancer therapies targeted against known proteins will eventually encounter significant resistance. The encouraging news is that such resistance is usually attributable to secondary mutations affecting the target protein—a situation that permits rational approaches to overcoming resistance through drug screens and protein design. The goal is to develop combination therapies, not unlike those now in wide use for the treatment of HIV/AIDS, which greatly diminish the probability that resistant clones of cancer cells will emerge by genetic selection.
Coping with Mutations in Multiple Proto-oncogenes
Most human cancers, if not all, are the products of multiple genetic changes, but we do not yet have a full account of the impact of additional oncogenic mutations on a tumor’s dependence on any single oncogene. Recent studies of mouse tumor models from Lewis Chodosh and his colleagues indicate that oncogene dependence (for instance, dependence of a mouse mammary tumor on a Myc transgene) can be abrogated by secondary mutations in other genes, such as endogenous Ras genes (D’Cruz et al. 2001).
These observations have stimulated us to seek secondary spontaneous mutations in mammary tumors arising in tumor-prone transgenic mice. One attractive feature of these experiments is that the detection of a secondary mutation by DNA sequencing implies that it has conferred a selective growth advantage, otherwise it would not be present in a large enough percentage of tumor cells to permit detection by sequencing of unfractionated tumor DNA. In this way, we have shown that secondary mutations in H-Ras are encountered in about half of breast tumors induced by an MMTV-Wnt1 transgene (Podsypanina et al. 2004). Such mutations are not found in tumors induced by an MMTV-Neu/ERBB2 transgene; this is presumably so because Ras proteins act in the same growth factor signaling pathway, and thus, mutants would confer no growth advantage. Ras mutations are also not found in the tumors that rapidly appear in bi-transgenic mice, expressing both Wnt-1 and Neu/ERBB2. Unexpectedly, however, the Wnt1 transgene removes the selective pressure for the secondary mutations in the Neu/ERBB2 gene that commonly enhance kinase activity in tumors arising in mono-transgenic mice (Siegel et al. 1994; Podsypanina et al. 2004). Also unexpectedly, Ras mutations do not occur in MMTV-Wnt1 tumors when they are induced in p53-deficient mice. An understanding of these genetic interactions could help to predict which mutant cancer genes are likely to coexist in tumors and which might be the best targets for therapeutic intervention.
Additional insight into the interactions among mutant cancer genes can be obtained by expressing two or more transgenic oncogenes, at least one of which can be transcriptionally controlled. For example, we have built transgenic mice that express Myc and mutant H-Ras in mammary epithelium, with one or the other under the control of doxycycline, in order to determine whether oncogene dependence of ensuing tumors pertains to one gene, both, or neither with this genetic combination (K. Podsypanina et al., unpubl.).
Assessing Tumor Progression
Approaches to targeted therapy remain limited by a poor understanding of metastasis, invasion, and angiogenesis—the aspects of tumor progression that are responsible for the lethal effects of most tumors. Several papers address these issues (see Egeblad et al.; Bissell et al.; Courtneidge et al.; G.P. Gupta et al.; all this volume; D. Hanahan; J.S. Brugge; both unpubl.). We have developed a novel experimental system in which the progression of phenotypes in a well-characterized model of pancreatic islet cell tumors (Hanahan 1985) can be used to measure the effects of genes (or inhibitory RNAs) that have been introduced into islet cells by cell-specific infection with retroviral vectors. In this strategy, bi-transgenic mice in which the rat insulin promoter drives production of the SV40 T antigen (RIP-Tag) and the avian retrovirus subgroup A receptor (RIP-TVA) are infected with avian RCAS vectors encoding a variety of factors that might influence tumor progression. Early results suggest that two suspected progression factors, an anti-apoptotic protein (Bcl-xL) and a dominant-negative version of E-cadherin (Naik et al. 1996; Perl et al. 1998), each accelerate the formation of islet cell tumors with invasive properties (Y.-C.N. Du et al., unpubl.).
CONCLUSIONS
As demonstrated in many papers in this volume, cancer research has progressed rapidly over the past three decades and is now well-positioned to contribute powerfully to the control of human cancer. However, the long-anticipated benefits for patients have appeared only recently and in limited sectors of clinical oncology. A more deliberate effort to catalog and comprehend the mutations that afflict all the common human cancers, and more innovative approaches to therapeutic development will now be required to achieve the long-range goals of molecular oncology.
