

Abstract
The neuronal soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex is essential for synaptic vesicle exocytosis, but its study has been limited by the neonatal lethality of murine SNARE knockouts. Here, we describe a viable mouse line carrying a mutation in the b-isoform of neuronal SNARE synaptosomal-associated protein of 25 kDa (SNAP-25). The causative I67T missense mutation results in increased binding affinities within the SNARE complex, impaired exocytotic vesicle recycling and granule exocytosis in pancreatic β-cells, and a reduction in the amplitude of evoked cortical excitatory postsynaptic potentials. The mice also display ataxia and impaired sensorimotor gating, a phenotype which has been associated with psychiatric disorders in humans. These studies therefore provide insights into the role of the SNARE complex in both diabetes and psychiatric disease.
Keywords: diabetes, mutagenesis, soluble N-ethylmaleimide-sensitive factor attachment protein receptor, schizophrenia, exocytosis
Intracellular membrane fusion events within eukaryotic cells are mediated by proteins of the soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) family. The prototypic SNARE-mediated fusion event is synaptic vesicle exocytosis, in which the membrane-associated SNAP-25 and syntaxin-1A interact with the vesicle-associated membrane protein (VAMP) to create a stable ternary core complex with 1:1:1 stoichiometry (1). This interaction has been extensively studied in various systems including neurons, neuroendocrine chromaffin cells, and pancreatic β-islet cells, all of which use the neuronal core complex proteins to regulate secretion (2, 3). In vivo knockout and mutagenesis studies in invertebrate systems have allowed SNARE function to be analyzed at the level of the synapse (4), but their scope has been limited by the failure of these organisms to model higher neurological functions in which SNARE function is strongly implicated. A major goal, therefore, has been the generation of a mammalian model of SNARE dysfunction in which the effects on complex neurological and behavioral phenotypes can be assessed.
Both Vamp2 and Snap25 knockouts are perinatally lethal in homozygous mice, and heterozygotes show no apparent phenotype, so detailed analysis of viable affected individuals has not been possible (5, 6). The spontaneous mouse mutant coloboma carries a heterozygous deletion spanning four genes including Snap25 (7), but its locomotor hyperactivity phenotype is likely to depend on the additional loss of one or more of the other genes within the deleted region. Most recently, a viable syntaxin-1a knockout mouse has been described with subtle defects in long-term potentiation and fear-memory behavior, although normal synaptic transmission is unaffected likely because of functional compensation by syntaxin-1b (8). Existing mammalian SNARE mutations have therefore offered only very limited insights into the consequences of SNARE dysfunction in an adult system and have yielded no information about possible involvement of SNARE proteins in human disease.
Here, we characterize the blind-drunk mouse, which possesses a mutation in a highly conserved domain of Snap25. We demonstrate the functional consequences of the mutation at both the molecular and cellular levels and describe a complex behavioral phenotype in mutant animals, which includes elements that have been linked to schizophrenia in humans. This new dominant mutation provides an opportunity to study the role of SNAP-25 in an adult mammalian system that would not have been possible by using a gene-knockout strategy.
Results
Identification of the Blind-Drunk (Bdr) Mutation.
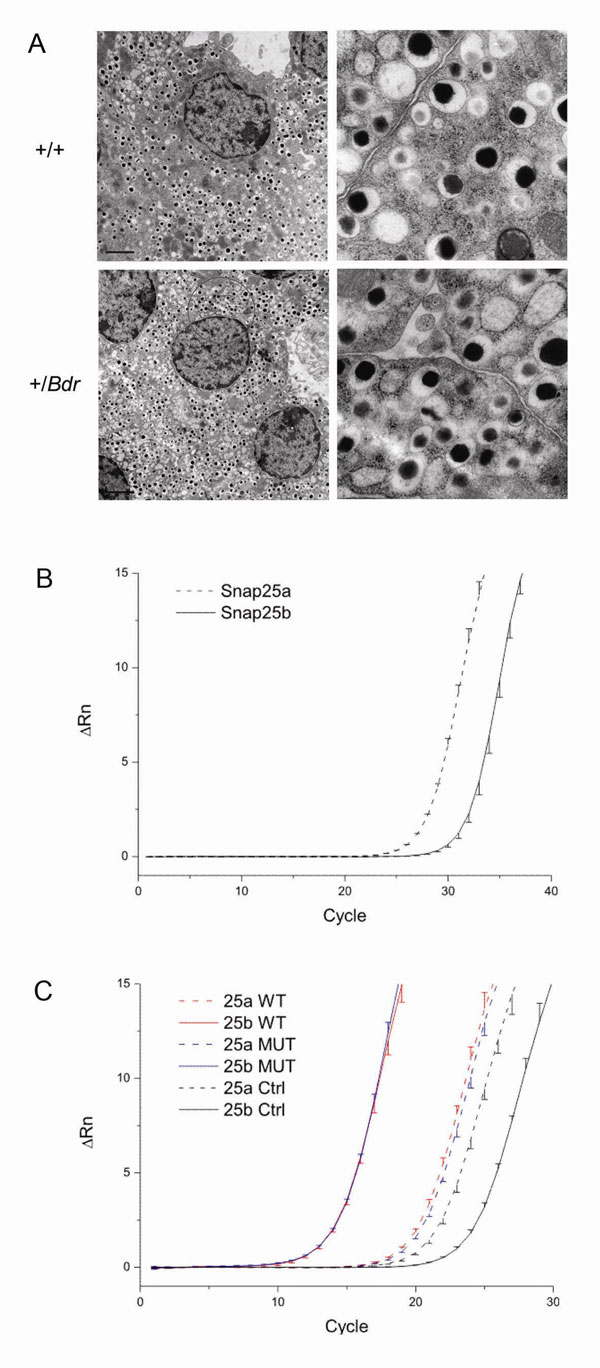
The blind-drunk mouse was originally identified from a large-scale, phenotype-driven dominant mutagenesis screen (9). Mutants (+/Bdr) are characterized by a subtle ataxic gait that is first detectable at ≈4 weeks of age. After an initial genome scan (9), subsequent genetic mapping reduced the minimal recombinant interval to <12 Mb between D2Mit106 and 145302160 (see Methods). Publicly available mouse and human genome sequence data identified Snap25 as the most promising candidate in this region. Both isoforms, SNAP-25a and SNAP-25b (10), were sequenced from genomic DNA and a T-to-C transversion was identified in codon 67 of SNAP-25b, leading to a nonconservative isoleucine-to-threonine amino acid substitution. This sequence change was not present in the parental strains, nor in five other inbred and two outbred mouse strains (data not shown). Ile-67 is conserved in all known SNAP-25 orthologues in addition to the paralogue SNAP-23 (11) (Fig. 1A). By using evidence from a gene-based screen of the Harwell mutant mouse archive, the probability of there being more than one potentially functional mutation in our study population of back-cross animals can be calculated as 0.00053 (12). However, to confirm that another mutation was not present in the recombinant interval, all known exons and intron–exon boundaries from all 39 genes and EST clones in the region were sequenced, and no other coding or potential splice-site mutations were discovered (data not shown).
