

Abstract
Technological advances over the past 10 years have generated powerful tools for parallel analysis of complex biological problems. Among these new technologies, DNA arrays have provided an important experimental approach for identifying changes in the levels of individual mRNA molecules during important cellular transitions. However, cellular behavior is dictated not by mRNA levels, but by the proteins translated from the individual mRNA species. We report a high-throughput method for simultaneously monitoring the translation state and level of individual mRNA species. Messenger RNAs from resting and mitogenically activated fibroblasts were separated, according to degree of ribosome loading, into well-translated and under-translated pools. cDNA probes generated from these fractions were used to interrogate cDNA arrays. Among approximately 1,200 genes analyzed, less than 1% were found to be translationally regulated in response to mitogenic activation, demonstrating the strong selectivity of this regulatory mechanism. This high-throughput approach is shown to be an effective tool for superimposing translation profile on mRNA level for large numbers of genes, as well as for identifying translationally regulated genes for further study.
Recent advances in technology have opened the way to massively parallel analysis of cellular mRNA levels (1–7, 30). However, it is generally protein molecules, and not mRNAs, that determine phenotype. Therefore, to gain a global understanding of the regulation of cellular phenotype, it is essential to know not just the levels of individual mRNA molecules, but whether they are being translated into their cognate proteins (i.e., mRNA translation state). Current methods for direct analysis of protein expression (“proteome” analysis) are cumbersome, insensitive, and not yet readily adapted to high-throughput analysis (8). This paper describes an approach to defining the translation state of individual mRNA species that has been adapted to large-scale analysis and has the sensitivity to detect mRNAs that are present in low abundance.
Messenger RNAs that are being actively translated usually have multiple ribosomes associated with them, forming large structures known as polyribosomes or polysomes. Translationally inactive mRNAs are often sequestered in messenger ribonucleoprotein (mRNP) particles or associated with a single ribosome (“monosome”). Polysomes and mRNP particles can be readily separated by sucrose gradient centrifugation, thus allowing an operational distinction between well-translated and under-translated mRNA molecules (9). We show in this paper that labeled cDNA copies of mRNAs from these two fractions can be used to interrogate DNA arrays, thereby forming the basis of a high-throughput assay for the translation state of individual messages.
We tested this experimental approach by analyzing the translation state of mRNAs in resting and mitogenically activated fibroblasts. There is strong evidence that considerable regulation at the translational level occurs during this cellular transition (10, 11). Additionally, the fact that deregulation of translation can lead to oncogenic transformation argues that key growth-control genes are under translational control (11, 12). In this study, we screened commercially available cDNA arrays and identified a set of mRNA molecules that change translation state in fibroblasts as a response to mitogenic signals.
MATERIALS AND METHODS
Cell Culture.
Swiss 3T3 D1 cells and human foreskin fibroblasts (a gift from M. W. White, Montana State University, Bozeman, MT) were grown in DMEM supplemented with nonessential amino acids (100 μM), pyruvate (1 mM), penicillin (100 units/ml), streptomycin (100 μg/ml), and 10% calf serum. Growth arrest was obtained by incubating 50% confluent cells in medium containing 0.5% serum for 3 days. Cells were activated by addition of serum to quiescent cells at a final concentration of 10%, followed by incubation for 6 h before harvesting.
Polysome Fractionation.
The following procedure is described in detail in ref. 9. Approximately 1 × 107 cells were first incubated with 100 μg of cycloheximide per ml for 10 min to arrest ribosome movement on polysomes before the cells were harvested from the plates. Cells were then lysed by detergent treatment. The cytoplasmic extracts were mixed with heparin and layered on 0.5–1.5 M sucrose gradients. After centrifugation at 164,000 × g in a Beckman SW40 rotor for 110 min, gradients were fractionated into 1-ml fractions, with continuous monitoring of A260. Total RNA was purified from each fraction by twice extracting with equal volumes of phenol/chloroform after incubation with SDS/proteinase K.
cDNA Probes.