Acknowledgments
We thank Jennifer Doherty for expert technical assistance, Maureen Zakowski for pathological evaluation of the murine lung tumors, and Jason Koutcher and Carl Le for magnetic resonance imaging of mice. This work was supported in part by a Mouse Models of Human Cancer Consortium Grant (to H. Varmus), P01 CA94060-02 (to H. Varmus), funds from the Martel Foundation (to H. Varmus) and the Carmel Hill Fund (to H. Varmus). K. Politi received support from the Labrecque Foundation and the American Cancer Society (PF-05-078-01-MGO); K. Podsypanina was supported by a Cancer Research Institute award; W. Pao received support from the CHEST and LUNGevity Foundations.
References
- Ahrendt SA, Decker PA, Alawi EA, Zhu YR, Sanchez-Cespedes M, Yang SC, Haasler GB, Kajdacsy-Balla A, Demeure MJ, Sidransky D. Cigarette smoking is strongly associated with mutation of the K-ras gene in patients with primary adenocarcinoma of the lung. Cancer. 2001;92:1525. doi: 10.1002/1097-0142(20010915)92:6<1525::aid-cncr1478>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- Amann J, Kalyankrishna S, Massion PP, Ohm JE, Girard L, Shigematsu H, Peyton M, Juroske D, Huang Y, Stuart Salmon J, Kim YH, Pollack JR, Yanagisawa K, Gazdar A, Minna JD, Kurie JM, Carbone DP. Aberrant epidermal growth factor receptor signaling and enhanced sensitivity to EGFR inhibitors in lung cancer. Cancer Res. 2005;65:226. [PubMed] [Google Scholar]
- Bardelli A, Parsons DW, Silliman N, Ptak J, Szabo S, Saha S, Markowitz S, Willson JK, Parmigiani G, Kinzler KW, Vogelstein B, Velculescu VE. Mutational analysis of the tyrosine kinome in colorectal cancers. Science. 2003;300:949. doi: 10.1126/science.1082596. [DOI] [PubMed] [Google Scholar]
- Brose MS, Volpe P, Feldman M, Kumar M, Rishi I, Gerrero R, Einhorn E, Herlyn M, Minna J, Nicholson A, Roth JA, Albelda SM, Davies H, Cox C, Brignell G, Stephens P, Futreal PA, Wooster R, Stratton MR, Weber BL. BRAF and RAS mutations in human lung cancer and melanoma. Cancer Res. 2002;62:6997. [PubMed] [Google Scholar]
- Burgess MR, Skaggs BJ, Shah NP, Lee FY, Sawyers CL. Comparative analysis of two clinically active BCR-ABL kinase inhibitors reveals the role of conformation-specific binding in resistance. Proc Natl Acad Sci. 2005;102:3395. doi: 10.1073/pnas.0409770102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappuzzo F, Hirsch FR, Rossi E, Bartolini S, Ceresoli GL, Bemis L, Haney J, Witta S, Danenberg K, Domenichini I, Ludovini V, Magrini E, Gregorc V, Doglioni C, Sidoni A, Tonato M, Franklin WA, Crino L, Bunn PA, Jr, Varella-Garcia M. Epidermal growth factor receptor gene and protein and gefitinib sensitivity in non-small-cell lung cancer. J Natl Cancer Inst. 2005;97:643. doi: 10.1093/jnci/dji112. [DOI] [PubMed] [Google Scholar]
- Carter TA, Wodicka LM, Shah NP, Velasco AM, Fabian MA, Treiber DK, Milanov ZV, Atteridge CE, Biggs WH, III, Edeen PT, Floyd M, Ford JM, Grotzfeld RM, Herrgard S, Insko DE, Mehta SA, Patel HK, Pao W, Sawyers CL, Varmus H, Zarrinkar PP, Lockhart DJ. Inhibition of drug-resistant mutants of ABL, KIT, and EGF receptor kinases. Proc Natl Acad Sci. 2005;102:11011. doi: 10.1073/pnas.0504952102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YR, Fu YN, Lin CH, Yang ST, Hu SF, Chen YT, Tsai SF, Huang SF. Distinctive activation patterns in constitutively active and gefitinib-sensitive EGFR mutants. Oncogene. 2006;25:1205. doi: 10.1038/sj.onc.1209159. [DOI] [PubMed] [Google Scholar]
- Chin L, Tam A, Pomerantz J, Wong M, Holash J, Bardeesy N, Shen Q, O’Hagan R, Pantginis J, Zhou H, Horner JW, 2nd, Cordon-Cardo C, Yancopoulos GD, DePinho RA. Essential role for oncogenic Ras in tumour maintenance. Nature. 1999;400:468. doi: 10.1038/22788. [DOI] [PubMed] [Google Scholar]
