

Abstract
Voltage-dependent K+ channels like Shaker use an intracellular gate to control ion flow through the pore. When the membrane voltage becomes more positive, these channels traverse a series of closed conformations before the final opening transition. Does the intracellular gate undergo conformational changes before channel opening? To answer this question we introduced cysteines into the intracellular end of the pore and studied their chemical modification in conditions favoring each of three distinct states, the open state, the resting closed state, and the activated-not-open state (the closed state adjacent to the open state). We used two independent ways to isolate the channels in the activated-not-open state. First, we used mutations in S4 (ILT; Smith-Maxwell, C.J., J.L. Ledwell, and R.W. Aldrich. 1998. J. Gen. Physiol. 111:421–439; Ledwell, J.L., and R.W. Aldrich. 1999. J. Gen. Physiol. 113:389–414) that separate the final opening step from earlier charge-movement steps. Second, we used the open channel blocker 4-aminopyridine (4-AP), which has been proposed to promote closure of the intracellular gate and thus specifically to stabilize the activated-not-open state of the channels. Supporting this proposed mechanism, we found that 4-AP enters channels only after opening, remaining trapped in closed channels, and that in the open state it competes with tetraethylammonium for binding. Using these tools, we found that in the activated-not-open state, a cysteine located at a position considered to form part of the gate (Shaker 478) showed higher reactivity than in either the open or the resting closed states. Additionally, we have found that in this activated state the intracellular gate continued to prevent access to the pore by molecules as small as Cd2+ ions. Our results suggest that the intracellular opening to the pore undergoes some rearrangements in the transition from the resting closed state to the activated-not-open state, but throughout this process the intracellular gate remains an effective barrier to the movement of potassium ions through the pore.
INTRODUCTION
Shaker channel proteins form a K+ selective pore that is gated by changes in the voltage across the membrane. Structurally, Shaker channels are homotetramers, with each subunit consisting of six transmembrane α-helices S1–S6. Of these, S1–S4 form the voltage sensing domains (one per subunit) with the highly charged S4 being the main component. The remaining S5-P region-S6 portion of each subunit come together to form the potassium selective pore. The P region forms the selectivity filter of the channel, and the S6 segments line the intracellular end of the pore and form the intracellular gate (Yellen et al., 1991; Liu et al., 1997; Doyle et al., 1998; Yellen, 2002). Based on the crystal structures of two non-voltage-dependent bacterial channels, KcsA (Doyle et al., 1998) and MthK (Jiang et al., 2002a), a general model for the motions of the S6 segments during gating was proposed (Jiang et al., 2002b). According to this model, the straight pore-lining helices arranged in a teepee-like structure in the closed state (as seen in the KcsA channel) bend at a conserved glycine hinge, leaving a wide open entrance to the pore (as seen in the MthK channel). In eukaryotic voltage-sensitive channels, both functional and direct structural studies agree that the bend occurs instead at a conserved Pro-X-Pro amino acid motif in the S6 (del Camino et al., 2000; del Camino and Yellen, 2001; Webster et al., 2004; Long et al., 2005a). This helix-breaking motif near the S6 bundle crossing introduces an ∼30° kink in the helix, which is likely to be at least partially maintained even in the closed state (del Camino and Yellen, 2001). Gating in Shaker channels may occur through a swivel type of motion in which the Pro-X-Pro motif acts as a hinge to move blocking residues away from the pore axis.
Our current understanding of the motions of gating is based mainly on studying two states of the channel: the resting closed state and the open state. During the process of activation, however, Shaker channels traverse several closed states before the final opening (Hoshi et al., 1994; Zagotta et al., 1994a,b; Schoppa and Sigworth, 1998). We were particularly interested in the closed state immediately adjacent to the open state, which we will refer to as the activated-not-open state. Does the intracellular end of the pore undergo conformational changes during the transition from the resting closed state to the activated-not-open state? Also, since it has been proposed that opening of the pore may be the result of the action of two gates, the S6 gate and the selectivity filter, does the intracellular gate remain tightly closed in the activated-not-open state? To answer these two questions, we used two different tools to isolate the channels in the activated-not-open state: the channel blocker 4-aminopyridine and the S4 ILT mutations.
4-Aminopyridine (4-AP) is a widely used blocker of many K+ channels, but its precise mechanism of action has remained elusive. Recently, Armstrong and Loboda (2001) proposed a general model for the mechanism of action of 4-AP based on their own characterization of the effect of the blocker on the gating currents of Shaker channels (Loboda and Armstrong, 2001) as well as previous results on the effect of 4-AP on the ionic currents (Kirsch et al., 1986; Kirsch and Drewe, 1993; McCormack et al., 1994). According to their model, 4-AP binds to a site located in the water-filled cavity formed by the S6 helices behind the intracellular gate. Only when the gate is open can the blocker reach its binding site, but once 4-AP is bound in the channel it strongly biases the last transition toward the activated-not-open state, thereby causing its own trapping in the cavity. This last feature of the mechanism of action of 4-AP is what differentiates this molecule from other well-characterized open-channel blockers such as the quaternary ammonium blockers (Armstrong, 1969; Holmgren et al., 1997), and it makes 4-AP a useful pharmacological tool to isolate the channels in the activated-not-open state.
The ILT mutations are three conservative changes of uncharged residues in the S4 of Shaker (V369I, I372L, S376T) that have marked effects on the channel gating properties (Smith-Maxwell et al., 1998; Ledwell and Aldrich, 1999). The voltage dependence of activation in this mutant is shallower and shifted to more positive voltages as compared with wild-type Shaker. Additionally, the activation kinetics of the ILT channels are slow, and more importantly single exponential, lacking the sigmoidicity of wild-type Shaker. Finally, analysis of the gating currents revealed that in this mutant, the voltage range of channel opening is clearly separated from the major charge-moving steps, which occur at more negative potentials. These effects can be explained if the ILT mutations slow down a final cooperative transition to the open state that then becomes rate limiting for channel opening. The ILT mutant thus simplifies the otherwise complex gating scheme of Shaker. Although in principle there are many partially “activated-not-open” states with different numbers of voltage sensors in various positions, the ability in these mutant channels to move nearly all of the charge before opening of any substantial fraction of the channels makes it possible to isolate the channels in a nearly unique “activated-not-open” state, which has all of the voltage sensors activated and immediately precedes the final opening step.
