

Abstract
Two members of the AAA+ superfamily, ClpB and Hsp104, collaborate with Hsp70 and Hsp40 to rescue aggregated proteins. However, the mechanisms that elicit and underlie their protein-remodeling activities remain unclear. We report that for both Hsp104 and ClpB, mixtures of ATP and ATPγS unexpectedly unleash activation, disaggregation, and unfolding activities independent of co-chaperones. Mutations reveal how remodeling activities are elicited by impaired hydrolysis at individual nucleotide binding domains. However, for some substrates, mixtures of ATP and ATPγS abolish remodeling, while for others ATP binding without hydrolysis is sufficient. Remodeling of different substrates necessitates a diverse balance of polypeptide holding (which requires ATP binding but not hydrolysis) and unfolding (which requires ATP hydrolysis). We suggest that this versatility in reaction mechanism enables ClpB and Hsp104 to reactivate the entire aggregated proteome after stress, and enables Hsp104 to control prion inheritance.
Life demands that members of the AAA+ ATPase superfamily (ATPases associated with various cellular activities) couple energy from ATP hydrolysis to the remodeling of a bewildering array of macromolecular structures, that range from protein to DNA and RNA1, 2. Typically, eukaryotic genomes encode 50–80 family members1, each of which occupies specific niches that require specialized modes of substrate selection and regulation. The extraordinary adaptive radiation of AAA+ proteins to function in a multitude of cellular reactions illustrates the versatility of their structurally conserved AAA+ domain. Subunits containing AAA+ domains assemble into oligomeric rings, and ATP binds at the interface between adjacent protomers1, 2. AAA+ oligomers undergo considerable conformational changes during ATP binding and hydrolysis, although how these events are regulated and transduced into productive substrate remodeling remains largely enigmatic. Furthermore, it remains unanswered whether individual AAA+ family members rely on a common reaction mechanism to remodel various macromolecular clients. It is also unclear whether different AAA+ members have evolved distinct methods to engage and restructure substrates, or if individual proteins can switch between distinct reaction mechanisms for different substrates.
Two members of the AAA+ superfamily separated by ~2 billion years of evolution3, yeast Hsp104, and its E. coli homolog, ClpB, allow cell survival after exposure to extreme environmental stress4–7. They function to dissolve and renature thousands of diverse substrates during reactivation of the aggregated proteome after multifarious stresses. They work in collaboration with the Hsp70/DnaK chaperone system8–10 and as a result cell survival can increase by 10,000-fold4–7. Under normal growth conditions Hsp104 is also essential for the formation and maintenance of prions, protein-based genetic elements comprised of amyloid fibers that self-perpetuate alterations in protein conformation and function11.
How does the structure of ClpB and Hsp104 facilitate these functions? Both are hexamers comprised of protomers containing two AAA+ ATPase domains (nucleotide binding domains, NBD-1 and NBD-2) and an N-terminal domain12 (Fig. 1a). Inserted in NBD-1 is a long coiled-coil middle domain that resembles in structure the shape of a two-bladed propeller12 and distinguishes ClpB and Hsp104 from other Hsp100 proteins that also contain two NBDs1. Electronmicroscopy and single particle reconstruction of Hsp104 and ClpB reveals an axial channel spanning the length of the hexamer7, 12. Although not completely visible, the coiled-coil middle domains are proposed to be located on the outside of the ClpB hexamer12. ATP is bound at the interface between adjacent subunits12 and hexamerization is stabilized by nucleotide13, 14. Substrate binding occurs when the hexamer is in its ATP-bound conformation and conserved pore residues may contact substrates directly15–17. The N- and C-terminal domains may also help engage substrates and co-factors18, 19. Cooperative ATPase activity occurs at both NBD-1 and NBD-2, and allosteric communication occurs within and between the two domains12, 19–23. Disruption of ATPase activity and consequent inhibition of conformational changes compromises protein-remodeling activity15–17, 19, 21. However, despite intense investigation, it remains unclear how the hexameric architecture of ClpB and Hsp104 contributes to the conformational changes associated with the dissolution and reactivation of aggregated proteins24.
Figure 1. Protein activation by ClpB or Hsp104 alone.
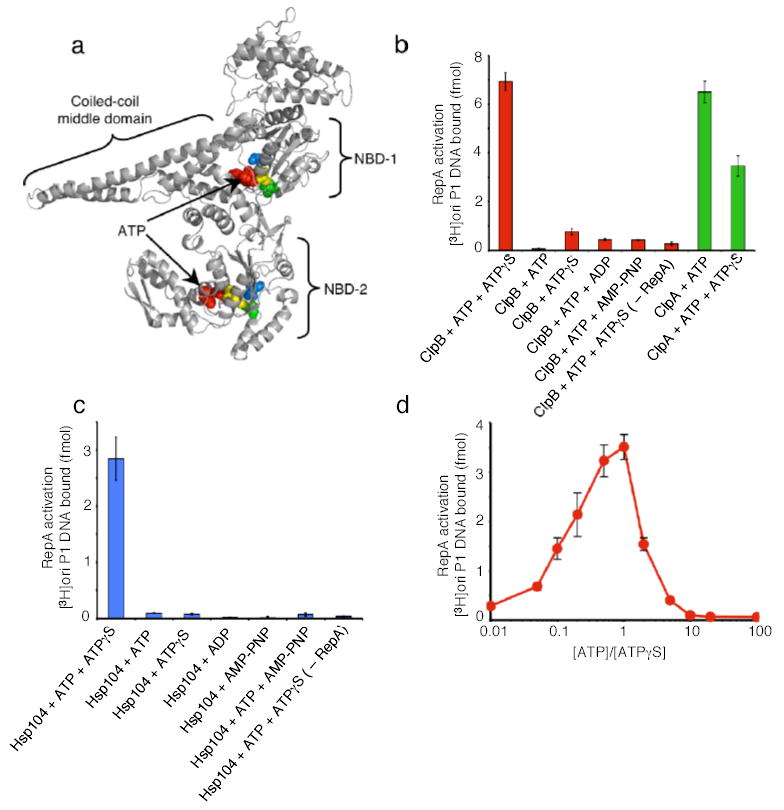
(a) Model of E. coli ClpB subunit. Each monomer possesses two nucleotide-binding domains (NBD), NBD-1 and NBD-2, which are separated by a large coiled-coil middle domain. Within each NBD is a Walker A, Walker B and sensor-1 motif. Note the position of ATP (red), Lys212 and Lys611 of the Walker A motif (yellow), Glu279 and Glu678 of the Walker B motif (blue), and the Thr315 and Asn719 of the sensor-1 motif (green) as CPK models. Model was generated using Swiss-Model53 and based on the crystal structure of T. thermophilus ClpB12.
(b) RepA activation by ClpB or ClpA.
(c) RepA activation by Hsp104.
(d) RepA activation by ClpB with varying ratios of ATP:ATPγS. In b–d, data are means ± SD (n=3).
The powerful remodeling activities of Hsp104 and ClpB must be tightly regulated and highly discriminating, because even very high levels of expression are not toxic5, 25. That is, despite a capacity to engage diverse substrates, ClpB and Hsp104 do not interfere with large functional protein complexes or filamentous structures. Most compellingly, a screen for mutations that perturb the regulated function of Hsp104 revealed that single amino acid substitutions in an NBD combined with single amino acid substitutions in the coiled-coil middle domain frequently caused Hsp104 to become highly toxic25. This suggests that the tight regulation of ClpB and Hsp104 may stem, in part, from the intrinsic properties of the hexamer itself. However, a clear understanding of how Hsp104 and ClpB are tightly regulated remains elusive.
In this study, we investigated the innate protein-remodeling activity of yeast Hsp104 and E. coli ClpB. We establish the completely unexpected ability of mixtures of ATP and ATPγS (a slowly hydrolyzable ATP analog) to elicit Hsp104 and ClpB activity in protein activation, disaggregation, and unfolding assays. Remarkably, Hsp104 and ClpB were elicited to function without assistance from co-chaperones. Thus far, ClpB has required DnaK/Hsp70 and DnaJ/Hsp40 in every protein-remodeling activity tested9, 10. Previously, Hsp104 has only remodeled the yeast prion proteins independent of co-chaperones26, 27. Hsp104 has also reactivated some protein aggregates with the assistance of the small heat shock protein, Hsp2628. However, with most substrates Hsp70 and Hsp40 chaperones are needed8. Hence, the effect of mixtures of ATP and ATPγS empowered us to study ClpB and Hsp104 remodeling activities without any confounding effects of co-chaperones. Our findings bring significant new insights into the mechanistic basis by which Hsp104 and ClpB hexamers function, and enlighten the coordinated roles of the two AAA+ domains in protein remodeling.