The purified RNA from sucrose gradient fractions was precipitated in 1.5 M LiCl with an equal volume of isopropanol before use in the labeling reactions. The complex 32P-labeled first-strand cDNA probes were synthesized and purified according to the protocol provided in the Altas cDNA Expression Arrays User Manual (CLONTECH). Briefly, 20 μg of total RNA was used as template in a 10-μl reverse transcription reaction. A gene-specific primer mixture (CLONTECH) was used to prime reverse transcription in the probe synthesis reaction.
Hybridization and Quantitation of cDNA Arrays.
The cDNA expression array filters were prehybridized in ExpressHyb (CLONTECH) for 30 min at 68°C and hybridized with 32P-labeled first strand cDNA probes (ca. 5 × 106 cpm) overnight at 68°C. After hybridization and washing, the array filters were sealed in plastic bags and exposed to a phosphorimaging screen for 24 h at room temperature. The exposed screen was scanned on PhosphorImager (Molecular Dynamics) and array elements on the array images were quantified by using the grid function of ImageQuant (Molecular Dynamics). The grid was superimposed over the array image with each box in the grid containing a single array element. The median count within the box was recorded and was corrected by subtracting its local background. The hybridization signal of each array element was then normalized to the median intensity of the hybridization signal on each array filter to allow comparison of the intensity of hybridization signals from different array filters. Only the array elements with specific hybridization signals on at least two filters (as assayed by the signal intensity and confirmed by visual inspection) were kept for further analysis.
RESULTS
The experimental approach is illustrated in Fig. 1. Growth-arrested and mitogenically activated mouse Swiss 3T3 cells and human foreskin fibroblasts were harvested, lysed, and fractionated by centrifugation through sucrose gradients. Fractions containing mRNP particles and monosomes were pooled as containing the under-translated mRNA species. Fractions containing mRNA with multiple ribosomes were collected as the translated mRNA pool. Total RNA from these fractions was labeled with 32P by reverse transcriptase reaction using a set of gene-specific primers to increase sensitivity and to reduce background of hybridization. CLONTECH Altas mouse and human Cancer Expression Arrays, which have a total of 1,194 double-spotted array elements (for 597 genes) on each array filter, were used to identify translationally controlled genes. The complex 32P-labeled first-strand cDNA probes from the mRNA fractions of resting and activated cells were hybridized to four identical array filters. The array filters were then washed at high stringency and exposed to phosphorimaging screens for analysis.
Figure 1.

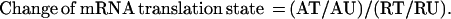 |
Significant hybridization signals were obtained for >50% of the array elements (Fig. 2). The signal of each array element was quantified and local background was subtracted. The intensities of the hybridization signals spanned two to three orders of magnitude for the 1,194 array elements on the filters. The signals were normalized to the median intensity of the hybridization signals of the particular array filter to correct for differences in global hybridization efficiency between filters. During the course of analysis, we found that an array element was undistinguishable from background if (i) the hybridization signal/background ratio was less than 1.1 and (ii) the adjusted signal (signal minus background) was less than 0.5-fold of the median signal intensity. The signal intensities of these unreliable low intensity array elements were assigned an arbitrary number equal to half of the median signal intensity to reduce the number of false-positive genes in the data analysis.
Figure 2.

Translation state of human genes monitored on cDNA arrays. Comparison of DNA arrays hybridized with complex 32P-labeled cDNA probes from the under-translated (Left) and translated (Right) RNA pools of both resting (Lower) and serum-activated human foreskin fibroblasts (Upper). Growth-arrested human foreskin fibroblasts were stimulated to enter the cell cycle by addition of serum and fractionated by centrifugation through a 0.5–1.5 M sucrose gradient (9). The RNA fractions containing one ribosome or less per mRNA were pooled as the under-translated fraction, and fractions loaded with two or more ribosomes per mRNA were pooled as translated fractions (Fig. 1). An equal percentage of the RNA in each fraction was used to prepare complex 32P-labeled first-strand cDNA probes as recommended by CLONTECH. CLONTECH Altas human cancer cDNA Expression Array filters were hybridized with the 32P-labeled cDNA probes. The arrows indicate the translationally regulated genes found in this study; 1, vimentin; 2, Stat1; and 3, 23-kDa highly basic protein.