Fig. 1.

Identification and modeling of the blind-drunk mutation. (A) Multiple amino acid sequence alignment, showing part of the highly conserved region within which the blind-drunk mutation (arrowhead) lies. (B and C) VMD visualization of molecular models of the SNARE complex (PDB ID 1KIL) containing WT (B) and I67T mutant (C) SNAP-25. Hydrogen bonds (shown in C) between N188 of the SNAP-25 C-terminal coiled-coil homology domain and both SNAP-25 T67 and VAMP L70 were predicted. (D) Varying amounts of lanthanide-labeled cytoplasmic domains of both syntaxin-1a (Stx1a) and VAMP-2 were added to either immobilized WT or mutant (I67T) SNAP-25b. The amount of protein bound is plotted as a function of amount of protein added; data are presented as means ± SD for three independent experiments. (E) Representative melting curves of assembled recombinant WT and mutant SNARE complexes containing SNAP-25 variants measured by CD spectroscopy.
Phenotype of the Bdr Mouse.
Blind-drunk mice are grossly normal in appearance and development, and brain morphology appeared normal up to 12 months of age by gross histological evaluation and more detailed examination by immunohistochemistry [supporting information (SI) Fig. 5A], including quantification of synaptophysin immunostaining as a measure of synaptic density. In situ hybridization showed no change in the level or pattern of expression of Snap25 in the brains of adult mutant mice compared with WT littermates (SI Fig. 5B), and a survey of SNAP-25, syntaxin-1a, and VAMP-2 presynaptic proteins by Western blot also revealed no significant differences in expression when quantified (SI Fig. 5C).
In Silico and in Vitro Modeling Predict Alterations in SNARE Complex Association in Blind-Drunk Mice.
Residue 67 of SNAP-25 is directly involved in interactions with both syntaxin and VAMP, forming, in part, one of the layers of the hydrophobic core of the neuronal SNARE complex (13). In silico modeling of the I67T substitution predicted the formation of two additional hydrogen bonds within the SNARE complex: one intramolecular bond involving the substituted Thr and Asn-188 in the C-terminal coiled-coil homology domain and the other between the same Asn-188 and Leu-70 of VAMP (Fig. 1 B and C). Formation of these bonds would be predicted to reduce free energy within the SNARE complex, increasing its stability. To confirm this finding in vitro, a quantifiable dissociation-enhanced, time-resolved lanthanide fluorescent binding assay (DELFIA) was performed in which cytoplasmic domains of both syntaxin-1a and VAMP-2 were bound to immobilized SNAP-25b. As expected, an ≈2-fold increase in affinity for syntaxin was seen in SNARE complex assembly with mutant SNAP-25 compared with WT protein; however, the affinity for VAMP was not significantly changed (Fig. 1D). The most likely explanation for this result is the previously described tendency of artifactual SNAP-25/syntaxin heterotrimers to form in vitro (14). To further quantify the stability of the mutant SNARE complex, CD spectroscopy was carried out by using recombinant proteins as described (15). In agreement with Nagy et al., our data show an increase in melting temperature of preassembled complexes containing WT SNAP-25b versus SNAP–25a of ≈4°C (Fig. 1E). Significantly, a similar reproducible increase in melting temperature (3–4°C) was observed for the complex containing the blind-drunk mutant SNAP-25b (Fig. 1E). As predicted by the in silico modeling and DELFIA assay, these data demonstrate further the influence of the mutation on stability of the core SNARE complex.
Impairment in the Spontaneous and Evoked Release of Glutamate Revealed by Cortical-Slice Electrophysiology.
To study the effect of the I67T mutation on neurotransmitter release, whole-cell voltage-clamp recordings (holding potential = −65 mV) were performed in layer 2–3 neurons of somatosensory cortical acute slices. The frequency, but not the amplitude, of miniature excitatory postsynaptic currents (mEPSCs) was lower in neurons from mutant mice compared with WT littermates (n = 8 each genotype, P < 0.0001; Fig. 2 A and B). Thus, blind-drunk mice have a clear impairment in the spontaneous, constitutive release of glutamate.