- Cools J, DeAngelo DJ, Gotlib J, Stover EH, Legare RD, Cortes J, Kutok J, Clark J, Galinsky I, Griffin JD, Cross NC, Tefferi A, Malone J, Alam R, Schrier SL, Schmid J, Rose M, Vandenberghe P, Verhoef G, Boogaerts M, Wlodarska I, Kantarjian H, Marynen P, Coutre SE, Stone R, Gilliland DG. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med. 2003;348:1201. doi: 10.1056/NEJMoa025217. [DOI] [PubMed] [Google Scholar]
- Davies H, Hunter C, Smith R, Stephens P, Greenman C, Bignell G, Teague J, Butler A, Edkins S, Stevens C, Parker A, O’Meara S, Avis T, Barthorpe S, Brackenbury L, Buck G, Clements J, Cole J, Dicks E, Edwards K, Forbes S, Gorton M, Gray K, Halliday K, Harrison R, Hills K, Hinton J, Jones D, Kosmidou V, Laman R, Lugg R, Menzies A, Perry J, Petty R, Raine K, Shepherd R, Small A, Solomon H, Stephens Y, Tofts C, Varian J, Webb A, West S, Widaa S, Yates A, Brasseur F, Cooper CS, Flanagan AM, Green A, Knowles M, Leung SY, Looijenga LH, Malkowicz B, Pierotti MA, Teh BT, Yuen ST, Lakhani SR, Easton DF, Weber BL, Goldstraw P, Nicholson AG, Wooster R, Stratton MR, Futreal PA. Somatic mutations of the protein kinase gene family in human lung cancer. Cancer Res. 2005;65:7591. doi: 10.1158/0008-5472.CAN-05-1855. [DOI] [PubMed] [Google Scholar]
- D’Cruz CM, Gunther EJ, Boxer RB, Hartman JL, Sintasath L, Moody SE, Cox JD, Ha SI, Belka GK, Golant A, Cardiff RD, Chodosh LA. c-MYC induces mammary tumorigenesis by means of a preferred pathway involving spontaneous Kras2 mutations. Nat Med. 2001;7:235. doi: 10.1038/84691. [DOI] [PubMed] [Google Scholar]
- Deininger M, Buchdunger E, Druker BJ. The development of imatinib as a therapeutic agent for chronic myeloid leukemia. Blood. 2005;105:2640. doi: 10.1182/blood-2004-08-3097. [DOI] [PubMed] [Google Scholar]
- Eberhard DA, Johnson BE, Amler LC, Goddard AD, Heldens SL, Herbst RS, Ince WL, Janne PA, Januario T, Johnson DH, Klein P, Miller VA, Ostland MA, Ramies DA, Sebisanovic D, Stinson JA, Zhang YR, Seshagiri S, Hillan KJ. Mutations in the epidermal growth factor receptor and in KRAS are predictive and prognostic indicators in patients with non-small-cell lung cancer treated with chemotherapy alone and in combination with erlotinib. J Clin Oncol. 2005;23:5900. doi: 10.1200/JCO.2005.02.857. [DOI] [PubMed] [Google Scholar]
- Engelman JA, Janne PA, Mermel C, Pearlberg J, Mokuhara T, Fleet C, Cichowski K, Johnson BE, Cantley LC. ErbB-3 mediates phosphoinositide 3-kinase activity in gefitinib-sensitive non-small cell lung cancer cell lines. Proc Natl Acad Sci. 2005;102:3788. doi: 10.1073/pnas.0409773102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsher DW, Bishop JM. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol Cell. 1999;4:199. doi: 10.1016/s1097-2765(00)80367-6. [DOI] [PubMed] [Google Scholar]
- Fisher GH, Wellen SL, Klimstra D, Lenczowski JM, Tichelaar JW, Lizak MJ, Whitsett JA, Koretsky A, Varmus HE. Induction and apoptotic regression of lung adenocarcinomas by regulation of a K-Ras transgene in the presence and absence of tumor suppressor genes. Genes Dev. 2001;15:3249. doi: 10.1101/gad.947701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores I, Murphy DJ, Swigart LB, Knies U, Evan GI. Defining the temporal requirements for Myc in the progression and maintenance of skin neoplasia. Oncogene. 2004;23:5923. doi: 10.1038/sj.onc.1207796. [DOI] [PubMed] [Google Scholar]
- Forgacs E, Zochbauer-Muller S, Olah E, Minna JD. Molecular genetic abnormalities in the pathogenesis of human lung cancer. Pathol Oncol Res. 2001;7:6. doi: 10.1007/BF03032598. [DOI] [PubMed] [Google Scholar]