We compared the translation states of mRNAs isolated from growth-arrested and activated fibroblasts by comparing the change of mRNA distribution on polysomes. The translation state of an mRNA from cells in a particular condition was defined by the ratio of normalized signal intensity between the translated and the under-translated RNA fractions. A well-translated gene, such as β-actin, has a value greater than 1.0, whereas translationally inactive mRNAs have values less than 1.0. The change of mRNA translation state between activated and resting cells was defined by the following formula:
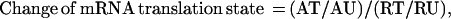 |
where AT, AU, RT, and RU represent the signal intensity of a particular array element on filters that were hybridized with complex cDNA probes made from the well-translated (T) or under-translated (U) RNA fractions of activated (A) and resting (R) cells. Thus, mRNAs with unchanged translation states during this cellular transition should have a value of 1.0. Indeed, 90% of the genes in this study have a value of 1.0 ± 1.3 on the array (Fig. 3A). As a conservative standard, messenger RNA translation state change was considered significant only if the following two criteria were met: (i) the signal intensity of the array element of the under-translated RNA filter of resting cells and the translated RNA filter of activated cells (or the reverse for translationally inactivated mRNAs) exceeded the median signal intensity of all the array elements and (ii) the value of the change in translation state was >2.5-fold of median value of the change of mRNA distribution of all the genes on the arrays. Because the DNA for each gene was double spotted on the CLONTECH arrays, only those genes for which the duplicates met the above criteria were chosen for further examination. By these criteria, eight genes on the mouse array and 10 genes on the human array were scored as potential translationally regulated genes. To confirm the array results, mRNA distribution across the polysome display was measured for each potentially positive gene by using Northern blot analysis. Examples are given in Fig. 4. Of potentially positive mRNAs that had changes in translation state greater than 3-fold on the arrays, 75% were confirmed to be altered in translation state by the Northern blot analysis. The quantitative change in mRNA distribution determined by the two methods differed by 1.4 ± 0.5-fold for the genes that were ultimately identified as positive in this study (Table 1).
Figure 3.

Comparing translation states (A) and relative levels (B) of mRNAs in growth-arrested and serum-activated human fibroblasts. The hybridization signal of each element on the human cancer cDNA expression array was quantified and normalized to the median value of all the elements on the filter. A subset of genes was selected for graphing according to the following selection criteria: (i) specific hybridization signals were found on all four human cancer expression array filters and (ii) hybridization signals were consistent among the duplicates of the particular gene. (A) The translation state of a particular gene was expressed as the ratio of the normalized hybridization signal intensities between the well-translated and under-translated RNA fractions. The change of mRNA translation state was defined in the legend to Fig. 1. The arrows indicate the genes that were identified as translationally regulated on the human DNA array and confirmed on the polysome display and Northern blot analysis in this study: 1, vimentin; 2, Stat1; and 3, 23-kDa highly basic protein. (B) The relative mRNA level was expressed as the ratio of the normalized total hybridization signal intensities between the serum-activated and growth-arrested cells. The total hybridization signal intensity was the sum of the signal on both translated and under-translated array filters.
Figure 4.

Polysome association in resting and activated human foreskin fibroblasts of mRNAs encoding vimentin, 23-kDa highly basic protein and β-actin. Cells were growth-arrested by serum starvation and stimulated by addition of serum (18). Cytoplasmic extracts were prepared and separated on sucrose gradients (9). (Upper) Optical density profiles of sucrose gradient. (Lower) Northern blot analysis of the RNA extracted from sucrose gradient fractions.
Table 1.