Fig. 2.

Blind-drunk cortical slice electrophysiology. (A) Representative example of mEPSCs recorded in layer 2–3 neurons of somatosensory cortical acute slices from WT and blind-drunk mutant mice under control conditions (1 μM TTX/50 μM picrotoxin/2.5 mM K+). (B) Pooled data. mEPSC frequency (Left) or amplitude (Right) in WT and mutant. Each value refers to one experiment in one slice; the average values are shown by the bars in the graph ± SEM (n = 8 for each histogram; ∗, P < 0.0001). (C–E) EPSCs recorded in neurons of layers 2 and 3 of somatosensory cortex elicited by intracortical stimulation. Graphs (Right) show pooled EPSC amplitude in WT and mutant. At 0.2 Hz (C), EPSCs peak amplitude was significantly smaller in neurons of mutant mice compared with WT (∗, P < 0.05). EPSCs elicited by 10 stimuli at 10 Hz (D) showed no difference between the groups (P > 0.05), but those elicited by 10 stimuli at 40 Hz (E) showed a different pattern of short-term plasticity in mutant mice (∗, P < 0.001), which lacked evidence of synaptic facilitation. EPSCs are shown normalized to those evoked by the first stimulus, and EPSCs elicited by the same stimulus number were compared between the two genotypes (n = 6 cells for each histogram). Student's t test and Mann–Whitney test.
Excitatory postsynaptic currents (EPSCs) were evoked by intracortical activation with a stimulating electrode in the presence of the GABAA receptor antagonist gabazine (0.4 μM). Stimulation at low frequency (0.2 Hz) elicited EPSCs on average 29% lower in amplitude in mutant mice compared with WT littermates (n = 6; P = 0.05; Fig. 2C); one possible cause of this result would be a failure to completely replenish the readily releasable pool (RRP) of synaptic vesicles between stimuli. Trains of high-frequency stimulation were then used to study short-term synaptic plasticity and rate of replenishment of the RRP (16, 17). Stimulation at 10 Hz evoked a short-term synaptic depression that was not significantly different in neurons from mice of either genotype (n = 6; P > 0.05; Fig. 2D). However, stimulation at 40 Hz, which mimics cortical γ-type oscillations, resulted in short-term facilitation, followed by depression in WT mice, whereas recordings from blind-drunk mice showed only depression without facilitation (n = 6; P < 0.001; Fig. 2E). These data demonstrate a rapid fall in synaptic transmission during repetitive high-frequency stimulation at γ-frequency and suggest that changes in action potential-evoked glutamate release in the blind-drunk cortex result from a failure in replenishment of the synaptic vesicle RRP.
Defective Vesicle Turnover in Blind-Drunk Mice Dissected by Electrophysiology in Pancreatic β-Cells.
To further investigate the possible impairment in vesicle trafficking, we used high-resolution capacitance measurements to study mouse pancreatic β-cells. Exocytosis of insulin-containing granules is SNARE-dependent and shares many features of synaptic vesicle release (18). Blind-drunk mice exhibited normal islet architecture (data not shown) and β cell ultrastructure; both total and docked secretory granules were unchanged in number in mutants compared with controls (SI Fig. 6A and Table 1). We also confirmed by quantitative RT-PCR that transcripts for both SNAP-25a and SNAP-25b are present in MIN6 mouse β-cells and whole mouse islets (SI Fig. 6 B and C). In concordance with previous studies, we conclude that, although SNAP-25a is predominant, mouse β-cells express both splice variants; moreover, it has been shown that both protein isoforms support Ca2+-stimulated insulin secretion (19).
Exocytosis was evoked by a train of 10 500-ms depolarizations during which the total capacitance increase was significantly reduced in blind-drunk β-cells (n = 9; P < 0.05; Fig. 3A). A second train was applied 1 min after completion of the first. Whereas exocytosis in control β-cells had recovered completely, the responses in mutant β-cells were further reduced (Fig. 3B). The responses to the first two depolarizations were taken to reflect the release of RRP granules, whereas the increase in capacitance during the subsequent eight depolarizations was taken to reflect replenishment of the RRP by mobilization of granules from the reserve pool (RP) (20). In mutant β-cells, mobilization is reduced by 50% and 70% during the first and second trains, respectively, whereas RRP is unaffected (Fig. 3C). If progressively longer depolarizations (5–350 ms) are applied (Fig. 3D), the responses plateau, providing an estimate of the size of the immediately releaseable pool (IRP), a subset of RRP vesicles that reside in the vicinity of the voltage-gated Ca2+-channels (21). The IRP was of similar size in WT and mutant β-cells, calculated as 37 ± 11 fF (n = 10) and 36 ± 7 (n = 10), respectively. The findings therefore support those of the neuronal electrophysiology, showing that after depletion of IRP and RRP, exocytosis becomes severely compromised by failure of mobilization of new vesicles from the RP.
Fig. 3.

