

Abstract
CCAAT enhancer binding protein β (C/EBPβ) plays an essential role in the cascade that triggers adipocyte differentiation. C/EBPβ activates transcription of C/EBPα and peroxisome proliferator-activated receptor-γ, transcriptional activators of genes that give rise to the adipocyte phenotype. Sequential phosphorylation of C/EBPβ/liver activating protein (LAP) on Thr188 by MAPK and on Ser184 or Thr179 by glycogen synthase kinase β (GSK3β) is required for acquisition of DNA binding activity and transcriptional activation. To investigate how phosphorylation and dimerization of C/EBPβ/LAP alter these activities, wild-type (Wt) and mutant rC/EBPβs were prepared and purified to assess DNA binding and transcription in cell-free systems. rC/EBPβ/LAP, phosphorylated by MAPK and GSK3β in vitro, produced a >100-fold increase in DNA binding activity. Mutation of the phosphorylation to Glu increased DNA binding activity. Using a cell-free transcription system with nuclear extract from 3T3-L1 preadipocytes and rC/EBPβ/LAP, only doubly phosphorylated rC/EBPβ/LAP (by MAPK and GSK3β) activated transcription driven by Wt C/EBPα, 422/aP2, and SCD1 promoters. Oxidation-induced dimerization of doubly phosphorylated Wt rC/EBPβ/LAP increased DNA binding, whereas unphosphorylated Wt rC/EBPβ/LAP lacked DNA binding activity. Mutation of the C-terminal Cys296 adjacent to the leucine zipper and Cys143 just upstream of the DNA binding domain eliminated phosphorylation-, oxidation-, and dimerization-dependent DNA binding activity, whereas mutation of Cys201 within the basic DNA binding domain had little effect on DNA binding. These findings indicate that dual phosphorylation of C/EBPβ/LAP caused a conformational change that facilitates S–S bond formation and dimerization, rendering the basic region accessible to the C/EBP regulatory element.
Keywords: adipocyte differentiation, disulfide bond, transactivation
Differentiation of growth-arrested 3T3-L1 preadipocytes into adipocytes is induced by hormonal stimuli that trigger expression of CCAAT enhancer binding protein β (C/EBPβ) (1–3) and, thereby, a transcriptional cascade that gives rise to the adipocyte phenotype. C/EBPβ activates the expression of the genes for two pleiotropic transcription factors, C/EBPα and PPARγ (4, 5), which in turn activate the expression of numerous adipogenic genes. Although maximal expression of C/EBPβ occurs rapidly (within 2–4 h) on induction of differentiation and remains at this level over the next 48 h, the acquisition of DNA binding activity by C/EBPβ required for the expression of C/EBPα and PPARγ only occurs after a long lag period of ≈12–14 h (1, 2, 6). At the end of the lag period, growth-arrested preadipocytes synchronously enter the cell cycle and undergo several rounds of mitotic clonal expansion (2, 3), a process required for subsequent differentiation. It is believed that chromatin remodeling occurs during mitotic clonal expansion and is a prerequisite of terminal differentiation. Presumably, this delay prevents the premature expression of C/EBPα, which is antimitotic, and allows mitotic clonal expansion and chromatin remodeling to occur before its expression (5, 7).
We recently showed that C/EBPβ must undergo sequential phosphorylation to acquire DNA binding ability (6). Immediately after the expression of C/EBPβ, a “priming” phosphorylation occurs on Thr188 catalyzed by MAP kinase, which is required but is insufficient for acquisition of DNA binding activity. After the lag period (at the G1 → S phase checkpoint), glycogen synthase kinase β (GSK3β) translocates from the cytoplasm into the nucleus and phosphorylates C/EBPβ on Ser184 or Thr179, giving rise to DNA binding activity (6). These and other findings suggest that dual phosphorylation of C/EBPβ induces a conformational change that leads to acquisition of DNA binding activity possibly by exposing the DNA binding domain.