After first characterizing the effect of 4-AP on Shaker channels to test the basic predictions of the model proposed by Armstrong and Loboda (2001), we combined the use of this blocker with the S4-ILT mutations to learn about the state-dependent cysteine accessibility at the intracellular end of the pore. We found that a cysteine introduced at position 478, located in the region supposed to form the gate (Liu et al., 1997; Hackos et al., 2002; Webster et al., 2004), shows a higher rate of reaction with MTS reagents in the activated-not-open than in the open or closed states. Additionally, we have found that in the activated-not-open state, the intracellular gate still prevents access of small ions to the pore. These results, in addition to validating the model for the mechanism of action of 4-AP proposed by Armstrong and Loboda, suggest that there are conformational changes in the intracellular mouth of the pore during the transition from the resting closed state to the activated-not-open state, but that the S6 gate continues to prevent ion permeation.
MATERIALS AND METHODS
Mutagenesis and Expression
All mutations were introduced by PCR mutagenesis into Shaker H4 K+ channels (Kamb et al., 1988) containing three additional modifications: a deletion of residues 6–46 to remove N-type inactivation (Hoshi et al., 1990), substitution of cysteines 301 and 308 by serines to minimize the wild-type effect of chemical modification (Holmgren et al., 1996), and a mutation of threonine 449 to valine to diminish C-type inactivation (López-Barneo et al., 1993). The mutations were confirmed by sequencing. Channels were transiently expressed in human embryonic kidney 293 cells and experiments were performed 1–3 d after transfection. Methods for transfection and identification of transfected cells have been described previously (Jurman et al., 1994).
For gating charge experiments, the nonconducting W434F mutation (Perozo et al., 1993; Yang et al., 1997) was introduced into the Shaker coding region in addition to the other mutations used. cRNAs were transcribed in vitro from linearized plasmid templates with the mMessage mMachine kit with T3 or T7 RNA polymerase (Ambion) and injected into Xenopus laevis oocytes 2–14 d before recording.
Solutions and Physiological Recording
All ionic current experiments were done with excised inside-out patches (Hamill et al., 1981). The pipette (extracellular) solution for most experiments contained 100 mM NaCl, 60 mM KCl, 3 mM CaCl2, 1 mM MgCl2, and 10 mM HEPES at pH 7.4, except as noted. For the experiments involving Cd2+ ions, the bath (intracellular) solution contained 160 mM KCl, 0.5 mM MgCl2, and 10 mM HEPES at pH 7.4; control solutions contained 20 or 50 μM EGTA, and solutions with Cd2+ contained no EGTA. The calculated free [Cd2+] in these solutions with 160 mM chloride ion is ∼0.13 times the total Cd2+ (calculated using data from Smith and Martell, 1998); the second-order rate constants in Fig. 4 are calculated with this correction. In other experiments, the intracellular solution contained 1 mM EGTA.
Figure 4.

The intracellular gate opens only when the channel finally opens, as reported by Cd2+ access to 474C. The plots show the voltage dependence for gating charge (Q-V; small symbols), channel opening (g-V), and chemical modification of 474C in 474C-ILT channels. The square symbols (mean ± SEM, n = 3–4) indicate the second order modification rate constant inferred from experiments using total [Cd2+] in the range 4–20 μM. The g-V is shown as smooth normalized Boltzmann functions with average values from 42 experiments for the midpoint and slope factor; dotted curves are ±1 SD for these values. The bottom plot shows the same g-V and modification rate data on a logarithmic scale, indicating the close match between inferred channel open probability and modification rate.
Trimethylaminoethyl methanethiosulfonate (MTSET) and hydroxyethyl methanethiosulfonate (MTSHE) were purchased from Toronto Research Chemicals Inc. Fresh stocks in water were prepared each day, held on ice, and mixed into the recording solution immediately before use. The methods for electrophysiological recordings and rapid perfusion switches were as previously described (Holmgren et al., 1996; Liu et al., 1997).
Gating current recordings were made using a high-performance cut-open oocyte clamp (CA-1; Dagan Inc.) (Taglialatela et al., 1992; Stefani and Bezanilla, 1998). Voltage control and dynamic response were optimized by permeabilizing the lower dome with 0.3% saponin solution and using agar bridges containing fine platinum-iridium wire and filled with 1 M Na methanesulfonate. Low-resistance (<1 MΩ) glass microelectrodes were filled with 3 M KCl. Online series-resistance compensation was used. Linear leak and capacitative currents were subtracted using a P/−5 to P/−8 protocol from a holding voltage of −140 mV. Records were low-pass filtered at 10 kHz. To reduce the slowly activating native oocyte chloride conductances when using the cut-open clamp, we perfused nominally chloride-free solutions containing (top and guard chambers, mM) 110 NaOH, 2 KOH, 2 Mg(OH) 2, 5 HEPES (MES), pH 7.1; (bottom chamber, mM) 110 KOH, 2 Mg(OH) 2, 1 Ca(OH) 2, 10 EGTA, 5 HEPES (MES), pH 7.1. Gating current records were analyzed further using Igor Pro (Wavemetrics, Inc.) and custom-written software.