- Fukuoka M, Yano S, Giaccone G, Tamura T, Nakagawa K, Douillard JY, Nishiwaki Y, Vansteenkiste J, Kudoh S, Rischin D, Eek R, Horai T, Noda K, Takata I, Smit E, Averbuch S, Macleod A, Feyereislova A, Dong RP, Baselga J. Multi-institutional randomized phase II trial of gefitinib for previously treated patients with advanced non-small-cell lung cancer. J Clin Oncol. 2003;21:2237. doi: 10.1200/JCO.2003.10.038. [DOI] [PubMed] [Google Scholar]
- Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, Sawyers CL. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293:876. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- Greulich H, Chen TH, Feng W, Janne PA, Alvarez JV, Zappaterra M, Bulmer SE, Frank DA, Hahn WC, Sellers WR, Meyerson M. Oncogenic transformation by inhibitor-sensitive and -resistant EGFR mutants. PLoS Med. 2005;2:e313. doi: 10.1371/journal.pmed.0020313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumireddy K, Baker SJ, Cosenza SC, John P, Kang AD, Robell KA, Reddy MV, Reddy EP. A non-ATP-competitive inhibitor of BCR-ABL overrides imatinib resistance. Proc Natl Acad Sci. 2005;102:1992. doi: 10.1073/pnas.0408283102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunther EJ, Moody SE, Belka GK, Hahn KT, Innocent N, Dugan KD, Cardiff RD, Chodosh LA. Impact of p53 loss on reversal and recurrence of conditional Wnt-induced tumorigenesis. Genes Dev. 2003;17:488. doi: 10.1101/gad.1051603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D. Heritable formation of pancreatic beta-cell tumours in transgenic mice expressing recombinant insulin/simian virus 40 oncogenes. Nature. 1985;315:115. doi: 10.1038/315115a0. [DOI] [PubMed] [Google Scholar]
- Huettner CS, Zhang P, Van Etten RA, Tenen DG. Reversibility of acute B-cell leukaemia induced by BCR-ABL1. Nat Genet. 2000;24:57–60. doi: 10.1038/71691. [DOI] [PubMed] [Google Scholar]
- Jain M, Arvanitis C, Chu K, Dewey W, Leonhardt E, Trinh M, Sundberg CD, Bishop JM, Felsher DW. Sustained loss of a neoplastic phenotype by brief inactivation of MYC. Science. 2002;297:102. doi: 10.1126/science.1071489. [DOI] [PubMed] [Google Scholar]
- Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, Halmos B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- Kris M, Natale RB, Herbst R, Lynch T, Jr, Prager D, Belani CP, Schiller JH, Kelly K, Spiridonidis H, Sandler A, Albain K, Cella D, Wolf M, Averbuch S, Ochs J, Kay A. Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non-small cell lung cancer. J Am Med Assoc. 2003;290:2149. doi: 10.1001/jama.290.16.2149. [DOI] [PubMed] [Google Scholar]
- Kwak EL, Sordella R, Bell DW, Godin-Heymann N, Okimoto RA, Brannigan BW, Harris PL, Driscoll DR, Fidias P, Lynch TJ, Rabindran SK, McGinnis JP, Wissner A, Sharma SV, Isselbacher KJ, Settleman J, Haber DA. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc Natl Acad Sci. 2005;102:7665. doi: 10.1073/pnas.0502860102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- Martin GS. Rous sarcoma virus: A function required for the maintenance of the transformed state. Nature. 1970;227:1021. doi: 10.1038/2271021a0. [DOI] [PubMed] [Google Scholar]
- Moody SE, Sarkisian CJ, Hahn KT, Gunther EJ, Pickup S, Dugan KD, Innocent N, Cardiff RD, Schnall MD, Chodosh LA. Conditional activation of Neu in the mammary epithelium of transgenic mice results in reversible pulmonary metastasis. Cancer Cell. 2002;2:451. doi: 10.1016/s1535-6108(02)00212-x. [DOI] [PubMed] [Google Scholar]
- Naik P, Karrim J, Hanahan D. The rise and fall of apoptosis during multistage tumorigenesis: Down-modulation contributes to tumor progression from angiogenic progenitors. Genes Dev. 1996;10:2105. doi: 10.1101/gad.10.17.2105. [DOI] [PubMed] [Google Scholar]