RESULTS
Eliciting ClpB and Hsp104 remodeling activity without Hsp70 and Hsp40
We sought conditions that might activate the remodeling activities of ClpB in the absence of co-chaperones. As a model substrate, we began with inactive dimers of RepA, a protein that initiates the replication of P1 plasmids in E. coli. RepA dimers are converted by either the DnaK chaperone system or by ClpA into active monomers that bind the plasmid replication origin, oriP1, with high affinity29, 30. ClpB had no capacity to activate RepA in the presence of ATP (Fig. 1b). Surprisingly, a 1:1 mixture of ATP and ATPγS (a non-physiological and slowly hydrolyzed ATP analog) profoundly stimulated RepA activation by ClpB (Fig. 1b). ATPγS alone did not support remodeling (Fig.1b).
Hsp104, also activated RepA when provided a mixture of ATP and ATPγS (Fig. 1c). As with ClpB, neither ATP nor ATPγS alone supported activation (Fig. 1c). The optimal ATP:ATPγS ratio was 1:1 for ClpB activity (Fig. 1d) and 3:1 for Hsp104 activity (data not shown). Moreover, for both ClpB and Hsp104, a mixture of ATP with ADP or of ATP with AMP-PNP (a non-hydrolyzable ATP analog) did not support the remodeling reaction (Fig. 1b, c). ClpA, another hexameric AAA+ ATPase with two AAA+ domains per monomer, but which lacks the coiled-coil middle domain present in Hsp104 and ClpB (Fig. 1a), behaved differently. ClpA activated RepA in the presence of ATP alone29 and a mixture of ATP with ATPγS was inhibitory (Fig. 1b). These results show that the remodeling activity of ClpB and Hsp104 is elicited by a combination of ATP and ATPγS. Although ATPγS is known to inhibit ATP hydrolysis, slowing hydrolysis of ATP is not its function here, since AMP-PNP and ADP, which are also expected to reduce ATP hydrolysis, were ineffective.
Protein disaggregation by ClpB and Hsp104
The observation that ClpB and Hsp104 have innate chaperone activity allowed us to examine, for the first time, the remodeling activity of these proteins without co-chaperones. We began by asking if mixtures of ATP and ATPγS also elicit the disaggregation of larger aggregates by ClpB or Hsp104. We used thermally denatured GFP (green fluorescent protein) aggregates of ~500kDa or greater in size31 as a substrate. Remarkably, both ClpB (Fig. 2a) and Hsp104 (Fig 2b) promoted reactivation of heat-aggregated GFP in the presence of ATP and ATPγS.
Figure 2. Reactivation of heat-aggregated proteins by ClpB or Hsp104 alone.
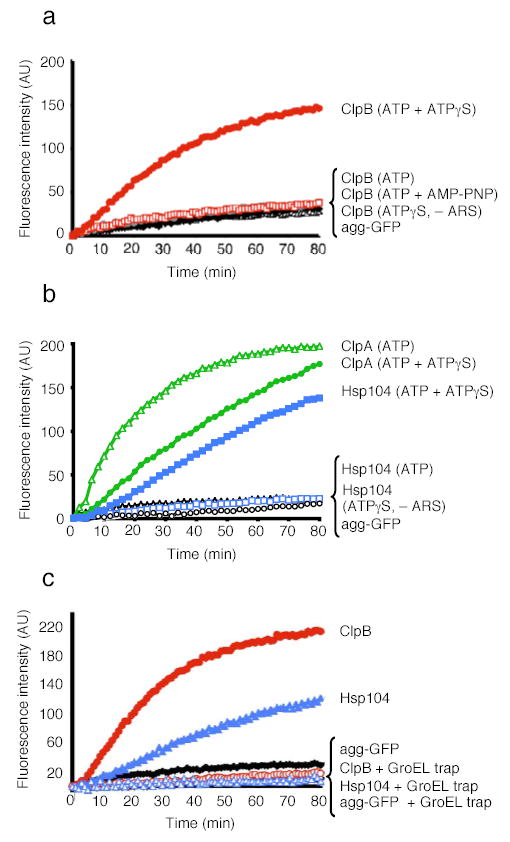
(a) Disaggregation of heat-aggregated GFP by ClpB. – ARS denotes absence of ATP regeneration system. Agg-GFP denotes aggregated GFP alone.
(b) Disaggregation of aggregated GFP by Hsp104 or ClpA.
(c) Disaggregation and release of unfolded GFP by ClpB or Hsp104. In a–c the initial fluorescence was set equal to 0 and a representative experiment is shown of three or more replicates.
Activation of ClpB and Hsp104 by mixtures of ATP and ATPγS was highly specific. ATP or ATPγS alone did not support disaggregation by ClpB or Hsp104 and neither did a mixture of ATP and AMP-PNP nor a mixture of ATP and ADP (Fig. 2a, b, data not shown). ClpA also reactivated aggregated GFP, an activity of ClpA not previously observed. In contrast to Hsp104 and ClpB, ClpA required ATP and the addition of ATPγS was inhibitory (Fig. 2b). Thus, for ~2 billion years of evolution3 a distinct mechanism for unleashing the protein-remodeling activities of the ClpB/Hsp104 class of proteins, which is mimicked in vitro by ATPγS, has been conserved.
ClpB and Hsp104 release unfolded polypeptides during disaggregation
The unexpected stimulation of ClpB and Hsp104 disaggregation activity by the mixture of ATP and ATPγS led us to investigate the nature of the disaggregation reaction products. To do so, we employed a mutant chaperonin, GroELtrap32 that captures unfolded proteins in the molten globule state33 and prevents refolding, but does not bind heat-aggregated GFP31 or native GFP32. GroELtrap inhibited reactivation of aggregated GFP by Hsp104 and ClpB (Fig. 2c). Thus, Hsp104 and ClpB released GFP in an unfolded state that could be captured by GroELtrap.
ClpB and Hsp104 can unfold natively structured proteins
ClpB and Hsp104 have never been found to unfold natively folded proteins. Might the combination of ATP and ATPγS elicit such an activity? To test this, we used GFP as our substrate, because it is extremely stable (Tm~70°C) and can withstand mechanical unfolding forces of ~100pN (ref. 34). Since GFP refolds rapidly once unfolded, we employed GroELtrap to capture potential unfolded products. However, native GFP was not unfolded by ClpB or Hsp104 (data not shown). We then tested a GFP fusion protein containing a fragment of RepA (amino acids 1–70) to provide a possible recognition signal. With both ClpB and Hsp104, a large loss of GFP fluorescence occurred when RepA(1–70)-GFP was incubated with a mixture ATP and ATPγS in the presence of GroELtrap (Fig. 3a, b). Thus, some feature of the RepA(1–70) fragment, likely the unstructured N-terminal end, provides a recognition signal for substrate engagement and allows ClpB and Hsp104 to unfold even highly stable proteins.
Figure 3. Protein unfolding by ClpB or Hsp104 alone.
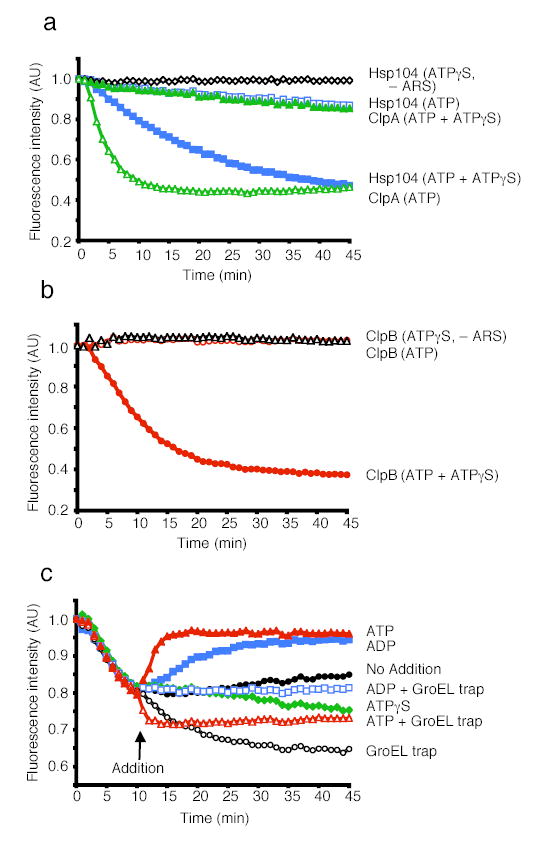
(a) Unfolding of RepA(1–70)-GFP by Hsp104 or ClpA alone.