RESULTS
4-AP Can Access its Binding Site Only after the Intracellular Gate Is Open
Armstrong and Loboda (2001) proposed that the 4-AP binding site is located in the cavity, and therefore it becomes accessible only when the intracellular gate is open. One basis for this idea was experiments by several groups on the onset of 4-AP block and subsequent recovery, showing that both of these are slower when channels are closed (Wagoner and Oxford, 1990; Choquet and Korn, 1992; Kirsch and Drewe, 1993; Russell et al., 1994). We performed several experiments to test the basic features of such state-dependent behavior for Shaker Δ6-46 channels, where the onset and recovery of blockade can be readily observed without interference from inactivation (Hoshi et al., 1990). We first studied the state dependence of block by this molecule using a fast perfusion system to rapidly apply 4-AP to inside-out patches expressing Shaker channels. Fig. 1 A shows that after opening the channels with a depolarizing pulse, the application of a saturating concentration of 4-AP caused a very rapid block of the current. Washout of 4-AP while maintaining the membrane at a depolarized voltage produced recovery of the current at a rate that reflects blocker unbinding from the channels. Alternatively, 4-AP could be trapped inside the channels (Fig. 1 B). After blocker was applied to open channel and a steady-state blockade was achieved, the voltage was returned to negative potentials in order to close channels securely before 4-AP was removed. The patch was then rinsed with a blocker-free solution for 2 min while maintaining the voltage at −90 mV. A second depolarizing test pulse was then applied. The current elicited by this second voltage step activated only slowly, at the rate previously seen for unbinding of the blocker, strongly indicating that 4-AP had remained trapped inside the channels during the 2-min washout at negative voltages. Finally, no effect on the current was observed when the blocker was applied to channels kept in the closed state, immediately before a depolarizing test voltage step (Fig. 1 C). These results clearly show that the 4-AP binding site is accessible from the intracellular side of the channel only when the channels are open, pointing toward the cavity as the likely location of the binding site.
Figure 1.

4-AP blocks Shaker only after channel opening, and it can be trapped by channel closure. Using a fast-perfusion system, 4-AP (3 mM) was applied intracellularly to an inside-out patch expressing Shaker channels. (A) Blocker was applied during a long depolarizing pulse to +60 mV and removed before returning to a holding potential of −90 mV. (B) 4-AP was perfused during a depolarizing pulse and washed out immediately after returning to a membrane potential of −90 mV. After a 2-min wash out at −90 mV, a new depolarizing step to +60 mV was applied. (C) blocker was applied for over 1 s at −90 mV and removed immediately before channel opening.
4-AP Competes with TEA for its Binding Site
The requirement of channel opening for 4-AP entry and the ability to trap the blocker inside the channel upon closing are consistent with 4-AP binding in the cavity behind the activation gate. Nonetheless, these results are also compatible with the binding site located elsewhere in the protein, provided that such a place becomes accessible only when the channels are open. To discriminate between these two possibilities, we performed competition experiments with TEA. This molecule is useful for this purpose because TEA is also an open channel blocker (Armstrong, 1966, 1969), and there is a great amount of experimental evidence indicating that its binding site is located in the cavity (Holmgren et al., 1997; Zhou et al., 2001a). Kirsch et al. (1993) showed for Kv3.1 channels that there was some antagonism between blockade by a TEA derivative, tetrapentylammonium, and 4-AP. In general, though, the interpretation of competition experiments can be complicated if 4-AP causes allosteric closure of the channel upon binding (which would result in the cavity not being accessible for TEA binding even if 4-AP binds elsewhere in the protein). To minimize this problem, we decided to perform these experiments in a Shaker channel carrying the mutation V476C. When Shaker V476C channels are exposed to Cd2+, this metal forms an intersubunit bridge between the introduced cysteine at position 476 and a native histidine located at position 486, and thereby locks the channels in the open conformation (Liu et al., 1997; Holmgren et al., 1998; Webster et al., 2004) (see Fig. 5 for the location of this bridge in the Kv1.2 structure).
Figure 5.

Stereogram of the Kv1.2 inner pore, illustrating the location of key pore positions in Shaker. Based on the crystal structure of Kv1.2 (Long et al., 2005a; Protein Data Bank entry 2A79). The view is from the intracellular side of the pore, after removal of the β subunit and T1 domain. Helices are shown as ribbons, with the S6 (inner) helices in magenta and the S4–S5 linker helices in blue. The pore-facing residues corresponding to Shaker V474 (yellow) and V478 (green) are shown as space-filling CPK models. The residues that participate in the Cd2+ lock-open bridge (Shaker V476C and H486) are shown in ball-and-stick models. Missing sidechains were restored automatically by SwissProt DeepView (http://www.expasy.org/spdbv), the H486 sidechains were manually reoriented, and the protein was displayed with a transparent Connolly surface (probe radius 1.4 A) using DS ViewerPro (Accelrys). A potassium ion with eight waters of hydration based on the high-resolution structure of KcsA (Zhou et al., 2001b; Protein Data Bank entry 1K4C) is shown in the cavity for reference.
When Shaker V476C channels were locked open with Cd2+, the two blockers behaved in a competitive manner (Fig. 2). Application of TEA alone (at 0.4 mM) produced the expected block of ∼69% (Yellen et al., 1991). Application of 4-AP to the locked-open channels produced inhibition with only very low affinity: 15 mM 4-AP produced ∼46% blockade (very weak compared with the complete blockade by 3 mM seen in Fig. 1, and the ∼60% blockade by 0.1 mM seen in McCormack et al., 1994). This is compatible with the notion that normal inhibition by 4-AP results from tight binding to the closed state of the channels. When this state is made inaccessible by the 476C-Cd2+-H486 lock-open bridge, the remaining interaction with 4-AP is quite weak. This weak interaction is seen to be competitive with TEA. In the constant presence of TEA, 4-AP produced even less fractional blockade: the combination of 4-AP and TEA blocked only ∼20% of the total current seen with TEA alone. Had the two blockers behaved noncompetitively, 4-AP would have blocked the same fraction of the current in both the presence and absence of TEA (arrows in Fig. 2).
Figure 2.