Functional effects on vesicle release kinetics of the blind-drunk mutation in pancreatic β-cells. (A) Capacitance increases (ΔCm; Lower) evoked by a train of 10 500-ms depolarizations from −70 mV to 0 mV (V; Upper) applied at 1 Hz in single β-cells from +/+ (Left) and +/Bdr (Right) mice. (B) Mean capacitance increase elicited by the individual pulses (ΔCm,n − ΔCm,n−1) of a first train (Left) and a second train (applied 1 min after the first; Right) displayed against the pulse number (n) in WT (■) and mutant (●) mice. (C) Mean capacitance increase during the last eight depolarizations as a measure of refilling of RRP. Data are mean values +SEM of 9 (+/+) and 11 (+/Bdr) experiments. (D) Relationship between pulse duration (t) and increase in cell capacitance (ΔCm) in WT and Bdr mutant mice; curves were derived by fitting equation 3 of Eliasson et al. (44) to the data points for depolarizations ≤350 ms. Data are mean values ± SEM of 5–10 experiments. (E) Capacitance increase (ΔCm) evoked by infusion of a Ca2+/EGTA buffer (free [Ca2+]i = 1.5 μM) in MIN6 cells overexpressing WT or blind-drunk mutant (Bdr) SNAP-25b as indicated. The whole-cell configuration was established at time 0. (F) Summary of experiments in E expressed as the rate of capacitance increase (ΔCm/Δt). Data are mean values ± SEM of 14 and 10 cells for the WT and Bdr transfected cells, respectively. ∗, P < 0.05; ∗∗, P < 0.01; Student's t test.
To confirm the effect of mutant SNAP-25 on granule trafficking, WT and blind-drunk SNAP-25b constructs were separately overexpressed in mouse insulinoma MIN6 β-cells (22), and replenishment of RRP granules from the RP was studied by infusion of a Ca2+/EGTA buffer as described (23) (Fig. 3E). Quantitative RT-PCR demonstrated that equivalent levels of both WT and mutant SNAP-25b were expressed in the transfected cells (SI Fig. 6C). The rate of capacitance increase in MIN6 cells overexpressing mutant SNAP-25b was significantly reduced (P < 0.05; Fig. 3F), suggesting that the effects on insulin secretion in transfected MIN6 cells are attributable to impaired replenishment of the RRP and providing further proof that the SNAP-25 mutation explains the vesicle-trafficking defects observed in blind-drunk mice.
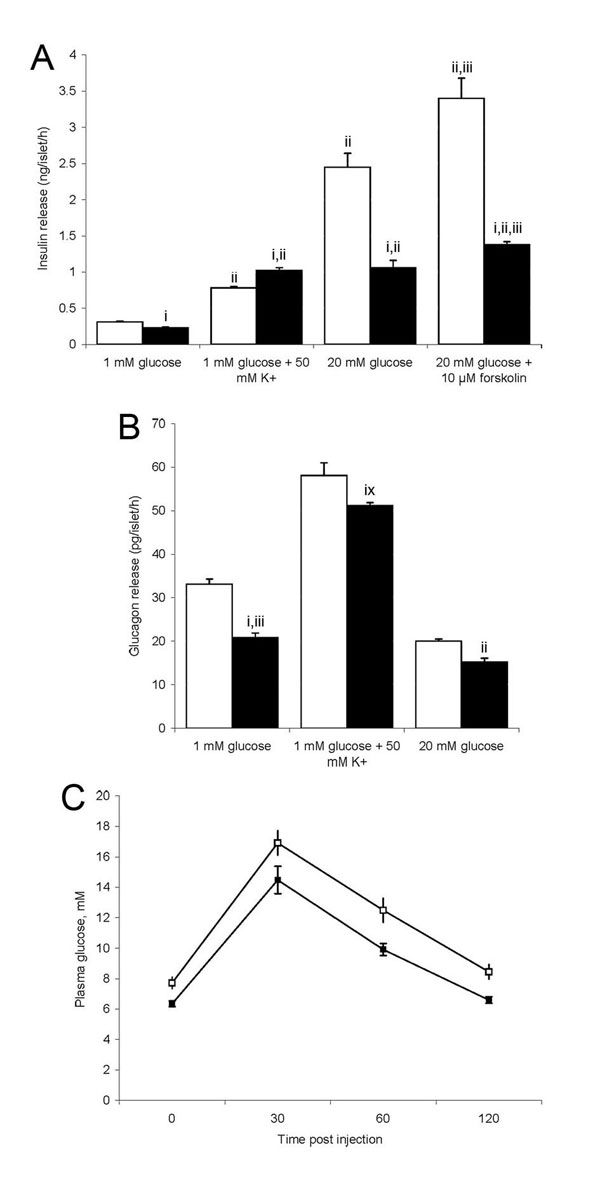
To explore possible in vivo consequences of these deficits, we studied the release of insulin and glucagon in intact pancreatic islets. There was a marked reduction in insulin secretion by mutant β-cells both under basal conditions and in the presence of a high glucose concentration that was accompanied by a reduction in glucagon secretion by α-cells (SI Fig. 7 A and B). However, baseline plasma glucose and insulin levels were not significantly different in freely fed blind-drunk mice compared with controls, and the response to the glucose challenge in a tolerance test was not impaired in mutants, although the initial glucose level in fasted mice was lower (SI Fig. 7C). This result is as expected, because secretion of both insulin and the antagonistically acting glucagon are impaired, the latter even more so under basal conditions.
Behavioral Testing of Blind-Drunk Mice.
To quantify the ataxic phenotype in blind-drunk animals, rotarod testing was performed at 7 weeks of age, and the mean latency to fall was significantly lower for mutants than for WT littermates (P < 0.001; Fig. 4A). The hyperactive phenotype of the coloboma mutant (7) led us to test the spontaneous locomotor activity of blind-drunk in an open-field apparatus; however, no significant difference between mutants and WT littermates was observed (P = 0.39; Fig. 4B). Prepulse inhibition (PPI) of the acoustic startle response, an important endophenotype for schizophrenia (24), was also tested in light of studies implicating SNAP-25 in the etiology of the disease (see Discussion). When a startle level of 110 db with a 90-db prepulse (9) was used, mutants showed significantly impaired PPI compared with their WT littermates (P = 0.01; Fig. 4C). We found no significant difference in the weight-corrected startle response amplitude (P > 0.1; data not shown).