(b) Unfolding of RepA(1–70)-GFP by ClpB.
(c) Steady-state unfolding of RepA(1–70)-GFP by ClpB. At the time indicated an excess of the component indicated was added. In a–c the initial fluorescence was set equal to 1. A representative experiment is shown of three or more replicates.
Again, the effect of ATP and ATPγS mixtures was specific. No unfolding occurred when ClpB or Hsp104 were incubated with ATPγS (Fig. 3a, b). Nor was RepA(1–70)-GFP unfolding observed when ClpB was incubated in the presence of ATP (Fig. 3b) and very little occurred with Hsp104 in the presence of ATP (Fig. 3a). Slowing ATP hydrolysis by limiting [ATP] over a range between 2mM and 5μM similarly failed to promote unfolding by ClpB (data not shown). Mixtures of ATP and ADP, or, ATP and AMP-PNP also failed to promote unfolding (data not shown). As with the activation and disaggregation reactions, the optimal ratio of ATP:ATPγS for ClpB was ~1:1 (data not shown). In contrast, ClpA catalyzed unfolding with ATP alone29 and ATPγS was inhibitory (Fig. 3a). Thus the coupling of ATP hydrolysis and protein unfolding occurs differently in ClpB/Hsp104 and ClpA, while the result of that hydrolysis, unfolded substrate, is the same.
Dissecting the mechanism of stimulation by mixtures of ATP and ATPγS
To investigate the mechanism by which the combination of ATP and ATPγS elicited ClpB remodeling activities, we sought to challenge a steady-state unfolding reaction with excess ATP, ATPγS or ADP. First, we established that steady-state unfolding could be achieved in the presence of ATP and ATPγS (Fig. 3c). When ClpB was incubated with both nucleotides in the absence of GroELtrap, a decline in RepA(1–70)-GFP fluorescence occurred, reaching a plateau after 10 min (Fig. 3c). This was due to a steady-state equilibrium between unfolding and refolding. Over the next 90 min as the energy supply was exhausted, the rate of unfolding slowed and the fluorescence slowly returned to the original level (data not shown). If GroELtrap was added at 10 min to capture released unfolded products, fluorescence slowly declined (Fig. 3c).
The addition of excess ATP after 10 min caused an immediate increase in fluorescence, indicating that the replacement of ATPγS with ATP in this steady-state reaction promotes substrate release (which is followed by spontaneous RepA(1–70)-GFP refolding) (Fig. 3c). When GroELtrap was added together with excess ATP, there was a rapid decrease in fluorescence followed by a plateau (Fig. 3c). This suggests that ATP does not simply trigger release of substrate, but additionally produces a burst of substrate unfolding and release as revealed by the capture of increased quantities of unfolded RepA(1–70)-GFP by GroELtrap. In contrast, the addition of excess ATPγS was followed by a gradual decrease in fluorescence, with or without GroELtrap, indicating that slow hydrolysis increases the residence time of the substrate with ClpB (Fig. 3c and data not shown).
The addition of excess ADP resulted in an increase in fluorescence, indicating substrate release followed by refolding (Fig. 3c). The addition of ADP with GroELtrap, unlike the addition of ATP with GroELtrap, stopped any further changes in fluorescence, revealing that substrate was released without additional unfolding (Fig. 3c). Therefore, ATPγS promotes substrate “holding” while ATP hydrolysis is required for substrate unfolding and release. These results suggest that the combination of ATPγS with ATP unleashes ClpB remodeling activities by allowing ClpB hexamers to achieve a balance of substrate binding, holding, and unfolding to forcibly unfold proteins.
Roles of the two AAA+ domains in substrate remodeling by Hsp104
To determine whether NBD-1, NBD-2, or both require restrained hydrolysis to promote substrate remodeling, we tested mutants in each domain. We used Hsp104 mutants with an amino-acid substitution in the Walker A box (a motif containing a lysine residue that directly contacts the phosphates of ATP1) of either NBD-1 or NBD-2 (Fig. 1a). The Walker A mutants are defective in ATP binding and thermotolerance35, 36. We also analyzed mutants with a substitution in the sensor-1 motif (a motif containing a threonine or asparagine that interacts with the γ-phosphate of ATP1) of NBD-1 or NBD-2 (Fig. 1a). The sensor-1 mutants bind nucleotide, although they are defective in ATP hydrolysis and thermotolerance20.
Mutants in NBD-2 of Hsp104, both in the Walker A (K620T) and sensor-1 (N728A) motifs could activate RepA dimers (Fig. 4a), reactivate heat-aggregated GFP (Fig. 4b), and unfold RepA(1–70)-GFP (Fig. 4c). In all three reactions the NBD-1 Walker A and sensor-1 mutants were inactive (Fig. 4a, data not shown). Furthermore, an Hsp104 mutant with both Walker A motifs mutated, Hsp104(K218T:K620T35), was also inactive (Fig. 4a, data not shown).
Figure 4. Protein remodeling by Hsp104 NBD mutants.
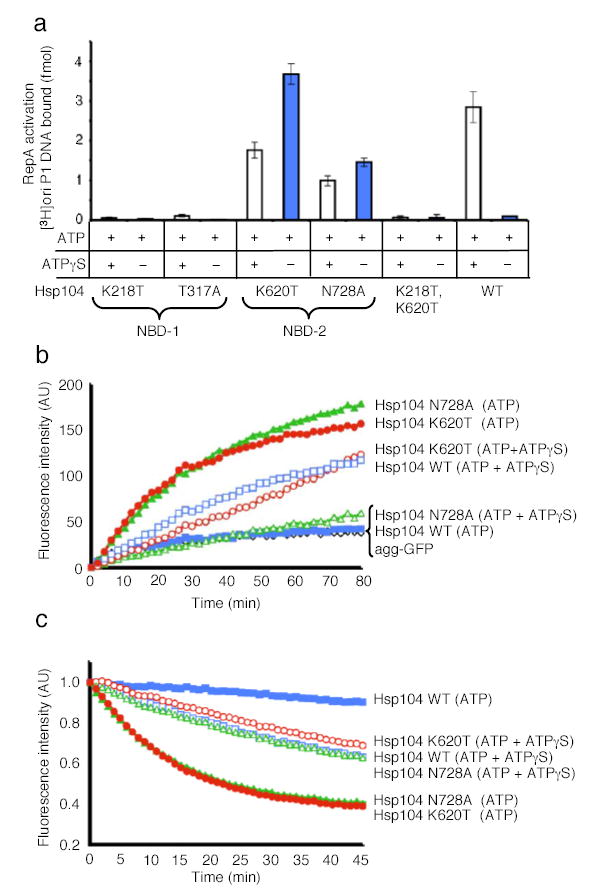
(a) RepA activation by Hsp104 and mutants in NBD-1 and NBD-2. Data are means ± SD (n=3).
(b) Reactivation of heat-aggregated GFP by Hsp104 mutants. The initial fluorescence was set equal to 0 and a representative experiment is shown of three or more replicates.
(c) Unfolding of RepA(1–70)-GFP by Hsp104 mutants. The initial fluorescence was set equal to 1 and a representative experiment is shown of three or more replicates.
Remarkably, in contrast to wild type Hsp104, the NBD-2 mutants utilized ATP as the sole nucleotide and ATPγS was inhibitory (Fig. 4a, b, c). These results indicate that remodeling of these substrates by Hsp104 is elicited when ATP hydrolysis is decelerated at NBD-2, but not at NBD-1. Deceleration of a subset of nucleotide binding sites can be accomplished either through utilization of ATPγS in combination with ATP or with mutants impaired in ATP hydrolysis at NBD-2.
Contribution of the two AAA+ domains in protein remodeling by ClpB
There were differences between ClpB and Hsp104 in the involvement of the two NBDs, as might have been expected from the known functional differences in the NBDs of the two proteins. For example, NBD-1 of Hsp104 hydrolyzes ATP rapidly and is primarily involved in substrate binding, whereas NBD-2 hydrolyzes ATP much more slowly and is mainly responsible for oligomerization14, 20, 36. In contrast, NBD-1 and NBD-2 of ClpB hydrolyze ATP at similar rates and NBD-1 primarily contributes to oligomerization21, 37, 38.
We tested ClpB mutants in residues comparable to those in Hsp104, including mutants in both Walker A motifs21, 37 that are known to be defective in ATP binding and thermotolerance as well as mutants in both sensor-1 motifs (M. Barnett and M. Zolkiewski, Kansas State University, Manhattan KS, unpublished results) that can bind nucleotide, although they are defective in ATP hydrolysis and thermotolerance (Fig. 1a). In addition, we tested mutants in the ClpB Walker B box (a motif containing a catalytic glutamate residue which activates a water molecule to hydrolyze ATP1) of NBD-1 or NBD-2 (Fig. 1a), that are defective in thermotolerance but bind and hydrolyze ATP21, 39.