TEA and 4-AP compete for blockade of locked-open Shaker channels. Shaker 476C channels were locked-open by application of 10 μM total Cd2+, and soon thereafter blocker was applied by solenoid switching during a long activating pulse. Fractional blockade was determined by dividing the current in blocker (after stabilization following perfusion) by the current immediately preceding the application of blocker. Results are shown for TEA (0.4 mM) and 4-AP (15 mM), with no other blocker present. Based on these single point estimates, the Kapparent for blockade is ∼0.18 mM for TEA and ∼17.6 mM for 4-AP. The third column shows the fractional blockade by 4-AP in the constant presence of TEA (i.e., current in 4-AP plus TEA, divided by current with TEA present). The arrows show the predicted outcomes for noncompetitive (NC) and competitive (C) blockade. Noncompetitive blockade would give the same fractional blockade as seen in the absence of TEA. Competitive blockade is expected to give a Kapparent for blockade by 4-AP in the presence of TEA equal to Kapparent(4-AP)·(1 + [TEA]/Kapparent(TEA)), ∼56.7 mM. The predicted relative fractional blockade at 15 mM 4-AP would thus be ∼0.21. For these experiments, [K+]out was 20 mM. Data are shown as mean ± SEM for three to four experiments.
These two sets of experiments showed that: (a) binding of 4-AP to Shaker channels requires opening of the intracellular gate, (b) once bound, 4-AP can be trapped inside the channel, and (c) 4-AP and TEA compete for their binding site to block the flow of current in Shaker channels. All of these results illustrate the common features of the blocking mechanisms of 4-AP and QA ions: both types of blockers reach their binding site in the cavity only when the intracellular gate is open. Their subsequent behavior is distinct, though; while QA ions hinder closure of the intracellular gate (Armstrong, 1969; Holmgren et al., 1997), 4-AP promotes closure of the channel. We were particularly interested in this last feature of the 4-AP mechanism of action since it could provide us with a tool (in addition to the ILT mutations) to isolate the channel in the activated-not-open state. We used both 4-AP and the ILT mutations to study cysteine accessibility at the intracellular end of the pore.
478C LT Shows a Distinct MTS Reactivity in the Closed, Activated-not-open, and Open States
To study whether there are conformational changes in the intracellular end of the pore during the transition from resting to activated-not-open states, we looked at changes in MTS reactivity with a cysteine introduced at position 478. This position is ideal for this purpose because, as the deepest S6 position still accessible in the closed state, it likely forms part of the gate (Liu et al., 1997; del Camino and Yellen, 2001; Hackos et al., 2002), but the change in reactivity between the closed and open states is nevertheless substantial (∼10-fold).
To isolate the activated-not-open state with a voltage protocol, we introduced two of the three ILT mutations (I372L and S376T; “LT”) into the S4 of 478C channels. The LT mutations are sufficient to produce a clear separation of opening from the major charge movement, but channel opening does not require the extreme positive voltages needed for ILT (Smith-Maxwell et al., 1998). We first confirmed that Shaker 478C LT channels maintain the basic features of the LT channel. As shown in Fig. 3 (A and B), 478C LT channels show a clear separation between the voltage range of major charge movement and channel opening; while the foot of the Q-V is at about −110 mV and saturates at around −30 mV, channel opening begins at voltages close to 0 mV and reaches the maximum open probability at voltages >100 mV. This leaves a window of voltages between −30 and −5 mV, where the channels are mostly in the activated-not-open state.
Figure 3.
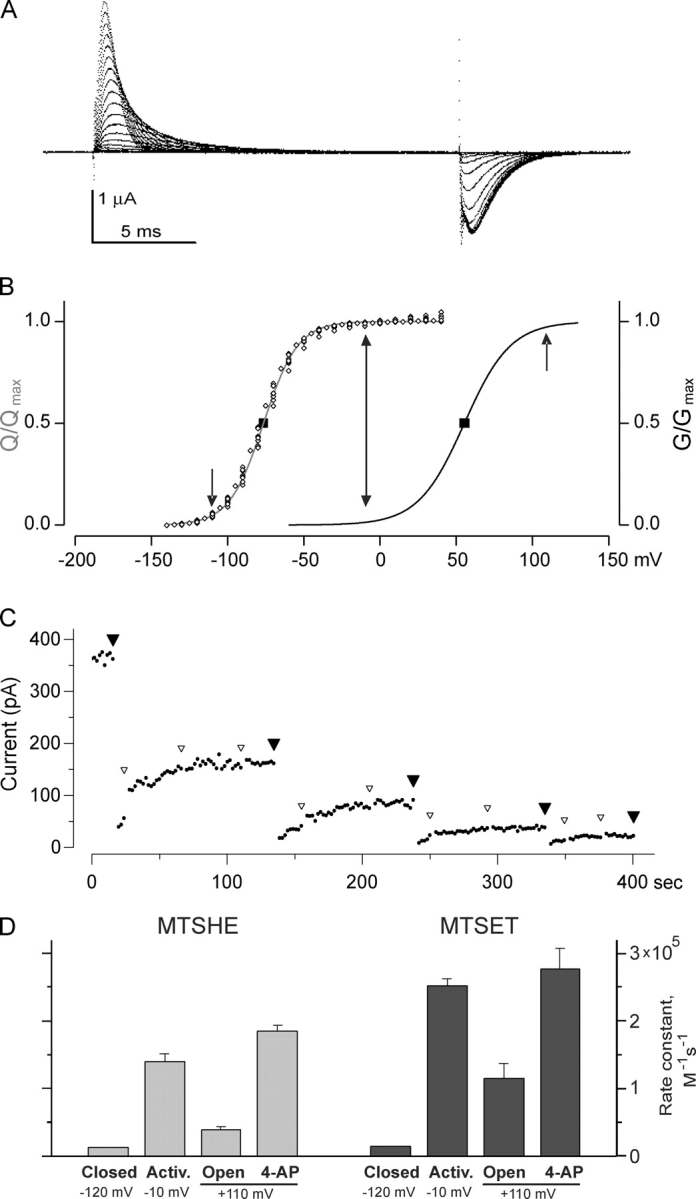
478C becomes accessible in the activated-not-open state. (A) Gating currents for 478C-LT-W434F channels expressed in cut-open oocytes, in response to voltage steps from a holding potential of −150 mV to a pulse potential of −140 to +50 mV, in steps of 10 mV, followed by a return to −150 mV. (B) Normalized Q-V and g-V data for 478C-LT channels, obtained as described in MATERIALS AND METHODS. The square symbols indicate the mean and SEM of the voltage midpoints, and the Boltzmann g-V function has the average mean and slope from five experiments. The arrows indicate the three voltages used for chemical modification measurements. (C) A typical measurement of MTSET reaction rate for 478C-LT channels in an inside-out patch, in the presence of 4-AP. Small dots are the steady-state current measured for an activating pulse to +120 mV in the absence of 4-AP. After each large filled arrowhead, a 590-ms pulse to +110 mV was applied; after the first 80 ms, a solenoid valve delivered a 500-ms pulse of MTSET (4 μM) and 4-AP (0.5 mM). The 4-AP block was complete within 25 ms, so that only a small amount of modification (<5%) occurred with unblocked channels. Small open arrowheads indicate 590 ms pulses to +110 mV with no drug application, applied to speed recovery from 4-AP application. To determine the rate constant of reaction, the points with the large arrowheads were plotted vs. cumulative modification time, and the reaction rate constant was calculated as (fitted time constant)−1 · [MTSET]−1. (D) Apparent second order rate constants for modification of 478C-LT channels by MTSHE and MTSET. The actual concentrations used ranged from 10 to 20 μM for MTSHE, and 4 to 10 μM for MTSET. Rates were determined for channels in various conditions: “closed” (−120 mV), “activated” (−10 mV), “open” (+110 mV), and 4-AP bound (at +110 mV, with 0.5 mM 4-AP). The mean ± SEM for at least three experiments is shown. For these experiments, [K+]out was 100 mM.