Translationally regulated genes revealed from comparative polysome distribution analysis of growth-arrested and serum-activated cells
| Gene | Entrez database ID | Change of mRNA level | Change of translation
|
|
|---|---|---|---|---|
| Array | Northern | |||
| 23-kDa highly basic protein* | X56932 | 0.97 | 5.7 | 3.2 |
| CACCC box-binding protein† | U36340 | 0.84 | 3.9 | 2.1 |
| Laminin receptor 1† | J02870 | 1.6 | 3.8 | 2.2 |
| Nur77 early response protein† | J04113 | 1.3 | 1.4 | 2.9 |
| Stat1* | M97935 | 0.85 | 3.3 | 2.6 |
| Human vimentin* | X56134 | 0.66 | 3.4 | 2.2 |
| Mouse vimentin† | X51438 | 0.61 | 2.1 | 3.0 |
Shown are changes translation state of the mRNAs that were identified on the human cancer expression array (∗) and on the mouse expression array (
).
A measure of the total level of any mRNA species could be obtained by summing the signal intensity obtained in the translated and under-translated fractions. The results are summarized for mRNAs isolated from growth-arrested and activated human fibroblasts in Fig. 3B. As expected (13) we found the levels of a number of mRNA species to be up-regulated after mitogenic activation. These included significant increases in the mRNAs corresponding to early growth response protein-1, mitogen-inducible gene-5, and c-fos. In addition to the data presented here, we provide the raw data sets, accessory data, and images of both cDNA arrays on our web site (http://weber.u.washington.edu/∼morrilab/). The relative levels of those mRNAs that were identified as being under translational control changed relatively little, if at all (Table 1). Because these samples were studied 6 h after mitogenic activation, the nur77 mRNA, which shows a transient early response in mRNA level (14), had already nearly returned to baseline level.
DISCUSSION
When quiescent mammalian cells are stimulated to re-enter the cell cycle, they exhibit a global increase of protein synthesis within the first several hours after activation. This large and rapid response in the rate of protein synthesis is due both to the recruitment of stored mRNA from the mRNP particles and to newly synthesized mRNA (10). Interestingly, mRNA arising from the under-translated pool may account for as much as 80% of the mRNA in polysomes during the initial 6 h after mitogenic activation (15). Messenger RNAs encoding the cytoskeletal protein vimentin and various components of the protein synthesis machinery, such as ribosomal proteins, elongation factor eEF1α, and polyA binding protein, were found previously to respond on the translational level to serum stimulation (16). A large part of the stored, untranslated mRNAs encode ribosome proteins and have an oligopyrimidine track (TOP element) adjacent to the 5′ cap structure of the mRNA (17). Messenger RNAs, such as that encoding ribosome protein L32, that contain the TOP element were known to be recruited from the stored mRNP particles and monosomes into polysomes following serum activation of quiescent Swiss 3T3 cells, whereas some mRNAs, such as β-actin, showed no significant change in distribution (18).
In the current study, two mRNAs with TOP elements were identified among the translationally up-regulated species. One of these TOP mRNAs, that encoding 37-kDa component of the laminin receptor, was identified as translationally up-regulated on the mouse expression array. The 37-kDa laminin receptor component is a bifunctional protein that is highly conserved in a wide spectrum of eukaryotic cells (19). It is incorporated into the cell-surface laminin receptor complex (20) and may also be a component of the protein synthesis machinery. This protein not only physically associates with the 40S ribosome subunit in both mammalian cells (21) and yeast (22), but it also is structurally related to the S2 family of ribosomal components (23). The other TOP mRNA found to be up-regulated is that encoding human highly basic protein (24). A search of Entrez databases revealed that human highly basic protein is highly conserved across species and also shares 87% identity with the ribosomal protein L13a of Rattus norvegicus. Not surprisingly, this mRNA also contains the signature TOP (5′-cttttcc-3′) sequence at its 5′ end.