Like the Hsp104 NBD-2 mutants, the ClpB NBD-2 sensor-1 mutant (N719A) and the Walker B mutant (E678A), could activate RepA, reactivate heat-aggregated GFP, and unfold RepA(1–70)-GFP (Fig. 5). Importantly, these ClpB mutants, like the Hsp104 NBD-2 mutants, utilized ATP as the sole nucleotide (Fig. 5). ATPγS inhibited activation, disaggregation and protein unfolding by the Walker B mutant and reactivation and protein unfolding by the sensor-1 mutant, although it did not inhibit RepA activation by the sensor-1 mutant (Fig. 5). In contrast to Hsp104, the ClpB NBD-2 Walker A mutant (K611T) was inactive in all assays (Fig. 5, data not shown). These data indicate that as for Hsp104, deceleration of the ATPase activity at NBD-2 of ClpB, when NBD-1 remains active, can activate diverse protein-remodeling activities.
Figure 5. Protein remodeling by ClpB NBD mutants.
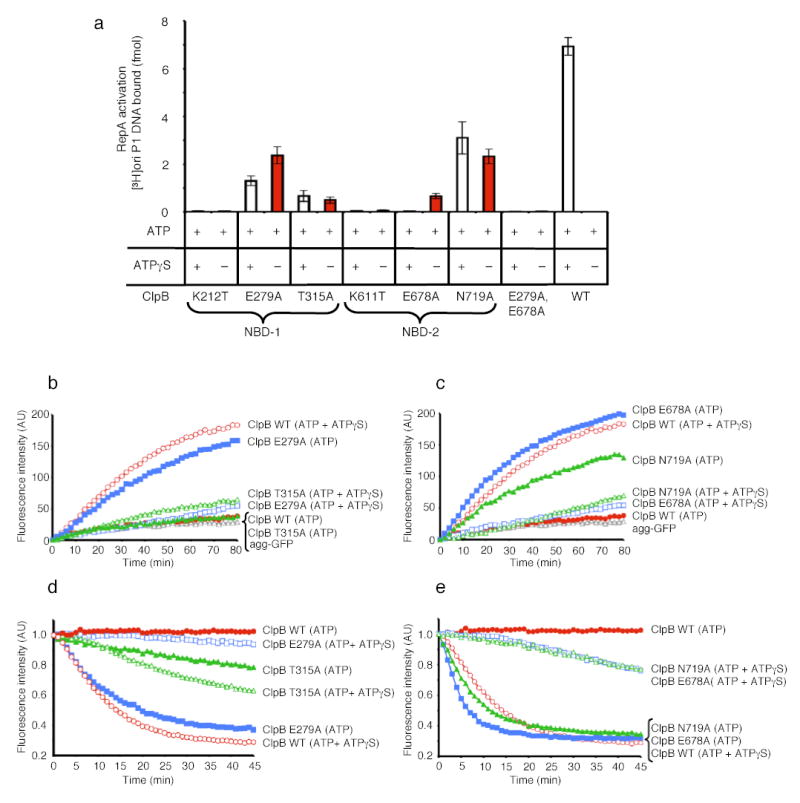
(a) RepA activation by ClpB mutants in NBD-1 and NBD-2. Data are means ± SD (n=3).
(b) Disaggregation of heat-aggreagted GFP by ClpB NBD-1 mutants. In b and c the initial fluorescence was set equal to 0 and a representative experiment is shown of three or more replicates.
(c) Disaggregation of heat-aggregated GFP by ClpB NBD-2 mutants.
(d) Unfolding of RepA(1–70)-GFP by ClpB NBD-1 mutants. In d and e the initial fluorescence was set equal to 1 and a representative experiment is shown of three or more replicates.
(e) Unfolding of RepA(1–70)-GFP by ClpB NBD-2 mutants.
Mutants in ClpB NBD-1 differed from those in Hsp104. The NBD-1 sensor-1 mutant (T315A) and the Walker B mutant (E279A) were functional in activation, disaggregation and protein unfolding (Fig. 5). In all cases the ClpB Walker B mutant utilized ATP alone and ATPγS was inhibitory (Fig. 5). The ClpB NBD-1 sensor-1 mutant possessed substantially reduced activity compared to wild type and utilized mixtures of ATP and ATPγS more efficiently than ATP (Fig. 5a, b, d). A Walker A mutant in NBD-1 (K212T), like the analogous Hsp104 mutant, was inactive in all remodeling reactions with ATP and ATPγS or ATP (Fig. 5a, data not shown). These data indicate that unlike Hsp104, deceleration of ClpB ATPase activity at NBD-1, when NBD-2 remains active, can also elicit protein-remodeling activity.
A ClpB double mutant in the Walker B motifs of NBD-1 and NBD-2 (E279A:E678A39) was also inactive (Fig. 5 and data not shown). The inactivity of both ClpB(E279A:E678A) and Hsp104(K218T:K620T) indicates that deceleration of ATPase activity at NBD-2 can be counterproductive if NBD-1 also has no ATPase activity. Importantly, together with the Hsp104 results these experiments demonstrate a fundamental similarity in the mechanism of Hsp104 and ClpB remodeling activities that was completely unexpected. Both proteins have an intrinsic capacity to remodel substrates on their own and this activity can be unleashed by blocking hydrolysis with mutations at all six sites within a single AAA+ domain.
Nucleotide hydrolysis by Hsp104 and ClpB
Next, we directly measured nucleotide hydrolysis by Hsp104 and ClpB using conditions where protein unfolding was apparent. Remarkably, mixtures of ATP and ATPγS did not inhibit nucleotide hydrolysis as would be expected for competition by a poorly hydrolyzable ATP analog. Instead, ATPγS stimulated nucleotide hydrolysis by ClpB about three-fold at a 1:1 mixture of ATP and ATPγS. Surprisingly, nucleotide hydrolysis by ClpB with a 9:1 mixture of ATPγS to ATP was approximately equal to that with ATP alone (Fig. 6a). Higher ratios of ATPγS to ATP eventually became inhibitory (Fig. 6a). ATPγS exhibited the predicted effect on nucleotide hydrolysis by ClpA; it was inhibitory at all ratios of ATP to ATPγS (Fig. 6a). There was a small inhibitory effect on nucleotide hydrolysis by the unfolding substrate, RepA(1–70)-GFP and no effect of the activation substrate, RepA (Fig. 6a, data not shown). These data unexpectedly demonstrate that ATPγS stimulates nucleotide hydrolysis by ClpB.
Figure 6. Nucleotide hydrolysis by ClpB and Hsp104.
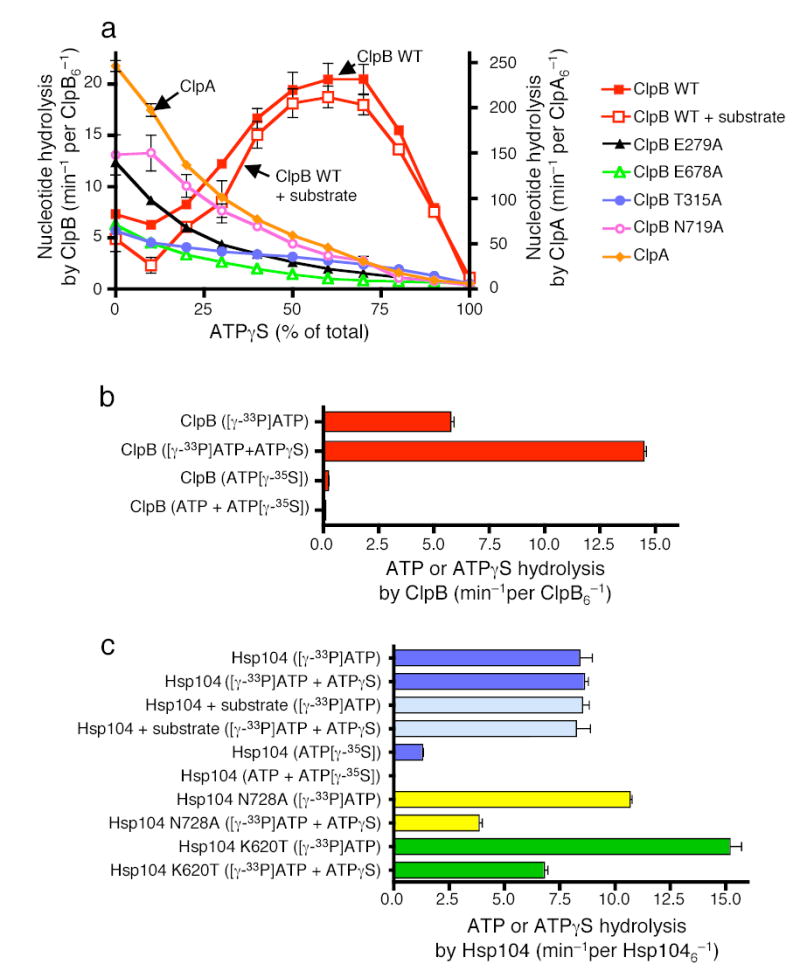
(a) Nucleotide hydrolysis by ClpB, ClpB mutants and ClpA in the presence of the indicated amounts of ATPγS. In a–c data are means ± SD (n=3).