- Naoki K, Chen TH, Richards WG, Sugarbaker DJ, Meyerson M. Missense mutations of the BRAF gene in human lung adenocarcinoma. Cancer Res. 2002;62:7001. [PubMed] [Google Scholar]
- Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, Eck MJ, Sellers WR, Johnson BE, Meyerson M. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science. 2004;304:1497. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- Pao W, Miller VA. EGFR mutations, small molecule kinase inhibitors, and non-small cell lung cancer: Current knowledge and future directions. J Clin Oncol. 2005;23:2556. doi: 10.1200/JCO.2005.07.799. [DOI] [PubMed] [Google Scholar]
- Pao W, Klimstra DS, Fisher GH, Varmus HE. Use of avian retroviral vectors to introduce transcriptional regulators into mammalian cells for analyses of tumor maintenance. Proc Natl Acad Sci. 2003;100:8764. doi: 10.1073/pnas.1133333100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005a;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pao W, Wang TY, Riely GJ, Miller VA, Pan Q, Landanyi M, Zakowski MF, Heelan RT, Kris MG, Varmus HE. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2005b;2:e17. doi: 10.1371/journal.pmed.0020017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, Singh B, Heelan R, Rusch V, Fulton L, Mardis E, Kupfer D, Wilson R, Kris M, Varmus H. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci. 2004;101:13306. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkin DM, Bray F, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- Parsons DW, Wang TL, Samuels Y, Bardelli A, Cummins JM, DeLong L, Silliman N, Ptak J, Szabo S, Willson JK, Markowitz S, Kinzler KW, Vogelstein B, Lengauer C, Velculescu VE. Colorectal cancer: Mutations in a signalling pathway. Nature. 2005;436:792. doi: 10.1038/436792a. [DOI] [PubMed] [Google Scholar]
- Pelengaris S, Khan M, Evan GI. Suppression of Myc-induced apoptosis in beta cells exposes multiple oncogenic properties of Myc and triggers carcinogenic progression. Cell. 2002;109:321. doi: 10.1016/s0092-8674(02)00738-9. [DOI] [PubMed] [Google Scholar]
- Perl AK, Wilgenbus P, Dahl U, Semb H, Christofori G. A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature. 1998;392:190. doi: 10.1038/32433. [DOI] [PubMed] [Google Scholar]
- Podsypanina K, Li Y, Varmus HE. Evolution of somatic mutations in mammary tumors in transgenic mice is influenced by the inherited genotype. BMC Med. 2004;2:24. doi: 10.1186/1741-7015-2-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg N, Jolicoeur P. Retroviral pathogenesis. In: JM Coffin, et al., editors. Retroviruses. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York: 1997. p. 475. [PubMed] [Google Scholar]
- Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, Willson JK, Markowitz S, Kinzler KW, Vogelstein B, Velculescu VE. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- Shachaf CM, Kopelman AM, Arvanitis C, Karlsson A, Beer S, Mandl S, Bachmann MH, Borowsky AD, Ruebner B, Cardiff RD, Yang Q, Bishop JM, Contag CH, Felsher DW. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature. 2004;431:1112. doi: 10.1038/nature03043. [DOI] [PubMed] [Google Scholar]
- Shah NP, Tran C, Lee FY, Chen P, Norris D, Sawyers CL. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science. 2004;305:399. doi: 10.1126/science.1099480. [DOI] [PubMed] [Google Scholar]
- Shigematsu H, Takahashi T, Nomura M, Majmudar K, Suzuki M, Lee H, Wistuba II, Fong KM, Toyooka S, Shimizu N, Fujisawa T, Minna JD, Gazdar AF. Somatic mutations of the HER2 kinase domain in lung adenocarcinomas. Cancer Res. 2005;65:1642. doi: 10.1158/0008-5472.CAN-04-4235. [DOI] [PubMed] [Google Scholar]