We used two different MTS reagents: positively charged MTSET and neutral but polar MTSHE. When applied to 478C channels, each of these produces almost complete current inhibition. Using both allowed us to control for direct effects of transmembrane voltage and electrostatic charge in the interpretation of the results. For both these reagents we determined the second-order reaction rate constants at three voltages: at −110 mV where the channels are in the resting closed state, at +110 mV to measure the reaction rate in the open state, and at −10 mV where the channels are in the activated-not-open state. Additionally, we measured the reaction rate at +110 mV while the channels were blocked with 4-AP. If the Armstrong-Loboda model is correct, 4-AP blocked channels should be in the activated-not-open state, and therefore the modification rate under these conditions should be comparable to that at −10 mV. Fig. 3 C shows that MTSET reacted with 478C LT channels 10 times more slowly in the resting closed state than in the open state, as previously found with 478C channels without the additional S4 LT mutations (Liu et al., 1997; del Camino and Yellen, 2001)1. Most importantly, the modification rate of 478C channels in the activated-not-open state was greater than twofold higher than that of the open state, in each of the two conditions used to isolate the activated-not-open state. We can rule out a possible charge effect for MTSET, as the MTSHE reaction rates exhibited a similar behavior.
Our measurement of the reaction rate for the activated-not-open state is a lower bound on the actual reaction rate. Under the measurement conditions, there will be a small amount of “contamination” by occupancy of the other states; for instance, at −10 mV, a few percent of the channel may be in the open state. Were we to correct for this small effect, the actual change of reaction rates between states would be even larger.
The ∼20-fold increase in reaction rate seen with the change from the resting closed state to the activated-not-open state suggests that this change involves conformational rearrangements that affect the intracellular end of the pore. Nevertheless, these experiments cannot differentiate whether such conformational rearrangements involve movement of the S6 helices themselves or changes in adjacent parts of the protein; possibilities include the S4–S5 linker apparently involved in coupling of voltage sensing to gating, or even the “windows” connecting the vestibule between the pore domain and T1 domain with the intracellular bathing solution (Long et al., 2005a,b). Finally, the same distinctively high reaction rate is seen regardless of which strategy is used to place the channels in the activated-not-open state, supporting the hypothesized mechanisms of action for both 4-AP and the ILT mutations.
The Intracellular Gate Continues to Prevent Cd2+ Access to 474C, even in the Activated-not-open State
Our previous work showed that in voltage-gated K+ channels, the lower S6 forms an effective barrier that regulates the flow of K+ ions through the pore (del Camino and Yellen, 2001). On the other hand, because of the existence of activation-coupled subconducting states in this type of channel (Chapman et al., 1997; Zheng and Sigworth, 1998; Chapman and VanDongen, 2005) and the apparent lack of an intracellular gate in the related CNG channels (Flynn and Zagotta, 2001), it has been proposed that the selectivity filter may also form an additional gate to control ion permeation (see also Soler-Llavina et al., 2003). In this context, the above evidence for a conformational change in S6 during the transition from the resting closed state to the activated-not-open state, while the channels remain nonconductive, raises an interesting question: might the S6 gate begin to open as the channel enters the activated-not-open state, with the channels remaining nonconductive because of continued closure at the selectivity filter? Some recent results show that the inactivation ball and organic molecules as small as DMPS can be excluded from the pore by the lower S6 gate in the activated-not-open state (Webster et al., 2004; Pathak et al., 2005), suggesting that this gate remains closed. Since both DMPS and the inactivation ball are clearly larger than K+ ions, we decided to use a smaller probe to further test how securely closed the intracellular gate is in the activated-not-open state.
We have previously shown that in Shaker channels a cysteine introduced at position 474C, right above the bundle crossing, reacts with Cd2+ ions in a strongly state-dependent manner (Liu et al., 1997; del Camino and Yellen, 2001). We therefore studied the voltage dependence of Cd2+ reactivity with 474C Shaker channels that also contained the ILT mutations (V369I, I372L, and S376T)2. Fig. 4 first shows that the Q-V and g-V in these channels are clearly separated, allowing for the isolation of the activated-not-open state. Additionally, this figure shows that the second order rate of 474C Cd2+ modification closely follows the voltage dependence of activation, even at the voltages where most of the gating charge has moved but the open probability remains very low. Thus, at −40 mV the reaction rate is ∼60-fold slower than at +100 mV, and at only 10 mV more negative this difference increases to over 100-fold. At the foot of the Q-V, the modification rate is >1,000-fold slower than in the open state. These results clearly show that even in the activated-not-open state the intracellular S6 gate is capable of excluding ions as small as Cd2+ from the pore, and suggest that most likely the intracellular gate remains securely closed throughout the entire activation process until the final transition to the open state.