An impediment to investigating the mechanism by which phosphorylation alters the DNA binding activity of C/EBPβ is the complexity of the mixture of leucine zipper proteins in nuclear extracts of 3T3-L1 cells. This mixture includes several C/EBP family members that are capable of forming homo- and heterodimers, including three C/EBPβ isoforms [i.e., flC/EBPβ (≈39 kDa), C/EBPβ/liver activating protein (LAP) (≈38 kDa), and dominant-negative C/EBPβ/liver inhibiting protein (LIP) (≈18 kDa)] (8); C/EBPδ (9, 10); and dominant-negative CHOP-10 (11). All of these factors are found in nuclear extract of 3T3-L1 cells after the induction of differentiation. Because these leucine zipper transcription factors can homo- and heterodimerize (12), interpretation of binding data with nuclear extracts becomes difficult. To circumvent these difficulties, the present investigation used purified rC/EBPβ/LAP to evaluate the effects of phosphorylation and -SS-bond-induced dimerization on the DNA binding activity and transactivation capacity. Our findings indicate that the dual phosphorylation of C/EBPβ/LAP leads to a conformational change that facilitates dimerization, rendering the DNA binding domain accessible to the C/EBP regulatory element in DNA.
Results and Discussion
Recombinant C/EBPβ/LAP Is Phosphorylated by MAPK and GSK3β.
GST-rC/EBPβ/LAP was prepared by using an expression system that produced a substantial quantity of the protein in the native state. The protein was partially purified by GSH-agarose chromatography, followed by cleavage with PreScission protease and final purification to homogeneity by CM-Sepharose ion exchange chromatography (Fig. 1A). The apparent molecular mass of rC/EBPβ/LAP was 34 kDa (or 35 kDa when phosphorylated by MAPK). Recombinant C/EBPβ/LAP is phosphorylated by MAPK alone and is further phosphorylated by GSK3β, but only after a “priming” phosphorylation by MAPK (Fig. 1B).
Fig. 1.
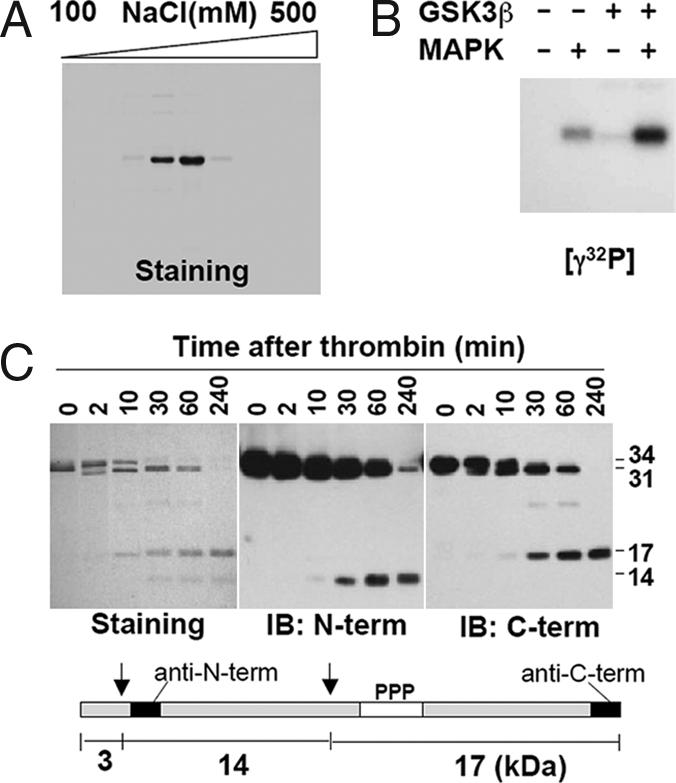
Purification and in vitro phosphorylation of rC/EBPβ/LAP. (A) After digestion of GST-rC/EBPβ/LAP with PreScission Protease, the rC/EBPβ/LAP was subjected to CM-Sepharose ion-exchange column chromatography and eluted by increasing [NaCl]. An aliquot (5 μl) of each fraction was subjected to SDS/12% PAGE, and the gel was stained with Coomassie Brilliant Blue G-250. (B) Purified rC/EBPβ/LAP was phosphorylated with [γ-32P]ATP, MAPK, and/or GSK3β and subjected to SDS/PAGE. (C) (Upper) rC/EBPβ/LAP was incubated with thrombin (1 unit) for the indicated time, subjected to SDS/12% PAGE, and stained or immunoblotted with anti-C/EBPβ/LAP N- or C-terminal antibody. (Lower) Schematic representation of the location of thrombin cleavage and phosphorylation sites.
In experiments designed to fragment rC/EBPβ/LAP, we determined that cleavage by thrombin occurs at two sites, giving rise to 3-, 14-, and 17-kDa fragments. As illustrated in Fig. 1C, three protein species of 34-, 31-, and 17-kDa molecular mass were evident by protein staining of SDS/PAGE gels during the course of proteolysis (Fig. 1C). The 31- and 14-kDa fragments were identified on SDS/PAGE gels with anti-C/EBPβ/LAP N-terminal antibody, whereas the 31- and 17-kDa fragments formed during proteolysis were identified with anti-C/EBPβ/LAP C-terminal antibody. The N- and C-terminal epitopes of the antibodies are shown at the bottom of Fig. 1C. The phosphorylation sites (Ser184, Thr179, and Thr188) are located in the C-terminal 17-kDa fragment.