Vimentin, a protein component of intermediate filaments, was independently identified as being under translational control on both the mouse and human arrays. Vimentin mRNA shifted from the under-translated fraction into large polysomes when both mouse Swiss 3T3 cells and human foreskin fibroblasts were re-activated to enter the cell cycle from the quiescent state (Fig. 4). Interestingly, the sum of the vimentin signals on the untranslated and translated arrays did not change significantly, or perhaps decreased slightly, after cells were re-activated (Table 1), although the average polysome loading number (an indicator of the relative translation efficiency of a particular mRNA) increased 1.6-fold. This change was comparable to change in loading number of the mRNA of ribosome protein L32 under the same cellular transition (data not shown). This increase in ribosome loading is consistent with the observation that actinomycin D did not block the elevation of vimentin protein synthesis when quiescent Swiss 3T3 cells are stimulated to proliferate (16).
Three medium to low abundance messenger RNAs, not previously known to be translationally regulated, were identified in this study. The early response protein nur77 (25), the CACCC-box-binding protein BKLF (26) and Stat 1 (27) (interferon-stimulated gene factor-3 protein) all encode transcription factors. Stat1, as a transcription factor that is directly coupled to signal transduction, is clearly of interest in the present context. Nur77 was identified as an immediate-early protein, whose mRNA was induced by serum and growth factors in mouse 3T3 fibroblasts (25). This inducible orphan receptor was found in our survey to be one of the translationally up-regulated mRNAs on the mouse array and this conclusion was confirmed by Northern blot analysis of RNA fractions from polysome displays of both mouse Swiss 3T3 cells and human foreskin fibroblasts. Nur77 mRNA was contained in the under-translated pool in quiescent cells, was recruited quickly (within 1 h) into large polysome fractions upon mitogenic activation, and had started to return to the small polysome fractions after 3 h (data not shown). As a control, the distribution of the mRNA encoding another early response protein, c-fos, which has similar mRNA expression kinetics to nur77 (28), did not score positively for translational control in this study.
The behavior of the nur77 and c-fos genes on these arrays underscores another attribute of the experimental approach taken here. By calculating the ratio of well-translated to under-translated pools of a mRNA species, the value for translation state should be independent of changes in overall cellular level of the mRNA. As products of early response genes, both nur77 and c-fos mRNAs undergo dramatic elevations in total level beginning within minutes after mitogenic activation. However, as illustrated here, only nur77 scored in our screen as being under translational control. Therefore, as anticipated, this method is robust enough to be insensitive to the orders-of-magnitude changes in total mRNA level seen with the early response genes and still detect those mRNAs that change in translation state. This is significant, since one suspects that expression of important regulatory genes could be controlled at multiple levels.
Combining polysome display and DNA array analysis to characterize the translation state of individual mRNA species greatly extends the biological capabilities of gene expression screens. The speed, effectiveness, and feasibility of simultaneously monitoring translational state and relative mRNA level was demonstrated in this study. Even in the experiments described here, which were designed only as a proof of concept, seven mRNAs out of 1,200 genes tested were identified as being growth-regulated at the translational level. This result suggests that the high throughput method developed in this study is suitable for efficaciously interrogating much larger sets of genes for translational regulation. Because of the way the polysome fractions were combined, the limitations of sensitivity, and the signal-to-background ratio of the filter-based DNA array technology, we were able to detect only medium to high abundance messenger RNA species which showed greater than a 2- to 3-fold change of its distribution between the under-translated and well-translated mRNA fractions. However, by separating the polysome fractions into light and heavy polysomal fractions, this array based method can be adapt to identify those translationally regulated mRNAs that only shift on the polysomal region. Recent developments in DNA array technology, based on two-color fluorescence labeling of probes, DNA arrays on glass slides (1, 5), and oligonucleotide-based DNA array chips (7, 29), has increased the sensitivity and pushed the detection limit to the level of a single mRNA copy per cell for genome-wide transcriptional analysis. By adopting the glass-based microarray technology, we expect to increase the sensitivity and accuracy of this method greatly.