Although the gate remains securely closed to ion passage in this intermediate state, it may be that the increase in the rate of chemical modification at the 478C position occurs because of an increased probability that some but not all of the lower S6 helices have undergone a conformational change associated with opening (Chapman and VanDongen, 2005).
DISCUSSION
In the present study we used the S4-ILT mutations and the channel blocker 4-AP to isolate the Shaker channels in the activated-not-open state and to learn about the accessibility of cysteines introduced in the S6 helices to react with MTS reagents. Using these two experimental approaches, we studied whether there are conformational changes in the intracellular mouth of the pore before channel opening, and additionally we further tested the recently proposed model for the mechanism of action of 4-AP. This model establishes that 4-AP binds to the cavity and, once bound, allosterically stabilizes the activated-not-open conformation of the channel. Our initial characterization of the mechanism of action of 4-AP showed that this blocker can access its binding site only when the channels are open, can be trapped inside the channel, and competes with TEA for a binding site known to be located in the cavity of the channel. Furthermore, we found that 478C LT channels blocked with 4-AP reacted with MTS reagents at a very similar rate as when these channels were isolated in the activated-not-open state by holding them at −10 mV. All of these results strongly agree with the model of the mechanism of action proposed by Armstrong and Loboda, and thus clearly suggest that 4-AP is an open channel blocker that binds with higher affinity to the closed state. Although this property distinguishes 4-AP from the widely used QAs, there are other open channel blockers of the voltage-gated ion channels that exhibit a similar behavior. This is probably the case for the bradycardic agent ZD7288, a commonly used blocker of hyperpolarization-activated channels (Shin et al., 2001). Also, the CNG channel blocker tetracaine constitutes another example of a blocker that binds in pore with higher affinity for the closed state (Fodor et al., 1997).
Our results also show that the modification rate of 478C LT channels is faster in the activated-not-open state than in the resting closed or open states. The 478 residue is considered to form the gate of the channel and therefore it probably is very sensitive to conformational changes that might occur in the process of activation. The fact that the modification rate in the activated-not-open state is faster than in any other state suggests that it specifically reflects the structural properties of the intracellular mouth of the gate in this state; because the rate is not intermediate between that of the fully closed and fully open states, it cannot reflect simply a linear combination of the reaction rates of these states. Nevertheless, our results do not allow us to determine whether it is a conformational change of the S6 segments or of other adjacent parts of the protein that determines the increase in accessibility of 478C to react with MTS reagents.
For reference, Fig. 5 shows the location of Shaker positions 474 and 478 (as well as the lock-open positions 476 and 486) based on the open state structure of the closely related Kv1.2 channel, which was very recently reported by Long et al. (2005a)(b). It is easy to imagine that a small change in the conformation of the lower S6 could produce the observed changes in 478C accessibility. Moreover, the very close match in size between the hydrated K+ ion and the opening at the bundle crossing means that very small differences in the size of the opening (between channel subtypes or between conformations) could dramatically alter the rate of K+ access to the pore, and thus the single channel conductance (Ding and Horn, 2002; Webster et al., 2004). This impression is supported by recent results of Brelidze and Magleby (2005), indicating that even the high conductance BK channel has a capture radius of only 2.2 Å. This seems surprising given the physically large entrance seen for the comparably large conductance MthK channel (Jiang et al., 2002b); they reconcile these two observations by supposing that the capture process involves the large hydrated K+ ion. The effective diffusional capture radius is approximately the difference between the radius of the diffusional target, the pore entrance, and that of the diffusing particle (Ferry, 1936; Läuger, 1976), though the actual effective value depends on the physical details of the capture process (Andersen, 1983; Andersen and Feldman, 1996). The close fit seen in Fig. 5 corresponds to a capture radius approaching zero, which would make the capture rate of hydrated K+ and probably the single channel conductance a very steep function of the entrance size. If a small entrance requires the entering ion to undergo partial dehydration for passage into the cavity, the energy barrier would be much higher and thus the rate of permeation even lower.
In the future, it should be possible to observe the structure of the activated-not-open state directly, by crystallizing channels in the presence of 4-AP or crystallizing channels with the ILT mutations.
Finally, we showed that a cysteine introduced at position 474, located above the gate, is not accessible to react with a probe as small as Cd2+ ions in the activated-not-open state. Although in the transition from the resting closed state to the activated-not-open state there are some anticipatory conformational changes that affect the intracellular entrance of the pore, the S6 gate most likely remains as the main barrier that regulates the movement of ions across the pore.
Acknowledgments
We are especially grateful to Mr. John Dekker and Dr. Sarah Webster for stimulating discussions.
This work was supported by a grant from the National Institutes of Health/National Institute of Neurological Disorders and Stroke (NS 29693) to G. Yellen.
Olaf S. Andersen served as editor.
D. del Camino's present address is Synta Pharmaceuticals, 45 Hartwell Ave., Lexington, MA 02421.
M. Kanevsky's present address is Department of Anesthesia, Stanford University School of Medicine, 300 Pasteur Drive, H3580, Stanford, CA 94305.
Abbreviations used in this paper: 4-AP, 4-aminopyridine; MTSET, trimethylaminoethyl methanethiosulfonate; MTSHE, hydroxyethyl methanethiosulfonate.
Footnotes
The values of the rate constants for the open state reaction also agree reasonably well for channels with and without the LT mutations (∼1.2 × 105 for 478C-LT vs. ∼0.8 × 105 for 478C; the latter value from Liu et al., 1997; del Camino and Yellen, 2001).
The 474C-ILT channel opens at less extreme depolarized voltages than the wild-type ILT channel, so we used the ILT version of the 474C channel (rather than the LT version used for 478C).