DNA Binding Activity Is Increased by Dual Phosphorylation by MAPK and GSK3β.
Recombinant C/EBPβ/LAP was phosphorylated by MAPK and/or GSK3β as described in Fig. 1B and then subjected to electrophoretic mobility shift assay (EMSA) with a labeled oligonucleotide corresponding to the C/EBP regulatory element in the C/EBPα gene promoter. Substantial DNA binding was detected with only 1 ng¶ of rC/EBPβ/LAP after dual phosphorylation by MAPK and GSK3β, with virtually no binding occurring in the absence of either (Fig. 2A). Although increasing the level of rC/EBPβ/LAP by 10-fold led to detectable DNA binding, even with either MAPK or GSK3β alone, dual phosphorylation of rC/EBPβ/LAP by both kinases led to a ≥100-fold in DNA binding than by either kinase alone. It should be noted that previous mass spectrometric analysis showed conclusively that dual phosphorylation by MAPK and GSK3β occurs at Thr188 and either Ser184 or Thr179 (6).
Fig. 2.

DNA binding activity is increased by dual phosphorylation. (A) rC/EBPβ/LAP was phosphorylated by MAPK and/or GSK3β and analyzed by EMSA by using a nucleotide corresponding to the C/EBP regulatory element in C/EBPα promoter as a probe. (B) EMSA with the C/EBP binding-site probes present in various adipocyte-specific promoters. Nuclear extract (NE) from 3T3-L1 cells (4 or 24 h after induction of differentiation) or rC/EBPβ/LAP was incubated with MAPK (M) and/or GSK3β (G) and different C/EBP-site probes and then subjected to EMSA. (C) Wt or phosphorylation-site mutants of rC/EBPβ/LAPs [Thr179, Ser184, or Thr188 mutated to Ala (A) or Glu (E)] were phosphorylated with MAPK and/or GSK3β as in A and B and subjected to EMSA.
These findings were verified with the C/EBP regulatory elements of several other promoters of genes that are transactivated by members of the C/EBP family of transcription factors. DNA binding activities were measured with nuclear extract from 3T3-L1 cells obtained at 4 h (before acquisition of DNA binding activity) or 24 h (after acquisition of DNA binding activity) (1, 6) after the induction of differentiation (Fig. 2B). The C/EBP regulatory elements of the C/EBPα, aP2/422, SCD1, PPARγ, and GLUT4 gene promoters exhibited patterns similar to that of the C/EBPα promoter element (i.e., stronger binding activity by 24- than by 4-h nuclear extracts). Further, as shown in Fig. 2B, rC/EBPβ/LAP exhibits DNA binding to this group of promoter elements only after dual phosphorylation by MAPK and GSK3β.
We also determined the effect of mutating the phosphorylation site Thr and Ser residues to Glu in an attempt to constitutively mimic the effect of phosphorylation. The three phosphorylation sites, Thr179, Ser184, and Thr188, were mutated to Ala or Glu. In a previous report we showed that mutation of Thr188 to Ala abolished the DNA binding response to dual phosphorylation by MAPK and GSK3β (6). Doubly mutated Thr179/Thr188 to Glu179/Glu188 (ESE mutant) provoked increased DNA binding relative to the AAA or TSA mutations, albeit less than that of the dually phosphorylated wild-type (Wt) construct (Fig. 2C). ESE mutant exhibited higher DNA binding activity than TEE mutant. As previously shown by LC-MS/MS analysis, C/EBPβ/LAP is dually phosphorylated on either Thr179/Thr188 or Ser184/Thr188. However, the triply phosphorylated form has never been detected (6). Therefore, of the two possible combinations of dual phosphorylation, Thr179/Thr188 appears to be more effective than Thr184/Thr188 in activating DNA binding. Although dual-Glu substitutions increase DNA binding, they do not achieve the activity of the dually phosphorylated form presumably because Glu mutants do not produce the optimal conformation required for maximal DNA binding activity. Taken together, however, these results show that dual phosphorylation of C/EBPβ/LAP is required for increased binding to adipocyte gene-specific promoters.
Cell-Free Transcription of Adipocyte-Specific Promoters Is Activated by Dual Phosphorylation.