Fig. 4.

Behavioral analysis of blind-drunk mutants (filled bars) and WT littermate controls (open bars). (A) Rotarod performance; the mean latency to fall from three consecutive trials is shown. (B) Spontaneous locomotor activity in open-field apparatus; the mean number of beam transgressions over a 30-min period is shown. (C) PPI testing; results shown as % PPI. (D–F) Social recognition testing; latency of interactions (D), time spent interacting with the intruder (E), and reduction in time spent interacting between the two trials (F) is shown. (G–I) Two-compartment light/dark testing; time spent in the light compartment (G), transitions between compartments (H), and the latency of transition from the light to dark area (I) is shown. (J–L) Exploration testing in a playground of toys; mice were scored as being inactive (J), using the toys (K), or exploring (L). Error bars + SEM; n = 16 each genotype; ∗, P < 0.05; ∗∗, P < 0.01; ∗∗∗, P < 0.001; ANOVA.
To further assess endophenotypes related to psychiatric disorders, social interaction and recognition testing was carried out. There was no significant difference in the latency of interactions by either genotype over two trials (P > 0.05; Fig. 4D), although blind-drunk mice spent a significantly greater amount of time interacting with the novel mice in the first trial (P < 0.05; Fig. 4E); however, there was no indication of a social-memory deficit in mutants, because although both groups showed a reduction in interaction time in the second trial, a greater and significant percentage reduction in was seen in blind-drunk mice (P < 0.03; Fig. 4F). Because of the importance of olfactory cues in such experiments, a buried chocolate chip location test was performed (25) which showed no significant impairment in blind-drunk (P > 0.1; data not shown). In the two-compartment light/dark test for anxiety (26), there was no difference between the two genotypes in the overall amount of time spent in the light compartments (P > 0.05; Fig. 4G); yet, there were significantly fewer transitions between the two compartments and a longer latency of transition to the dark compartment in blind-drunk mutant mice (P < 0.01; P < 0.02; Fig. 4 H and I). Blind-drunk mice did not, however, show signs of anxiety when tested in the elevated-plus maze (P > 0.1 in all cases; data not shown). In the “playground” paradigm (27), mutant mice demonstrated a marginally greater number of inactive episodes (P < 0.02) and exploring episodes (P < 0.01; Fig. 4 J and K) but interacted with the toys almost 20% less than WT littermate controls (P < 0.001, Fig. 4L). No significant differences were found in any of the measurements for the “emergence” paradigm (P = 0.15) or in spatial learning by examination of spontaneous alternation in the T maze or spatial memory in the Y maze (P = 0.22, P = 0.09, respectively; data not shown).
Discussion
The work presented here describes an ataxic mutant mouse with a complex phenotype caused by a missense mutation in a highly conserved codon of Snap25. This mutant has allowed the specific functional study of one SNAP-25 isoform, SNAP-25b, which varies from SNAP-25a at 9 aa residues because of alternative splicing of duplicated, tandemly arranged copies of exon 5. SNAP-25a is the major isoform expressed in the embryonic brain; however, this predominance switches at weaning age so that ultimately more than three-quarters of the expression in adult brains is derived from SNAP-25b, whereas SNAP-25a remains the principal isoform in neuroendocrine cells (10, 28, 29).
Here, we show that the blind-drunk mutation enhances the affinity of SNAP-25 for its binding partners and is therefore likely to cause an increase in association of SNARE proteins into complexes. This may also affect the association of SNAP-25 with regulatory factors; however, in silico modeling shows that the side chain of I67 is orientated toward the core of the complex. The mutation results in a reduction in the frequency of spontaneous mEPSCs, although these were of normal amplitude, consistent with a presynaptic defect, and a marked short-term depression of EPSCs at γ-frequency stimulation, suggesting a functional depletion of synaptic vesicles due to defective synaptic vesicle mobilization and replenishment of the RRP. High-resolution capacitance measurements in pancreatic islet β-cells support these findings, showing that whereas release of the IRP/RRP granules is essentially normal, subsequent release is severely reduced, indicating that replenishment of the RRP is impaired. In neurons, a combination of fast local recycling of exocytosed vesicles and recruitment of vesicles from the RP replenishes the RRP after its depletion; the RP itself is refilled via slower pathways of vesicle recycling (30). In β-cells, as in other peptide-secreting cells, the secretory granules cannot be refilled with insulin and must recycle via the Golgi complex (31). A defect in vesicle recycling due to increased SNARE complex stability and consequent impaired SNARE complex dissociation, an essential step in all these pathways of recycling, would account for our observations in both cell types. Consistent with this idea, Drosophila carrying a mutation in N-ethylmaleimide-sensitive factor (NSF), the major effector of SNARE complex dissociation (32), show an activity-dependent decrease in neurotransmitter release nearly identical to that seen in blind-drunk mice (33). Impaired vesicle recycling would be expected particularly to affect neural circuits containing very rapidly firing cells, for example cortical inhibitory interneurons, which may explain specific features of the behavioral phenotype, such as impaired sensorimotor gating, which depend on circuits such as these.