References
- Andersen, O.S. 1983. Ion movement through gramicidin A channels: single-channel measurements at very high potentials. Biophys. J. 41:119–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen, O.S., and S.W. Feldman. 1996. The heterogeneous collision velocity for hydrated ions in aqueous solutions is 104 cm/s. J. Physiol. Chem. 100:4622–4629. [Google Scholar]
- Armstrong, C.M. 1966. Time course of TEA+-induced anomalous rectification in squid giant axons. J. Gen. Physiol. 50:491–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong, C.M. 1969. Inactivation of the potassium conductance and related phenomena caused by quaternary ammonium ion injection in squid axons. J. Gen. Physiol. 54:553–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong, C.M., and A. Loboda. 2001. A model for 4-aminopyridine action on K channels: similarities to tetraethylammonium ion action. Biophys. J. 81:895–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brelidze, T.I., and K.L. Magleby. 2005. Probing the geometry of the inner vestibule of BK channels with sugars. J. Gen. Physiol. 126:105–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman, M.L., and A.M. VanDongen. 2005. K channel subconductance levels result from heteromeric pore conformations. J. Gen. Physiol. 126:87–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman, M.L., H.M. VanDongen, and A.M. VanDongen. 1997. Activation-dependent subconductance levels in the drk1 K channel suggest a subunit basis for ion permeation and gating. Biophys. J. 72:708–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choquet, D., and H. Korn. 1992. Mechanism of 4-aminopyridine action on voltage-gated potassium channels in lymphocytes. J. Gen. Physiol. 99:217–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Camino, D., and G. Yellen. 2001. Tight steric closure at the intracellular activation gate of a voltage-gated K+ channel. Neuron. 32:649–656. [DOI] [PubMed] [Google Scholar]
- del Camino, D., M. Holmgren, Y. Liu, and G. Yellen. 2000. Blocker protection in the pore of a voltage-gated K+ channel and its structural implications. Nature. 403:321–325. [DOI] [PubMed] [Google Scholar]
- Ding, S., and R. Horn. 2002. Tail end of the s6 segment: role in permeation in shaker potassium channels. J. Gen. Physiol. 120:87–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle, D.A., J.H. Morais Cabral, R.A. Pfuetzner, A. Kuo, J. Gulbis, S. Cohen, B.T. Chait, and R. MacKinnon. 1998. The structure of the potassium channel: molecular basis of potassium conduction and selectivity. Science. 280:69–77. [DOI] [PubMed] [Google Scholar]
- Ferry, J.D. 1936. Statistical evaluation of sieve constants in ultrafiltration. J. Gen. Physiol. 20:95–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn, G.E., and W.N. Zagotta. 2001. Conformational changes in S6 coupled to the opening of cyclic nucleotide-gated channels. Neuron. 30:689–698. [DOI] [PubMed] [Google Scholar]
- Fodor, A.A., S.E. Gordon, and W.N. Zagotta. 1997. Mechanism of tetracaine block of cyclic nucleotide-gated channels. J. Gen. Physiol. 109:3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackos, D.H., T.H. Chang, and K.J. Swartz. 2002. Scanning the intracellular S6 activation gate in the Shaker K+ channel. J. Gen. Physiol. 119:521–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill, O.P., A. Marty, E. Neher, B. Sakmann, and F.J. Sigworth. 1981. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 391:85–100. [DOI] [PubMed] [Google Scholar]
- Holmgren, M., M.E. Jurman, and G. Yellen. 1996. N-type inactivation and the S4-S5 region of the Shaker K+ channel. J. Gen. Physiol. 108:195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmgren, M., P.L. Smith, and G. Yellen. 1997. Trapping of organic blockers by closing of voltage-dependent K+ channels: evidence for a trap door mechanism of activation gating. J. Gen. Physiol. 109:527–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmgren, M., K.S. Shin, and G. Yellen. 1998. The activation gate of a voltage-gated K+ channel can be trapped in the open state by an intersubunit metal bridge. Neuron. 21:617–621. [DOI] [PubMed] [Google Scholar]
- Hoshi, T., W.N. Zagotta, and R.W. Aldrich. 1990. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 250:533–538. [DOI] [PubMed] [Google Scholar]
- Hoshi, T., W.N. Zagotta, and R.W. Aldrich. 1994. Shaker potassium channel gating. I: Transitions near the open state. J. Gen. Physiol. 103:249–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, Y., A. Lee, J. Chen, M. Cadene, B.T. Chait, and R. MacKinnon. 2002. a. Crystal structure and mechanism of a calcium-gated potassium channel. Nature. 417:515–522. [DOI] [PubMed] [Google Scholar]
- Jiang, Y., A. Lee, J. Chen, M. Cadene, B.T. Chait, and R. MacKinnon. 2002. b. The open pore conformation of potassium channels. Nature. 417:523–526. [DOI] [PubMed] [Google Scholar]
- Jurman, M.E., L.M. Boland, Y. Liu, and G. Yellen. 1994. Visual identification of individual transfected cells for electrophysiology using antibody-coated beads. Biotechniques. 17:876–881. [PubMed] [Google Scholar]
- Kamb, A., J.C.L. Tseng-Crank, and M.A. Tanouye. 1988. Multiple products of the Drosophila Shaker gene may contribute to potassium channel diversity. Neuron. 1:421–430. [DOI] [PubMed] [Google Scholar]
- Kirsch, G.E., and J.A. Drewe. 1993. Gating-dependent mechanism of 4-aminopyridine block in two related potassium channels. J. Gen. Physiol. 102:797–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsch, G.E., J.Z. Yeh, and G.S. Oxford. 1986. Modulation of aminopyridine block of potassium currents in squid axon. Biophys. J. 50:637–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsch, G.E., C.C. Shieh, J.A. Drewe, D.F. Vener, and A.M. Brown. 1993. Segmental exchanges define 4-aminopyridine binding and the inner mouth of K+ pores. Neuron. 11:503–512. [DOI] [PubMed] [Google Scholar]