As discussed in the introduction, translation of mRNA into protein defines cellular phenotype. In the future, one anticipates that proteomics will achieve the throughput capacity and sensitivity necessary for large-scale analysis and thereby provide a measure both of protein level and of posttranslational regulation. However, analysis of translation state by the approach presented here can provide direct information on translational regulation of mRNAs, whereas changes in protein levels identified through proteomic analysis could arise from alterations in rates of either protein synthesis or protein degradation. The simultaneous monitoring of both cellular level and translation state of all messenger RNAs at a genome-wide level will provide a much more complete description of global mRNA expression than was hitherto possible.
Acknowledgments
We are grateful to Dr. Peter Nelson for his continuing advice on array analysis during this study and to Mr. Gregory Mize for sharing his expertise on polysome analysis. D.R.M. thanks Dr. Robert Franza for first bringing to his attention the potential power of DNA arrays as applied to analysis of translational control. The initial phase of this work was supported by a grant from the University of Washington Royalty Research Fund and continuing support was from National Institutes of Health Grants CA71453, GM57912, and HG01713 and from National Science Foundation Grant BIR9214821/9423347.
ABBREVIATION
- mRNP
messenger ribonucleoprotein
References
- 1.Schena M, Shalon D, Heller R, Chai A, Brown P O, Davis R W. Proc Natl Acad Sci USA. 1996;93:10614–10619. doi: 10.1073/pnas.93.20.10614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.DeRisi J, Penland L, Brown P O, Bittner M L, Meltzer P S, Ray M, Chen Y, Su Y A, Trent J M. Nat Genet. 1996;14:457–460. doi: 10.1038/ng1296-457. [DOI] [PubMed] [Google Scholar]
- 3.Zhang L, Zhou W, Velculescu V E, Kern S E, Hruban R H, Hamilton S R, Vogelstein B, Kinzler K W. Science. 1997;276:1268–1272. doi: 10.1126/science.276.5316.1268. [DOI] [PubMed] [Google Scholar]
- 4.Chee M, Yang R, Hubbell E, Berno A, Huang X C, Stern D, Winkler J, Lockhart D J, Morris M S, Fodor S P. Science. 1996;274:610–614. doi: 10.1126/science.274.5287.610. [DOI] [PubMed] [Google Scholar]
- 5.DeRisi J L, Iyer V R, Brown P O. Science. 1997;278:680–686. doi: 10.1126/science.278.5338.680. [DOI] [PubMed] [Google Scholar]
- 6.Wodicka L, Dong H, Mittmann M, Ho M H, Lockhart D J. Nat Biotechnol. 1997;15:1359–1367. doi: 10.1038/nbt1297-1359. [DOI] [PubMed] [Google Scholar]
- 7.Cho R J, Campbell M J, Winzeler E A, Steinmetz L, Conway A, Wodicka L, Wolfsberg T G, Gabrielian A E, Landsman D, Lockhart D J, et al. Mol Cell. 1998;2:65–73. doi: 10.1016/s1097-2765(00)80114-8. [DOI] [PubMed] [Google Scholar]
- 8.Garrels J I, McLaughlin C S, Warner J R, Futcher B, Latter G I, Kobayashi R, Schwender B, Volpe T, Anderson D S, Mesquita-Fuentes R, et al. Electrophoresis. 1997;18:1347–1360. doi: 10.1002/elps.1150180810. [DOI] [PubMed] [Google Scholar]
- 9.Ruan H J, Brown C Y, Morris D R. In: Analysis of mRNA Formation and Function. Richter J D, editor. New York: Academic; 1997. pp. 305–321. [Google Scholar]