The reduction of glucose-induced insulin secretion observed in β-cells in vitro does not result in measurably altered glucose tolerance in vivo. It is possible that the parallel reduction of glucagon secretion obscures the hyperglycemic effect of insufficient insulin secretion. Nonetheless, it is becoming increasingly clear that defective insulin secretion due to β-islet cell dysfunction is the major genetic factor predisposing to the eventual development of type 2 diabetes mellitus (34), so mutations or polymorphisms of SNAP25 may well play a role in type-2 diabetes susceptibility. Indeed, recent studies have shown that SNAP-25 expression is decreased in β-cells from GK-rats (a nonobese rodent model of type-2 diabetes) compared with control rats (35) and in cells from diabetic patients (36).
Blind-drunk mice were assessed over a number of behavioral domains, and the resultant data demonstrate a complex phenotype. Several elements of the blind-drunk phenotype suggest that it may be a model for psychiatric disorders, in particular schizophrenia. Impaired sensorimotor gating, an important component of the schizophrenia phenotype related to altered sensory processing, is augmented by anxiety behavior exhibited in the light/dark test and by the apathetic behavior observed in the playground paradigm test, which replicates aspects of the negative symptomatology of the disease. There is already evidence from several immunohistochemical and Western blot studies in postmortem brains implicating reductions in SNAP-25 expression in the etiology of schizophrenia (37, 38); our data, however, provides supporting evidence from in vivo and in vitro functional studies. Moreover, a recent metaanalysis of 20 separate genome-wide linkage scans for schizophrenia-susceptibility genes reported significant linkage to the chromosomal region 20p12.3–11, which contains SNAP25, and suggested 20p12.3–11 as a strong candidate region for the disease (39); this interval does not contain any other known candidate susceptibility loci.
Our data show that germ-line missense Snap25 mutations can give rise to phenotypes important in both human psychiatric disease and diabetes mellitus. It is, therefore, of interest that unaffected first-degree relatives of people with schizophrenia have high rates of type-2 diabetes (19–30% compared with 4–5% in the general population) (40, 41); this observation, together with the findings presented here, raises the interesting possibility that SNAP25 may represent a common genetic link between these two complex polygenic diseases.
Methods
Genetic Mapping and Mutation Detection.
Genotyping was carried out by PCR using standard polymorphic Mit markers or newly identified di-, tri- and tetranucleotide repeats. Marker 145302160 was amplified by using primers 145302160F (GAGCGACAATATAGCCGTAC) and 145302160R (GTACAGATCATAGAAGACTG). For mutation detection, all exons and exon/intron boundaries of Snap25 were amplified by PCR from +/Bdr, +/+ littermate and BALB/c genomic DNA and the resulting products sequenced in both directions.
In Situ Hybridization.
A 309-bp region of Snap25 (172 to 497 bp of GenBank accession no. NM_011428) was used for digoxigenin-labeled riboprobe synthesis and hybridization as described (42).
Western Blot Analysis.
Equal amounts of protein extract from homogenized brain tissue were analyzed by using standard methods using anti-SNAP-25 (Sternberger Monoclonals), anti-syntaxin-1a (Sigma, St. Louis, MO) or anti-VAMP-2 (Synaptic Systems) all at 1 in a 4,000 dilution. After exposure, membranes were stripped and reprobed with anti-β-tubulin (Sigma) at 1 in 10,000 as above as a loading control.
In Silico Modeling of the SNARE Complex.
The model mutant SNARE complex was generated by using Protein Data Base data file 1KIL (43). Ile 67 of chain C was mutated to Thr by using Modeller 4.10 and evaluated by using Procheck. The model was energy-minimized by using <1,000 steps of steepest descents to relax any steric conflicts. Energy minimization was performed by using the GROMACS 3.14 simulation package and The Visual Molecular Dynamics (Sunnyvale, CA) (VMD) software was used for visualization.
Protein Purification and in Vitro Binding Assay.
For information about protein purification and the in vitro binding assay, see SI Methods.
CD Measurements.
Recombinant SNARE proteins were purified as above, complexes were assembled overnight, and CD measurements were carried out in a J-720 machine (Jasco, Easton, MD) in the presence of 20 mM guanidine hydrochloride with a 30°C per hour temperature gradient and assay conditions as described (15). Recordings were taken at 1°C intervals and each assay was carried out in triplicate.
Cortical-Slice Electrophysiology.
For information on cortical-slice electrophysiology, see SI Methods.
β-Cell Preparation, Capacitance Measurements, and Pancreatic Hormone-Release Measurements.
Exocytosis was measured as an increase in membrane capacitance by using standard whole-cell configurations as described (44). The intracellular solution contained 0.1 mM cAMP to enhance the exocytotic response. Insulin and glucagon secretion were determined as reported (45).
MIN6 Cell Transfections.
MIN6 cells (46) were cultured in DMEM containing 4.5 mM glucose. Before (≈48 h) the experiments, the cells were transiently cotransfected with pEGFP-N1 and pCDNA3-wt SNAP-25b or pCDNA3-mut SNAP-25b. Recordings from eGFP-positive cells and exocytosis were triggered by intracellular dialysis with a buffer with a free [Ca2+]i of 1.5 μM (23).
Quantitative RT-PCR of Neuroendocrine Cells and Behavioral Testing.
For information on quantitative RT-PCR of neuroendocrine cells and behavioral testing, see SI Methods.
Supplementary Material
Acknowledgments
We thank Syma Khalid for expert assistance in creating the in silico models, Medical Research Council (MRC) Harwell for maintaining the blind-drunk line, Kristina Borglid for valuable technical support, and Jo Madge and Roger Cox at MRC Harwell for assistance with the glucose-tolerance testing.