- Läuger, P. 1976. Diffusion-limited ion flow through pores. Biochim. Biophys. Acta. 455:493–509. [DOI] [PubMed] [Google Scholar]
- Ledwell, J.L., and R.W. Aldrich. 1999. Mutations in the S4 region isolate the final voltage-dependent cooperative step in potassium channel activation. J. Gen. Physiol. 113:389–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y., M. Holmgren, M.E. Jurman, and G. Yellen. 1997. Gated access to the pore of a voltage-dependent K+ channel. Neuron. 19:175–184. [DOI] [PubMed] [Google Scholar]
- Loboda, A., and C.M. Armstrong. 2001. Resolving the gating charge movement associated with late transitions in K channel activation. Biophys. J. 81:905–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long, S.B., E.B. Campbell, and R. MacKinnon. 2005. a. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 309:897–903. [DOI] [PubMed] [Google Scholar]
- Long, S.B., E.B. Campbell, and R. MacKinnon. 2005. b. Voltage sensor of Kv1.2: structural basis of electromechanical coupling. Science. 309:903–908. [DOI] [PubMed] [Google Scholar]
- López-Barneo, J., T. Hoshi, S.H. Heinemann, and R.W. Aldrich. 1993. Effects of external cations and mutations in the pore region on C-type inactivation of Shaker potassium channels. Receptors Channels. 1:61–71. [PubMed] [Google Scholar]
- McCormack, K., W.J. Joiner, and S.H. Heinemann. 1994. A characterization of the activating structural rearrangements in voltage-dependent Shaker K+ channels. Neuron. 12:301–315. [DOI] [PubMed] [Google Scholar]
- Pathak, M., L. Kurtz, F. Tombola, and E. Isacoff. 2005. The cooperative voltage sensor motion that gates a potassium channel. J. Gen. Physiol. 125:57–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perozo, E., R. MacKinnon, F. Bezanilla, and E. Stefani. 1993. Gating currents from a nonconducting mutant reveal open-closed conformations in Shaker K+ channels. Neuron. 11:353–358. [DOI] [PubMed] [Google Scholar]
- Russell, S.N., N.G. Publicover, P.J. Hart, A. Carl, J.R. Hume, K.M. Sanders, and B. Horowitz. 1994. Block by 4-aminopyridine of a Kv1.2 delayed rectifier K+ current expressed in Xenopus oocytes. J. Physiol. 481(Pt 3):571–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoppa, N.E., and F.J. Sigworth. 1998. Activation of Shaker potassium channels. III. An activation gating model for wild-type and V2 mutant channels. J. Gen. Physiol. 111:313–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin, K.S., B.S. Rothberg, and G. Yellen. 2001. Blocker state dependence and trapping in hyperpolarization-activated cation channels: evidence for an intracellular activation gate. J. Gen. Physiol. 117:91–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, R. M., and A.E. Martell. 1998. NIST Critically Selected Stability Constants of Metal Complexes Database. NIST Standard Reference Database 46 [v. 5.0]. US Department of Commerce.
- Smith-Maxwell, C.J., J.L. Ledwell, and R.W. Aldrich. 1998. Uncharged S4 residues and cooperativity in voltage-dependent potassium channel activation. J. Gen. Physiol. 111:421–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soler-Llavina, G.J., M. Holmgren, and K.J. Swartz. 2003. Defining the conductance of the closed state in a voltage-gated K+ channel. Neuron. 38:61–67. [DOI] [PubMed] [Google Scholar]
- Stefani, E., and F. Bezanilla. 1998. Cut-open oocyte voltage-clamp technique. Methods Enzymol. 293:300–318. [DOI] [PubMed] [Google Scholar]
- Taglialatela, M., L. Toro, and E. Stefani. 1992. Novel voltage clamp to record small, fast currents from ion channels expressed in Xenopus oocytes. Biophys. J. 61:78–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagoner, P.K., and G.S. Oxford. 1990. Aminopyridines block an inactivating potassium current having slow recovery kinetics. Biophys. J. 58:1481–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster, S.M., D. del Camino, J.P. Dekker, and G. Yellen. 2004. Intracellular gate opening in Shaker K+ channels defined by high-affinity metal bridges. Nature. 428:864–868. [DOI] [PubMed] [Google Scholar]
- Yang, Y., Y. Yan, and F.J. Sigworth. 1997. How does the W434F mutation block current in Shaker potassium channels? J. Gen. Physiol. 109:779–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yellen, G. 2002. The voltage-gated potassium channels and their relatives. Nature. 419:35–42. [DOI] [PubMed] [Google Scholar]
- Yellen, G., M.E. Jurman, T. Abramson, and R. MacKinnon. 1991. Mutations affecting internal TEA blockade identify the probable pore forming region of a K+ channel. Science. 251:939–942. [DOI] [PubMed] [Google Scholar]
- Zagotta, W.N., T. Hoshi, and R.W. Aldrich. 1994. a. Shaker potassium channel gating. III: Evaluation of kinetic models for activation. J. Gen. Physiol. 103:321–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagotta, W.N., T. Hoshi, J. Dittman, and R.W. Aldrich. 1994. b. Shaker potassium channel gating. II: Transitions in the activation pathway. J. Gen. Physiol. 103:279–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, J., and F.J. Sigworth. 1998. Intermediate conductances during deactivation of heteromultimeric shaker potassium channels. J. Gen. Physiol. 112:457–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, M., J.H. Morais-Cabral, S. Mann, and R. MacKinnon. 2001. a. Potassium channel receptor site for the inactivation gate and quaternary amine inhibitors. Nature. 411:657–661. [DOI] [PubMed] [Google Scholar]
- Zhou, Y., J.H. Morais Cabral, A. Kaufman, and R. MacKinnon. 2001. b. Chemistry of ion hydration and coordination revealed by a K+ channel-Fab complex at 2.0 Å resolution. Nature. 414:43–48. [DOI] [PubMed] [Google Scholar]