(b) ATP and ATPγS hydrolysis by ClpB.
(c) ATP and ATPγS hydrolysis by Hsp104 and Hsp104 mutants.
To demonstrate whether ATPγS stimulated hydrolysis of ATP or was it self hydrolyzed, we separately measured ATP and ATPγS hydrolysis by ClpB using radiolabeled nucleotides. The rate of ClpB ATP hydrolysis increased about three-fold when the nucleotide source was a 1:1 mixture of ATP and ATPγS compared to ATP alone and the rate was similar to that of total nucleotide hydrolysis (Fig. 6b). The rate of ATPγS hydrolysis by ClpB was 30-fold slower than that of ATP and was inhibited further when in a 1:1 mixture with ATP (Fig. 6b). Thus, ATPγS specifically stimulated ATP hydrolysis by ClpB.
In contrast, ATP hydrolysis by Hsp104 was not stimulated by ATPγS at ratios of ATP to ATPγS that activated remodeling, although it was not inhibited (Fig. 6c). The unfolding substrate, RepA(1–70)-GFP, did not appreciably effect the rate of ATP hydrolysis by Hsp104 in the absence or presence of ATPγS (Fig. 6c). Thus, for both Hsp104 and ClpB the effect of ATPγS on ATP hydrolysis is a direct effect on ClpB/Hsp104 and is independent of interactions of the substrate with ClpB/Hsp104.
For Hsp104, like ClpB, the rate of ATPγS hydrolysis was much slower than that of ATP and was inhibited further when in a mixture with ATP (Fig. 6c). Taken together these results suggest that the binding or very slow hydrolysis of ATPγS at some of the 12 ATP binding sites of Hsp104 and ClpB may stimulate ATP hydrolysis at other sites by a mechanism of allosteric regulation that relieves the negative cooperativity in ATP hydrolysis. Thus as is often the case with perturbations caused by drugs and mutations, the non-physiological nucleotide, ATPγS, revealed an aspect of the reaction mechanism for these proteins that would otherwise have remained undiscovered.
We also measured nucleotide hydrolysis by ClpB and Hsp104 mutants that are active in protein remodeling in the presence of ATP and are inhibited by mixtures of ATP and ATPγS. With the two ClpB sensor-1 mutants (T315A; T719A) and the two Walker B mutants (E279A; E678A), as the ratio of ATPγS to ATP was increased, the rate of nucleotide hydrolysis slowed (Fig. 6a). Both ClpB(E279A) and ClpB(N719A) hydrolyzed ATP at a two-fold faster rate than wild type ClpB for unknown reasons. With Hsp104 mutants in NBD-2 (N728A; K620T), the ATPase activity was inhibited about 50% when the ATP to ATPγS ratio was 3:1, in contrast to the lack of inhibition of the wild type Hsp104 at this ratio of ATP:ATPγS (Fig. 6c). With Hsp104(K620T) the rate of ATP hydrolysis was markedly faster than wild type. Thus, while there is not a simple correlation between the rates of ATP hydrolysis and the rates of remodeling, for both ClpB and Hsp104 mutants whose remodeling activities were inhibited by ATPγS, their ATPase activities were similarly inhibited by ATPγS.
Effects of ATP and ATPγS on prion-remodeling by Hsp104
Finally, we asked how mixtures of ATP and ATPγS would affect Hsp104 remodeling reactions involving an unusual substrate that can exist stably in two different extremes of protein conformation. Hsp104 controls the inheritance of the yeast prion [PSI+], which consists of self-propagating amyloid fibers generated by Sup35’s prion domain, NM11. In its prion state, NM adopts an incredibly stable β-sheet rich amyloid conformation40,41. At high concentrations, Hsp104 rapidly disassembled these unusually stable fibers in an ATP-dependent reaction (Fig. 7a). The disassembly reaction was not supported by ATPγS alone and mixtures of ATP and ATPγS were inhibitory. Even an ATP:ATPγS ratio of 6:1 inhibited, and a 1:1 ratio abolished fiber disassembly (Fig. 7a). In keeping with these observations, neither Hsp104(N728A) nor Hsp104(K620T) are able to disassemble NM fibers26 even though they can resolve small GFP aggregates (Fig. 4b). Thus, Hsp104 has an absolute requirement for ATP to remodel the stable conformation of NM fibers.
Figure 7. Effects of ATP and ATPγS on prion remodeling by Hsp104.

(a) Prion disassembly by Hsp104. In a and b values represent means ± SD (n=3).
(b) Prion assembly in the presence of Hsp104.
(c) Prion assembly in the presence of Hsp104 probed for the presence of either oligomer or NM.
In the non-prion state, NM has the unusual capacity to populate a predominantly random-coil conformation for extended periods40. At low concentrations, Hsp104 catalyzed the folding of these largely unstructured proteins into the β-sheet rich prion state in two ways (Fig. 7b, c). First, it promoted the formation of a critical oligomeric intermediate of NM, detected using a conformation-specific antibody26, which is essential to nucleate amyloid assembly and eliminate the lag phase (the time before the first appearance of amyloid) (Fig. 7c). Second, it accelerated the assembly phase (the time between the first appearance of amyloid and the completion of assembly) by severing NM fibers to create additional polymerization surfaces for NM assembly26 (Fig. 7b). The latter activity, like fiber disassembly, was supported by ATP but not ATPγS (Fig. 7b). At all ratios of ATP:ATPγS the acceleration of assembly was inhibited (Fig. 7b). Thus, as with fiber disassembly, the acceleration of assembly phase by fiber severing is not stimulated by the mixture of ATP and ATPγS, but rather is inhibited.
By contrast, Hsp104 eliminated the lag phase by catalyzing the formation of the critical oligomeric species that nucleate assembly in the presence of ATP or ATPγS, but not with ADP or without nucleotide (Fig. 7b, c). Thus, ATP or ATPγS binding promotes this prion-folding activity. This illuminates the versatility of Hsp104 hexamers, in that they can promote protein folding or unfolding, depending on the reaction conditions and nature of the substrate.
DISCUSSION
Our surprising finding that a mixture of ATP and ATPγS unleashes the protein remodeling activities of ClpB and Hsp104 has led to unexpected insights into their reaction mechanism. ATPγS allowed us to discover unsuspected features inherent to the ATPase domains that control their remodeling activities. For the first time, it allowed us to study ClpB and Hsp104 remodeling activities without complicating effects of co-chaperones and to do so with substrates that were tractable for dynamic analysis.
Effects of ATPγS have been examined on a large number of ATPases, with which it acts as a classic competitive inhibitor of ATP. To our knowledge, this is the first case in which ATPγS stimulates a reaction requiring ATP hydrolysis. The absence of literature on activating effects of ATP and ATPγS is not because the mixture has not been tested on other proteins. When we examined the effects of such mixtures on ClpA, another Hsp100 protein with two AAA+ domains per monomer, its remodeling activities were not activated. Rather ATPγS inhibited remodeling. Thus, the unexpected effects of mixed nucleotides in unleashing the otherwise tightly restricted remodeling capacity of ClpB and Hsp104 tells us there is something distinctive about their reaction cycle.
Using Hsp104 mutants in various Walker A, Walker B, and sensor-1 motifs we discovered that remodeling can be triggered by impeding ATP hydrolysis at NBD-2. Since there is known allosteric communication between the two NBDs19, 20, it is likely that signals originating in the altered NBD-2 of the mutants are propagated through the coiled-coil middle domain to the distant functional NBD-1, resulting in a conformational state of the hexamer conducive for protein remodeling. In contrast to Hsp104, ClpB mutants with defective ATPase activity at either NBD-1 or NBD-2 could trigger protein-remodeling activity. This difference most likely reflects alterations in the properties of NBD-1 and NBD-2 in the respective protein that have accrued, perhaps as a consequence of genetic drift or selective pressures. What is most remarkable is that protein remodeling by ClpB and Hsp104 is controlled by an amazingly similar mechanism and yet distinct from ClpA.