- 10.Morris D R. Prog Nucleic Acids Res Mol Biol. 1995;51:339–363. doi: 10.1016/s0079-6603(08)60883-1. [DOI] [PubMed] [Google Scholar]
- 11.Sonenberg N. In: Translational Control. Hershey J W B, Mathews M B, Sonenberg N, editors. Plainview, NY: Cold Spring Harbor Lab. Press; 1996. pp. 245–270. [Google Scholar]
- 12.Korth M J, Katze M G. In: mRNA Metabolism & Post-transcriptional Gene Regulation. Harford J B, Morris D R, editors. New York: Wiley-Liss; 1997. pp. 265–280. [Google Scholar]
- 13.Iyer V R, Eisen M B, Ross D T, Schuler G, Moore T, Lee J C F, Trent J M, Staudt L M, Hudson J, Jr, Boguski M S, et al. Science. 1999;283:83–87. doi: 10.1126/science.283.5398.83. [DOI] [PubMed] [Google Scholar]
- 14.Yoon J K, Lau L F. J Biol Chem. 1993;268:9148–9155. [PubMed] [Google Scholar]
- 15.Rudland P S, Weil S, Hunter A R. J Mol Biol. 1975;96:745–766. doi: 10.1016/0022-2836(75)90150-3. [DOI] [PubMed] [Google Scholar]
- 16.Thomas G, Thomas G. J Cell Biol. 1986;103:2137–2144. doi: 10.1083/jcb.103.6.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hammond M L, Bowman L H. J Biol Chem. 1988;263:17785–17791. [PubMed] [Google Scholar]
- 18.Kaspar R L, Rychlik W, White M W, Rhoads R E, Morris D R. J Biol Chem. 1990;265:3619–3622. [PubMed] [Google Scholar]
- 19.Garcia-Hernandez M, Davies E, Staswick P E. J Biol Chem. 1994;269:20744–20749. [PubMed] [Google Scholar]
- 20.Castronovo V, Taraboletti G, Sobel M E. J Biol Chem. 1991;266:20440–20446. [PubMed] [Google Scholar]
- 21.Rosenthal E T, Wordeman L. J Cell Sci. 1995;108:245–256. doi: 10.1242/jcs.108.1.245. [DOI] [PubMed] [Google Scholar]
- 22.Demianova M, Formosa T G, Ellis S R. J Biol Chem. 1996;271:11383–11391. doi: 10.1074/jbc.271.19.11383. [DOI] [PubMed] [Google Scholar]
- 23.Davis S C, Tzagoloff A, Ellis S R. J Biol Chem. 1992;267:5508–5514. [PubMed] [Google Scholar]
- 24.Kato S, Sekine S, Oh S W, Kim N S, Umezawa Y, Abe N, Yokoyama-Kobayashi M, Aoki T. Gene. 1995;150:243–250. doi: 10.1016/0378-1119(94)90433-2. [DOI] [PubMed] [Google Scholar]
- 25.Ryseck R P, Bravo H M, Mattei M G, Ruppert S, Bravo R. EMBO J. 1989;8:3327–3335. doi: 10.1002/j.1460-2075.1989.tb08494.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crossley M, Whitelaw E, Perkins A, Williams G, Fujiwara Y, Orkin S H. Mol Cell Biol. 1996;16:1695–1705. doi: 10.1128/mcb.16.4.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumar R, Korutla L. Exp Cell Res. 1995;216:143–148. doi: 10.1006/excr.1995.1018. [DOI] [PubMed] [Google Scholar]
- 28.Williams G T, Lau L F. Mol Cell Biol. 1993;13:6124–6136. doi: 10.1128/mcb.13.10.6124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lockhart D J, Dong H, Byrne M C, Follettie M T, Gallo M V, Chee M S, Mittmann M, Wang C, Kobayashi M, Horton H, et al. Nat Biotechnol. 1996;14:1675–1680. doi: 10.1038/nbt1296-1675. [DOI] [PubMed] [Google Scholar]
- 30.Velculescu V E, Zhang L, Zhou W, Vogelstein J, Basrai M A, Bassett D E, Jr, Hieter P, Vogelstein B, Kinzler K W. Cell. 1997;88:243–251. doi: 10.1016/s0092-8674(00)81845-0. [DOI] [PubMed] [Google Scholar]