- Siegel PM, Dankort DL, Hardy WR, Muller WJ. Novel activating mutations in the neu proto-oncogene involved in induction of mammary tumors. Mol Cell Biol. 1994;14:7068. doi: 10.1128/mcb.14.11.7068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sordella R, Bell DW, Haber DA, Settleman J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science. 2004;305:1163. doi: 10.1126/science.1101637. [DOI] [PubMed] [Google Scholar]
- Stehelin D, Varmus HE, Bishop JM, Vogt PK. DNA related to the transforming gene(s) of avian sarcoma viruses is present in normal avian DNA. Nature. 1976;260:170. doi: 10.1038/260170a0. [DOI] [PubMed] [Google Scholar]
- Stephens P, Hunter C, Bignell G, Edkins S, Davies H, Teague J, Stevens C, O’Meara S, Smith R, Parker A, Barthorpe A, Blow M, Brackenbury L, Butler A, Clarke O, Cole J, Dicks E, Dike A, Drozd A, Edwards K, Forbes S, Foster R, Gray K, Greenman C, Halliday K, Hills K, Kosmidou V, Lugg R, Menzies A, Perry J, Petty R, Raine K, Ratford L, Shepherd R, Small A, Stephens Y, Tofts C, Varian J, West S, Widaa S, Yates A, Brasseur F, Cooper CS, Flanagan AM, Knowles M, Leung SY, Louis DN, Looijenga LH, Malkowicz B, Pierotti MA, Teh B, Chenevix-Trench G, Weber BL, Yuen ST, Harris G, Goldstraw P, Nicholson AG, Futreal PA, Wooster R, Stratton MR. Lung cancer: Intragenic ERBB2 kinase mutations in tumours. Nature. 2004;431:525. doi: 10.1038/431525b. [DOI] [PubMed] [Google Scholar]
- Stephens P, Edkins S, Davies H, Greenman C, Cox C, Hunter C, Bignell G, Teague J, Smith R, Stevens C, O’Meara S, Parker A, Tarpey P, Avis T, Barthorpe A, Brackenbury L, Buck G, Butler A, Clements J, Cole J, Dicks E, Edwards K, Forbes S, Gorton M, Gray K, Halliday K, Harrison R, Hills K, Hinton J, Jones D, Kosmidou V, Laman R, Lugg R, Menzies A, Perry J, Petty R, Raine K, Shepherd R, Small A, Solomon H, Stephens Y, Tofts C, Varian J, Webb A, West S, Widaa S, Yates A, Brasseur F, Cooper CS, Flanagan AM, Green A, Knowles M, Leung SY, Looijenga LH, Malkowicz B, Pierotti MA, Teh B, Yuen ST, Nicholson AG, Lakhani S, Easton DF, Weber BL, Stratton MR, Futreal PA, Wooster R. A screen of the complete protein kinase gene family identifies diverse patterns of somatic mutations in human breast cancer. Nat Genet. 2005;37:590. doi: 10.1038/ng1571. [DOI] [PubMed] [Google Scholar]
- Tamborini E, Bonadiman L, Greco A, Albertini V, Negri T, Gronchi A, Bertulli R, Colecchia M, Casali PG, Pierotti MA, Pilotti S. A new mutation in the KIT ATP pocket causes acquired resistance to imatinib in a gastrointestinal stromal tumor patient. Gastroenterology. 2004;127:294. doi: 10.1053/j.gastro.2004.02.021. [DOI] [PubMed] [Google Scholar]
- Tracy S, Mukohara T, Hansen M, Meyerson M, Johnson BE, Janne PA. Gefitinib induces apoptosis in the EGFRL858R non-small-cell lung cancer cell line H3255. Cancer Res. 2004;64:7241. doi: 10.1158/0008-5472.CAN-04-1905. [DOI] [PubMed] [Google Scholar]
- Tsao MS, Sakurada A, Cutz JC, Zhu CQ, Kamel-Reid S, Squire J, Lorimer I, Zhang T, Liu N, Daneshmand M, Marrano P, da Cunha Santos G, Lagarde A, Richardson F, Seymour L, Whitehead M, Ding K, Pater J, Shepherd FA. Erlotinib in lung cancer: Molecular and clinical predictors of outcome. N Engl J Med. 2005;353:133. doi: 10.1056/NEJMoa050736. [DOI] [PubMed] [Google Scholar]
- Wang R, Ferrell LD, Faouzi S, Maher JJ, Bishop JM. Activation of the Met receptor by cell attachment induces and sustains hepatocellular carcinomas in transgenic mice. J Cell Biol. 2001;153:1023. doi: 10.1083/jcb.153.5.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]