Analyses of various nucleotide combinations revealed that ATPase activity must be asymmetrically decelerated in a specific manner to trigger ClpB and Hsp104 protein-remodeling activities. Only mixtures of ATP and ATPγS could elicit the protein-remodeling activities. Mixtures of ATP and ADP or ATP and AMP-PNP failed to promote remodeling, even though addition of ADP or AMP-PNP would also decelerate ATPase activity. In addition, slowing hydrolysis by limiting ATP did not elicit remodeling activities. These data imply that it is not simply slow hydrolysis that is essential to elicit the remodeling activity. This implication was clarified by the direct measurements of ATP hydrolysis. ATPγS stimulated ATP hydrolysis by ClpB and did not inhibit ATP hydrolysis by Hsp104, while it inhibited hydrolysis by the ClpB and Hsp104 mutants. Together these results suggest that the binding and/or slow hydrolysis of ATPγS at a subset of NBDs actually translates through inter-domain communication, into increased ATP hydrolysis at the other.
Why should there be a requirement for slow hydrolysis at one NBD to elicit remodeling activities, as demonstrated by the results with mutant proteins? We suggest that the multiple ATP binding sites of the Hsp104/ClpB hexamer must bind nucleotide, such that the ATP conformation is induced. In addition, ATP hydrolysis must be slow at NBD-2 of Hsp104 and at either NBD of ClpB to ensure that substrates are not prematurely released without unfolding, while hydrolysis at the other NBD must proceed at a rate sufficient to promote energy-dependent remodeling. Although it is difficult to compare AAA+ proteins with one AAA+ domain per monomer to those with two AAA+ domains per monomer, this need for holding and unfolding is slightly reminiscent of that proposed for ClpX where there is asymmetric nucleotide occupancy of the six ATP binding sites within the single AAA+ domain42. However, for Hsp104 and ClpB, the mutants demonstrate that a balanced asymmetry between the two sets of AAA+ domains stimulates the remodeling activities.
This unexpected reaction mechanism may apply more broadly to other hexameric AAA+ ATPases with two AAA+ domains per monomer. For example, it would explain the otherwise puzzling finding that the NBD-2 of NSF (N-ethylmaleimide-sensitive fusion protein) has been conserved for nucleotide binding activity but has little or no ATPase activity43, yet is potently able to disassemble a large number of protein complexes involved in vesicle trafficking1. Similarly, NBD-1 of p97 is also catalytically inactive44, yet p97 assists the retrotranslocation of diverse polypeptides from the ER to the cytosol for proteasomal degradation1.
Because ATPγS is a non-physiological nucleotide, it is likely that other factors mimic the stimulatory effects of ATPγS in vivo. ClpB and Hsp104 usually cooperate with Hsp70 and Hsp40 chaperones8–10 as well as small Hsps28, 45, in various disaggregation scenarios. Our results suggest that a major function of the co-factors is to coordinate the ATPase cycles of the two NBDs during substrate remodeling. Co-chaperones might interact directly with ClpB or Hsp1048, 19, 46, 47 such that they attain an optimal mode of ATPase activity for protein disaggregation. Alternatively, initial remodeling of the aggregate surface by co-chaperones17, 31, 48 might ensure that the substrate itself exerts the appropriate alterations in ClpB or Hsp104 ATPase activity. In either way, co-chaperones would directly or indirectly facilitate the asymmetric deceleration of ATPase activity at an NBD during the protein-remodeling process. Such tight regulation by Hsp70/DnaK or other co-factors would ensure successful deployment and activation of Hsp104/ClpB remodeling activity whenever or wherever it is needed most. However, although this putative role for Hsp70/DnaK may be critical for maximal synergy between Hsp104/ClpB and Hsp70/DnaK during disaggregation, Hsp70/DnaK must be doing more than simply coordinating the Hsp104/ClpB ATPase cycle since mutants that are active in protein remodeling in vitro in the absence of co-chaperones do not convey thermotolerance in vivo20, 35, 37. The Hsp70/DnaK system may also help present aggregated polypeptides to Hsp104/ClpB at an early stage of disaggregation8, 17, and undoubtedly help newly solubilized polypeptides to refold once they are released from Hsp104/ClpB8, 9.
Notably, the asymmetric deceleration of ATPase activity by ATPγS or point mutations cannot be tolerated for the remodeling of some substrates, as with the disassembly of Sup35 prions by Hsp10425, 26. In these instances, the substrate may itself impose the requisite changes on Hsp104 ATPase activity. Alternatively, the remodeling of some substrates may require all protomers of the hexamer to bind and hydrolyze ATP in a sequential or concerted manner. This is in contrast to restricted probabilistic events where only select protomers hydrolyze ATP during substrate remodeling, which can be sufficient for many functions of another AAA+ ATPase, ClpX49. Indeed, in some situations Hsp104 and ClpB hexamers may even need to switch between probabilistic and concerted modes of ATP hydrolysis. This may also explain why Hsp104 NBD-2 mutants and ClpB NBD-1 or NBD-2 mutants do not provide thermotolerance20, 35, 37.
Alternatively, these mutants may be less able to collaborate productively or synergistically with co-chaperones. We suggest that it is the versatility of reaction mechanisms that ClpB and Hsp104 hexamers can bring to bear on an amazing variety of different substrates, which guarantees successful reactivation of the entire aggregated proteome following environmental stress.
METHODS
Plasmids
To generate a plasmid expressing ClpB, a clpB PCR product containing a 5′ NdeI and a 3′ EcoRI site was cut and ligated into NdeI- and EcoRI-digested pET24b (Novagen). Mutants were created by site-directed mutagenesis using the QuikChange Kit (Stratagene).
Proteins and DNA
P1 RepA50, ClpA50, GFP51, RepA(1–70)-GFP51, GroELtrap32 (GroELD87K), Hsp104 and Hsp104 mutants26, ClpB and ClpB mutants37, and [3H]oriP1 DNA30 (2200 cpm/fmol) were prepared as described. Protein concentrations given are for monomeric GFP and RepA(1–70)-GFP, dimeric RepA, tetradecameric GroELtrap, and hexameric ClpA, ClpB and Hsp104.
Assays
RepA activation assays (20 μl) contained buffer A [20 mM Tris-HCl, pH 7.5/100 mM KCl/5 mM DTT/0.1mM EDTA/10% glycerol (vol/vol)], 4 mM total nucleotide where indicated, 10mM MgCl2, 50μg/ml BSA, 0.005% Triton X-100 (vol/vol), 4nM RepA, and either 1μM ClpB or ClpB mutant, 1μM Hsp104 or Hsp104 mutant, or 30 nM ClpA, where indicated. When two nucleotides were added, the ratio of ATP to the second nucleotide was 1:1 for ClpB, ClpB mutants, and ClpA and 3:1 for Hsp104 and Hsp104 mutants. When the ratio of ATP:ATPγS was varied with ClpB, 350nM ClpB was used. After 10 min at 23°C, 12mM EDTA was added and reactions were chilled to 0°C. Calf thymus DNA (1μg) and 13fmol of [3H]oriP1 plasmid DNA were added. After 5 min at 0°C, the mixtures were filtered through nitrocellulose filters and retained radioactivity was measured.
GFP reactivation assays (100μL) contained buffer A, 4mM total nucleotide where indicated, an ATP regenerating system (ARS; 20mM creatine phosphate and 6μg creatine kinase), 20mM MgCl2, 0.45μM heat-aggregated GFP (heated 10min at 80°C at 4.5μM) and 1μM of either ClpB, Hsp104 or ClpA, as indicated. When two nucleotides were added, the ratio of ATP to the second nucleotide was 1:1 for ClpB, ClpB mutants, and ClpA and 3:1 for Hsp104 and Hsp104 mutants. When the ARS was omitted, 5 units hexokinase and 5mM glucose were added. Where indicated, 2.5μM GroELtrap was added. Reactivation was monitored with time at 23°C using a Perkin-Elmer LS50B luminescence spectrophotometer with a well plate reader. Excitation and emission wavelengths were 395nm and 510nm, respectively.