Abbreviations
- ESPC
excitatory postsynaptic current
- mEPSC
miniature EPSC
- PPI
prepulse inhibition
- RRP
readily releasable pool
- SNARE
soluble N-ethylmaleimide-sensitive factor attachment protein receptor
- VAMP
vesicle-associated membrane protein.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS direct submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0610222104/DC1.
References
- 1.Sollner T, Bennett MK, Whiteheart SW, Scheller RH, Rothman JE. Cell. 1993;75:409–418. doi: 10.1016/0092-8674(93)90376-2. [DOI] [PubMed] [Google Scholar]
- 2.Glenn DE, Burgoyne RD. FEBS Lett. 1996;386:137–140. doi: 10.1016/0014-5793(96)00432-2. [DOI] [PubMed] [Google Scholar]
- 3.Wheeler MB, Sheu L, Ghai M, Bouquillon A, Grondin G, Weller U, Beaudoin AR, Bennett MK, Trimble WS, Gaisano HY. Endocrinology. 1996;137:1340–1348. doi: 10.1210/endo.137.4.8625909. [DOI] [PubMed] [Google Scholar]
- 4.Kidokoro Y. NeuroSignals. 2003;12:13–30. doi: 10.1159/000068912. [DOI] [PubMed] [Google Scholar]
- 5.Schoch S, Deak F, Konigstorfer A, Mozhayeva M, Sara Y, Sudhof TC, Kavalali ET. Science. 2001;294:1117–1122. doi: 10.1126/science.1064335. [DOI] [PubMed] [Google Scholar]
- 6.Washbourne P, Thompson PM, Carta M, Costa ET, Mathews JR, Lopez-Bendito G, Molnar Z, Becher MW, Valenzuela CF, Partridge LD, Wilson MC. Nat Neurosci. 2002;5:19–26. doi: 10.1038/nn783. [DOI] [PubMed] [Google Scholar]
- 7.Hess EJ, Collins KA, Wilson MC. J Neurosci. 1996;16:3104–3111. doi: 10.1523/JNEUROSCI.16-09-03104.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fujiwara T, Mishima T, Kofuji T, Chiba T, Tanaka K, Yamamoto A, Akagawa K. J Neurosci. 2006;26:5767–5776. doi: 10.1523/JNEUROSCI.0289-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nolan PM, Peters J, Strivens M, Rogers D, Hagan J, Spurr N, Gray IC, Vizor L, Brooker D, Whitehill E, et al. Nat Genet. 2000;25:440–443. doi: 10.1038/78140. [DOI] [PubMed] [Google Scholar]
- 10.Bark IC, Hahn KM, Ryabinin AE, Wilson MC. Proc Natl Acad Sci USA. 1995;92:1510–1514. doi: 10.1073/pnas.92.5.1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ravichandran V, Chawla A, Roche PA. J Biol Chem. 1996;271:13300–13303. doi: 10.1074/jbc.271.23.13300. [DOI] [PubMed] [Google Scholar]
- 12.Keays DA, Clark TG, Flint J. Mamm Genome. 2006;17:230–238. doi: 10.1007/s00335-005-0101-4. [DOI] [PubMed] [Google Scholar]
- 13.Fasshauer D, Sutton RB, Brunger AT, Jahn R. Proc Natl Acad Sci USA. 1998;95:15781–15786. doi: 10.1073/pnas.95.26.15781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fasshauer D, Margittai M. J Biol Chem. 2004;279:7613–7621. doi: 10.1074/jbc.M312064200. [DOI] [PubMed] [Google Scholar]
- 15.Nagy G, Milosevic I, Fasshauer D, Muller EM, de Groot BL, Lang T, Wilson MC, Sorensen JB. Mol Biol Cell. 2005;16:5675–5685. doi: 10.1091/mbc.E05-07-0595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dobrunz LE, Stevens CF. Neuron. 1997;18:995–1008. doi: 10.1016/s0896-6273(00)80338-4. [DOI] [PubMed] [Google Scholar]
- 17.Brager DH, Capogna M, Thompson SM. J Physiol. 2002;541:545–559. doi: 10.1113/jphysiol.2001.015842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rorsman P, Renstrom E. Diabetologia. 2003;46:1029–1045. doi: 10.1007/s00125-003-1153-1. [DOI] [PubMed] [Google Scholar]
- 19.Gonelle-Gispert C, Halban PA, Niemann H, Palmer M, Catsicas S, Sadoul K. Biochem J. 1999;339:159–165. [PMC free article] [PubMed] [Google Scholar]
- 20.Barg S, Eliasson L, Renstrom E, Rorsman P. Diabetes. 2002;51(Suppl. 1):S74–S82. doi: 10.2337/diabetes.51.2007.s74. [DOI] [PubMed] [Google Scholar]
- 21.Voets T, Neher E, Moser T. Neuron. 1999;23:607–615. doi: 10.1016/s0896-6273(00)80812-0. [DOI] [PubMed] [Google Scholar]
- 22.Ishihara H, Asano T, Tsukuda K, Katagiri H, Inukai K, Anai M, Kikuchi M, Yazaki Y, Miyazaki JI, Oka Y. Diabetologia. 1993;36:1139–1145. doi: 10.1007/BF00401058. [DOI] [PubMed] [Google Scholar]