Unfolding assays (100μL) contained buffer A, 20μg/ml BSA, 0.005% Triton X-100 (vol/vol), 20mM creatine phosphate, 6μg creatine kinase, 10mM MgCl2, 4mM nucleotide as indicated, 0.7μM RepA(1–70)-GFP, and 2.1μM ClpB, ClpB mutants, Hsp104 or hsp104 mutants, or ClpA, as indicated. GroELtrap (2.5μM) was added where indicated. When two nucleotides were added, the ratio of ATP to the second nucleotide was 1:1 for ClpB, ClpB mutants, and ClpA and 3:1 for Hsp104 and Hsp104 mutants. When the ARS was omitted, 5 units hexokinase and 5mM glucose were added. Fluorescence was monitored as above.
Steady-state unfolding assays were as described for unfolding but with 1mM ATP and 1mM ATPγS. After 10 min of incubation one of the following was added: 10 mM ATP, 10mM ATP with GroELtrap, 10mM ADP, 10mM ADP with GroELtrap, 10mM ATPγS, or GroELtrap. ATPase assays (25μL) contained buffer A, 0.005% Triton X-100 (vol/vol), 4mM total nucleotide where indicated, 20mM MgCl2, 0.4μM RepA(1–70)-GFP (where indicated), and either 2μM ClpB, ClpB mutant, Hsp104, Hsp104 mutant, or ClpA, as indicated. When ATP and ATPγS were used, the ratio was 1:1 for ClpB, ClpB mutants, and ClpA, 3:1 for Hsp104 and Hsp104 mutants, or varied as indicated. For the assays containing non-radioactive nucleotides, phosphate production was determined using a malachite green phosphate detection kit (Biomol). For the radioactivity assays, 1μCi of ATP[γ-35S] (>1000 Ci/mM; GE Healthcare) or 0.1μCi of [γ-33P]ATP(>3000 Ci/mM; GE Healthcare) was used. Reaction mixtures with radioactive ATP or ATPγS were incubated 15 min or 90 min at 23ºC, respectively. Those with [γ-33P]ATP were analyzed as described52. Those with ATP[γ-35S] were stopped with the addition of 37mM EDTA; aliquots (5μL) were spotted on polyethyleneimine thin layer plates, developed in 0.5M formic acid and 0.5M LiCl, air-dried and analyzed using a phosphoimaging device.
Prion assembly and disassembly assays were performed as described26. For disassembly, 2.5μM NM was incubated for 6h with rotation (80rpm) to generate prions and then incubated with 0.3 μM Hsp104 at 25°C and 5mM total nucleotide as indicated. Where indicated, the ratio of ATP:ATPγS was varied. Disassembly was monitored by Thioflavin-T (ThT) fluorescence. To measure assembly, 2.5μM unseeded, rotated (80rpm) NM was incubated with 0.03μM Hsp104, plus 5mM total nucleotide as indicated using the ATP:ATPγS ratios indicated. Prion assembly was monitored by ThT fluorescence. At various times during the assembly reaction, samples were applied to nitrocellulose and probed with anti-oligomer antibody26 or anti-NM antibody26.
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, NCI, CCR, an American Heart Association scientist development grant to J.S, NIH grant (number GM58626) to M.Z., and NIH grant (number GM25874) to S.L. We thank Charles Glabe for the anti-oligomer antibody, Kiyoshi Mizuuchi and Keith McKenney for helpful discussions.
Author Contributions: S. M. D., J. S. and J. R. H., designed experiments, performed experiments, interpreted data, and wrote manuscript; M. Z., interpreted data, and wrote paper; S. W. and S. L. designed experiments, interpreted data, and wrote paper.
Footnotes
Competing interests statement: The authors declare that they have no competing financial interests.
References
- 1.Hanson PI, Whiteheart SW. AAA+ proteins: have engine, will work. Nat Rev Mol Cell Biol. 2005;6:519–29. doi: 10.1038/nrm1684. [DOI] [PubMed] [Google Scholar]
- 2.Erzberger JP, Berger JM. Evolutionary relationships and structural mechanisms of AAA+ proteins. Annu Rev Biophys Biomol Struct. 2006;35:93–114. doi: 10.1146/annurev.biophys.35.040405.101933. [DOI] [PubMed] [Google Scholar]
- 3.Hedges SB, Blair JE, Venturi ML, Shoe JL. A molecular timescale of eukaryote evolution and the rise of complex multicellular life. BMC Evol Biol. 2004;4 doi: 10.1186/1471-2148-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sanchez Y, Lindquist SL. HSP104 required for induced thermotolerance. Science. 1990;248:1112–5. doi: 10.1126/science.2188365. [DOI] [PubMed] [Google Scholar]
- 5.Squires CL, Pedersen S, Ross BM, Squires C. ClpB is the Escherichia coli heat shock protein F84.1. J Bacteriol. 1991;173:4254–62. doi: 10.1128/jb.173.14.4254-4262.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sanchez Y, Taulien J, Borkovich KA, Lindquist S. Hsp104 is required for tolerance to many forms of stress. EMBO J. 1992;11:2357–64. doi: 10.1002/j.1460-2075.1992.tb05295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parsell DA, Kowal AS, Singer MA, Lindquist S. Protein disaggregation mediated by heat-shock protein Hsp104. Nature. 1994;372:475–8. doi: 10.1038/372475a0. [DOI] [PubMed] [Google Scholar]
- 8.Glover JR, Lindquist S. Hsp104, Hsp70, and Hsp40: a novel chaperone system that rescues previously aggregated proteins. Cell. 1998;94:73–82. doi: 10.1016/s0092-8674(00)81223-4. [DOI] [PubMed] [Google Scholar]
- 9.Goloubinoff P, Mogk A, Zvi AP, Tomoyasu T, Bukau B. Sequential mechanism of solubilization and refolding of stable protein aggregates by a bichaperone network. Proc Natl Acad Sci U S A. 1999;96:13732–7. doi: 10.1073/pnas.96.24.13732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zolkiewski M. ClpB cooperates with DnaK, DnaJ, and GrpE in suppressing protein aggregation. A novel multi-chaperone system from Escherichia coli. J Biol Chem. 1999;274:28083–6. doi: 10.1074/jbc.274.40.28083. [DOI] [PubMed] [Google Scholar]
- 11.Shorter J, Lindquist S. Prions as adaptive conduits of memory and inheritance. Nat Rev Genet. 2005;6:435–50. doi: 10.1038/nrg1616. [DOI] [PubMed] [Google Scholar]
- 12.Lee S, et al. The structure of ClpB: a molecular chaperone that rescues proteins from an aggregated state. Cell. 2003;115:229–40. doi: 10.1016/s0092-8674(03)00807-9. [DOI] [PubMed] [Google Scholar]
- 13.Akoev V, Gogol EP, Barnett ME, Zolkiewski M. Nucleotide-induced switch in oligomerization of the AAA+ ATPase ClpB. Protein Sci. 2004;13:567–74. doi: 10.1110/ps.03422604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parsell DA, Kowal AS, Lindquist S. Saccharomyces cerevisiae Hsp104 protein Purification and characterization of ATP-induced structural changes . J Biol Chem. 1994;269:4480–7. [PubMed] [Google Scholar]
- 15.Lum R, Tkach JM, Vierling E, Glover JR. Evidence for an Unfolding/Threading Mechanism for Protein Disaggregation by Saccharomyces cerevisiae Hsp104. J Biol Chem. 2004;279:29139–46. doi: 10.1074/jbc.M403777200. [DOI] [PubMed] [Google Scholar]
- 16.Schlieker C, et al. Substrate recognition by the AAA+ chaperone ClpB. Nat Struct Mol Biol. 2004;11:607–15. doi: 10.1038/nsmb787. [DOI] [PubMed] [Google Scholar]
- 17.Weibezahn J, et al. Thermotolerance requires refolding of aggregated proteins by substrate translocation through the central pore of ClpB. Cell. 2004;119:653–65. doi: 10.1016/j.cell.2004.11.027. [DOI] [PubMed] [Google Scholar]
- 18.Barnett ME, Nagy M, Kedzierska S, Zolkiewski M. The amino-terminal domain of ClpB supports binding to strongly aggregated proteins. J Biol Chem. 2005;280:34940–5. doi: 10.1074/jbc.M505653200. [DOI] [PubMed] [Google Scholar]
- 19.Cashikar AG, et al. Defining a pathway of communication from the C-terminal peptide binding domain to the N-terminal ATPase domain in a AAA protein. Mol Cell. 2002;9:751–60. doi: 10.1016/s1097-2765(02)00499-9. [DOI] [PubMed] [Google Scholar]
- 20.Hattendorf DA, Lindquist SL. Cooperative kinetics of both Hsp104 ATPase domains and interdomain communication revealed by AAA sensor-1 mutants. EMBO J. 2002;21:12–21. doi: 10.1093/emboj/21.1.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mogk A, et al. Roles of Individual Domains and Conserved Motifs of the AAA+ Chaperone ClpB in Oligomerization, ATP Hydrolysis, and Chaperone Activity. J Biol Chem. 2003;278:17615–24. doi: 10.1074/jbc.M209686200. [DOI] [PubMed] [Google Scholar]
- 22.Schirmer EC, Ware DM, Queitsch C, Kowal AS, Lindquist SL. Subunit interactions influence the biochemical and biological properties of Hsp104. Proc Natl Acad Sci U S A. 2001;98:914–9. doi: 10.1073/pnas.031568098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schlee S, Groemping Y, Herde P, Seidel R, Reinstein J. The chaperone function of ClpB from Thermus thermophilus depends on allosteric interactions of its two ATP-binding sites. J Mol Biol. 2001;306:889–99. doi: 10.1006/jmbi.2001.4455. [DOI] [PubMed] [Google Scholar]
- 24.Shorter J, Lindquist S. Navigating the ClpB channel to solution. Nat Struct Mol Biol. 2005;12:4–6. doi: 10.1038/nsmb0105-4. [DOI] [PubMed] [Google Scholar]
- 25.Schirmer EC, Homann OR, Kowal AS, Lindquist S. Dominant gain-of-function mutations in Hsp104p reveal crucial roles for the middle region. Mol Biol Cell. 2004;15:2061–72. doi: 10.1091/mbc.E02-08-0502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shorter J, Lindquist S. Hsp104 catalyzes formation and elimination of self-replicating Sup35 prion conformers. Science. 2004;304:1793–7. doi: 10.1126/science.1098007. [DOI] [PubMed] [Google Scholar]
- 27.Shorter J, Lindquist S. Destruction or potentiation of different prions catalyzed by similar hsp104 remodeling activities. Mol Cell. 2006;23:425–38. doi: 10.1016/j.molcel.2006.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haslbeck M, Miess A, Stromer T, Walter S, Buchner J. Disassembling protein aggregates in the yeast cytosol. The cooperation of Hsp26 with Ssa1 and Hsp104. J Biol Chem. 2005;280:23861–8. doi: 10.1074/jbc.M502697200. [DOI] [PubMed] [Google Scholar]
- 29.Wickner S, et al. A molecular chaperone, ClpA, functions like DnaK and DnaJ. Proc Natl Acad Sci U S A. 1994;91:12218–12222. doi: 10.1073/pnas.91.25.12218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wickner S, Hoskins J, McKenney K. Function of DnaJ and DnaK as chaperones in origin-specific DNA binding by RepA. Nature. 1991;350:165–167. doi: 10.1038/350165a0. [DOI] [PubMed] [Google Scholar]
- 31.Zietkiewicz S, Lewandowska A, Stocki P, Liberek K. Hsp70 chaperone machine remodels protein aggregates at the initial step of Hsp70-Hsp100-dependent disaggregation. J Biol Chem. 2006;281:7022–9. doi: 10.1074/jbc.M507893200. [DOI] [PubMed] [Google Scholar]
- 32.Weber-Ban EU, Reid BG, Miranker AD, Horwich AL. Global unfolding of a substrate protein by the Hsp100 chaperone ClpA. Nature. 1999;401:90–93. doi: 10.1038/43481. [DOI] [PubMed] [Google Scholar]
- 33.Martin J, et al. Chaperonin-mediated protein folding at the surface of groEL through a ‘molten globule’-like intermediate. Nature. 1991;352:36–42. doi: 10.1038/352036a0. [DOI] [PubMed] [Google Scholar]
- 34.Dietz H, Rief M. Exploring the energy landscape of GFP by single-molecule mechanical experiments. Proc Natl Acad Sci U S A. 2004;101:16192–7. doi: 10.1073/pnas.0404549101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Parsell DA, Sanchez Y, Stitzel JD, Lindquist S. Hsp104 is a highly conserved protein with two essential nucleotide-binding sites. Nature. 1991;353:270–3. doi: 10.1038/353270a0. [DOI] [PubMed] [Google Scholar]
- 36.Schirmer EC, Queitsch C, Kowal AS, Parsell DA, Lindquist S. The ATPase activity of Hsp104, effects of environmental conditions and mutations. J Biol Chem. 1998;273:15546–52. doi: 10.1074/jbc.273.25.15546. [DOI] [PubMed] [Google Scholar]
- 37.Barnett ME, Zolkiewski M. Site-directed mutagenesis of conserved charged amino acid residues in ClpB from Escherichia coli. Biochemistry. 2002;41:11277–83. doi: 10.1021/bi026161s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Watanabe YH, Motohashi K, Yoshida M. Roles of the two ATP binding sites of ClpB from Thermus thermophilus. J Biol Chem. 2002;277:5804–9. doi: 10.1074/jbc.M109349200. [DOI] [PubMed] [Google Scholar]
- 39.Weibezahn J, Schlieker C, Bukau B, Mogk A. Characterization of a trap mutant of the AAA+ chaperone ClpB. J Biol Chem. 2003;278:32608–17. doi: 10.1074/jbc.M303653200. [DOI] [PubMed] [Google Scholar]
- 40.Scheibel T, Lindquist SL. The role of conformational flexibility in prion propagation and maintenance for Sup35p. Nat Struct Biol. 2001;8:958–62. doi: 10.1038/nsb1101-958. [DOI] [PubMed] [Google Scholar]
- 41.Scheibel T, et al. Conducting nanowires built by controlled self-assembly of amyloid fibers and selective metal deposition. Proc Natl Acad Sci U S A. 2003;100:4527–32. doi: 10.1073/pnas.0431081100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hersch GL, Burton RE, Bolon DN, Baker TA, Sauer RT. Asymmetric interactions of ATP with the AAA+ ClpX6 unfoldase: allosteric control of a protein machine. Cell. 2005;121:1017–27. doi: 10.1016/j.cell.2005.05.024. [DOI] [PubMed] [Google Scholar]
- 43.Whiteheart SW, et al. N-ethylmaleimide-sensitive fusion protein: a trimeric ATPase whose hydrolysis of ATP is required for membrane fusion. J Cell Biol. 1994;126:945–54. doi: 10.1083/jcb.126.4.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Q, Song C, Li CC. Molecular perspectives on p97-VCP: progress in understanding its structure and diverse biological functions. J Struct Biol. 2004;146:44–57. doi: 10.1016/j.jsb.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 45.Mogk A, et al. Refolding of substrates bound to small Hsps relies on a disaggregation reaction mediated most efficiently by ClpB/DnaK. J Biol Chem. 2003;278:31033–42. doi: 10.1074/jbc.M303587200. [DOI] [PubMed] [Google Scholar]
- 46.Schlee S, Beinker P, Akhrymuk A, Reinstein J. A chaperone network for the resolubilization of protein aggregates: direct interaction of ClpB and DnaK. J Mol Biol. 2004;336:275–85. doi: 10.1016/j.jmb.2003.12.013. [DOI] [PubMed] [Google Scholar]
- 47.Kedzierska S, Chesnokova LS, Witt SN, Zolkiewski M. Interactions within the ClpB/DnaK bi-chaperone system from Escherichia coli. Arch Biochem Biophys. 2005;444:61–5. doi: 10.1016/j.abb.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 48.Zietkiewicz S, Krzewska J, Liberek K. Successive and synergistic action of the Hsp70 and Hsp100 chaperones in protein disaggregation. J Biol Chem. 2004;279:44376–83. doi: 10.1074/jbc.M402405200. [DOI] [PubMed] [Google Scholar]
- 49.Martin A, Baker TA, Sauer RT. Rebuilt AAA + motors reveal operating principles for ATP-fuelled machines. Nature. 2005;437:1115–20. doi: 10.1038/nature04031. [DOI] [PubMed] [Google Scholar]
- 50.Hoskins JR, Wickner S. Two peptide sequences can function cooperatively to facilitate binding and unfolding by ClpA and degradation by ClpAP. Proc Natl Acad Sci U S A. 2006;103:909–14. doi: 10.1073/pnas.0509154103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hoskins JR, Kim SY, Wickner S. Substrate recognition by the ClpA chaperone component of ClpAP protease. J Biol Chem. 2000;275:35361–35367. doi: 10.1074/jbc.M006288200. [DOI] [PubMed] [Google Scholar]
- 52.Shacter E. Organic extraction of Pi with isobutanol/toluene. Anal Biochem. 1984;138:416–20. doi: 10.1016/0003-2697(84)90831-5. [DOI] [PubMed] [Google Scholar]
- 53.Schwede T, Kopp J, Guex N, Peitsch MC. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003;31:3381–5. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]