- 23.Eliasson L, Renstrom E, Ding WG, Proks P, Rorsman P. J Physiol. 1997;503:399–412. doi: 10.1111/j.1469-7793.1997.399bh.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Braff DL, Geyer MA, Swerdlow NR. Psychopharmacology (Berlin) 2001;156:234–258. doi: 10.1007/s002130100810. [DOI] [PubMed] [Google Scholar]
- 25.Moy SS, Nadler JJ, Perez A, Barbaro RP, Johns JM, Magnuson TR, Piven J, Crawley JN. Genes Brain Behav. 2004;3:287–302. doi: 10.1111/j.1601-1848.2004.00076.x. [DOI] [PubMed] [Google Scholar]
- 26.Crawley J, Goodwin FK. Pharmacol Biochem Behav. 1980;13:167–170. doi: 10.1016/0091-3057(80)90067-2. [DOI] [PubMed] [Google Scholar]
- 27.Glynn D, Drew CJ, Reim K, Brose N, Morton AJ. Hum Mol Genet. 2005;14:2369–2385. doi: 10.1093/hmg/ddi239. [DOI] [PubMed] [Google Scholar]
- 28.Boschert U, O'Shaughnessy C, Dickinson R, Tessari M, Bendotti C, Catsicas S, Pich EM. J Comp Neurol. 1996;367:177–193. doi: 10.1002/(SICI)1096-9861(19960401)367:2<177::AID-CNE2>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 29.Bark C, Bellinger FP, Kaushal A, Mathews JR, Partridge LD, Wilson MC. J Neurosci. 2004;24:8796–8805. doi: 10.1523/JNEUROSCI.1940-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sudhof TC. Annu Rev Neurosci. 2004;27:509–547. doi: 10.1146/annurev.neuro.26.041002.131412. [DOI] [PubMed] [Google Scholar]
- 31.Vo YP, Hutton JC, Angleson JK. Biochem Biophys Res Commun. 2004;324:1004–1010. doi: 10.1016/j.bbrc.2004.09.147. [DOI] [PubMed] [Google Scholar]
- 32.Hanson PI, Heuser JE, Jahn R. Curr Opin Neurobiol. 1997;7:310–315. doi: 10.1016/s0959-4388(97)80057-8. [DOI] [PubMed] [Google Scholar]
- 33.Kawasaki F, Mattiuz AM, Ordway RW. J Neurosci. 1998;18:10241–10249. doi: 10.1523/JNEUROSCI.18-24-10241.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pimenta W, Korytkowski M, Mitrakou A, Jenssen T, Yki-Jarvinen H, Evron W, Dailey G, Gerich J. J Am Med Assoc. 1995;273:1855–1861. [PubMed] [Google Scholar]
- 35.Nagamatsu S, Nakamichi Y, Yamamura C, Matsushima S, Watanabe T, Ozawa S, Furukawa H, Ishida H. Diabetes. 1999;48:2367–2373. doi: 10.2337/diabetes.48.12.2367. [DOI] [PubMed] [Google Scholar]
- 36.Ostenson CG, Gaisano H, Sheu L, Tibell A, Bartfai T. Diabetes. 2006;55:435–440. doi: 10.2337/diabetes.55.02.06.db04-1575. [DOI] [PubMed] [Google Scholar]
- 37.Thompson PM, Egbufoama S, Vawter MP. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:411–417. doi: 10.1016/S0278-5846(03)00027-7. [DOI] [PubMed] [Google Scholar]
- 38.Mukaetova-Ladinska EB, Hurt J, Honer WG, Harrington CR, Wischik CM. Neurosci Lett. 2002;317:161–165. doi: 10.1016/s0304-3940(01)02458-2. [DOI] [PubMed] [Google Scholar]
- 39.Lewis CM, Levinson DF, Wise LH, DeLisi LE, Straub RE, Hovatta I, Williams NM, Schwab SG, Pulver AE, Faraone SV, et al. Am J Hum Genet. 2003;73:34–48. doi: 10.1086/376549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mukherjee S, Schnur DB, Reddy R. Lancet. 1989;1:495. doi: 10.1016/s0140-6736(89)91392-5. [DOI] [PubMed] [Google Scholar]
- 41.Thakore JH. Br J Psychiatry. 2005;186:455–456. doi: 10.1192/bjp.186.6.455. [DOI] [PubMed] [Google Scholar]
- 42.Isaacs AM, Oliver PL, Jones EL, Jeans A, Potter A, Hovik BH, Nolan PM, Vizor L, Glenister P, Simon AK, et al. J Neurosci. 2003;23:1631–1637. doi: 10.1523/JNEUROSCI.23-05-01631.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen X, Tomchick DR, Kovrigin E, Arac D, Machius M, Sudhof TC, Rizo J. Neuron. 2002;33:397–409. doi: 10.1016/s0896-6273(02)00583-4. [DOI] [PubMed] [Google Scholar]
- 44.Eliasson L, Ma X, Renstrom E, Barg S, Berggren PO, Galvanovskis J, Gromada J, Jing X, Lundquist I, Salehi A, et al. J Gen Physiol. 2003;121:181–197. doi: 10.1085/jgp.20028707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jing X, Li DQ, Olofsson CS, Salehi A, Surve VV, Caballero J, Ivarsson R, Lundquist I, Pereverzev A, Schneider T, et al. J Clin Invest. 2005;115:146–154. doi: 10.1172/JCI22518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miyazaki J, Araki K, Yamato E, Ikegami H, Asano T, Shibasaki Y, Oka Y, Yamamura K. Endocrinology. 1990;127:126–132. doi: 10.1210/endo-127-1-126. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}