Although exposure of 3T3-L1 preadipocytes to inhibitors of MAPK or GSK3β prevents C/EBPβ/LAP-dependent processes (i.e., mitotic clonal expansion and terminal differentiation) (6), questions remain as to whether these effects are due solely to a blockade of C/EBPβ phosphorylation. To directly assess the effect of phosphorylation of C/EBPβ/LAP on transcriptional activation, we used the cell-free transcription system used previously in our laboratory (13). Nuclear extracts from 3T3-L1 preadipocytes or adipocytes provided the basal transcriptional machinery along with rC/EBPβ/LAP that had (or had not) been phosphorylated by MAPK and/or GSK3β. The promoters of several adipocyte-specific genes [i.e., the C/EBPα (4), aP2/422 (13), and SCD1 (14) genes] were fused to a luciferase reporter and served as DNA templates. The resulting RNA transcripts were analyzed by PCR. In the absence of nuclear extract or rC/EBPβ/LAP, no PCR product was detected (Fig. 3A), indicating the effectiveness of removal of the luciferase template with DNase1. In the presence of nuclear extract from day-2 3T3-L1 cells, transcription driven by the aP2/422 promoter was increased compared with that from day-0 cells, consistent with the fact that aP2/422 is expressed by 3T3-L1 preadipocytes 2 days after the induction of differentiation (5). To determine the effect of dual phosphorylation, rC/EBPβ/LAP was first incubated with and/or without MAPK and GSK3β and then with day-0 preadipocyte nuclear extract and the other components of the cell-free transcription system. As shown in Fig. 3A, transcription was activated by ≥10-fold after phosphorylation by MAPK and GSK3β. Thus, dual phosphorylation of rC/EBPβ/LAP (Fig. 1B) markedly increased transcription. An unrelated viral promoter (SV40-driven transcript), included as an internal control, exhibited ±1.5-fold variation between the groups based on the real-time PCR (data not shown). Other adipocyte-specific promoters, including those from the C/EBPα or SCD1 genes, were also tested and exhibited substantially increased transcription only when rC/EBPβ/LAP was phosphorylated by both MAPK and GSK3β (Fig. 3B). These findings indicate that dual phosphorylation of C/EBPβ/LAP by MAPK and GSK3β is not only required for DNA binding activity, but also for transcriptional activation.
Fig. 3.
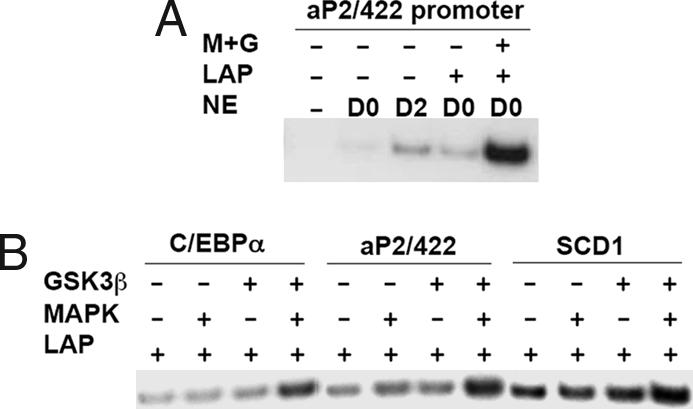
Cell-free transcription of promoter-luciferase genes in the presence of rC/EBPβ/LAP (phosphorylated or not by MAPK and/or GSK3β) and nuclear extract. Nuclear extracts (NE) were from 3T3-L1 cells before (day 0) or after (day 2) induction of differentiation. (A) After incubation with the aP2/422 promoter-luciferase construct, the resulting transcript was isolated and quantified by RT-PCR. (B) Promoter-reporter genes in which the promoters of various adipocyte genes (the C/EBPα, aP2/422, and SCD1 genes) were subjected to cell-free transcription analysis with day-0 nuclear extract and doubly phosphorylated rC/EBPβ/LAP as described in A.
Dual Phosphorylation Induces a Conformational Change That Facilitates Dimerization and DNA Binding.
In studies with an analogous model system, McKnight's group used a short C-terminal segment of C/EBPα encompassing the leucine zipper and adjacent DNA binding domain. Their findings suggested that C/EBPα undergoes conformation-induced dimerization of the leucine zipper during the binding and release of DNA (15). We considered the possibility that dual phosphorylation of C/EBPβ/LAP might induce a similar conformational change that facilitates dimerization through the C-terminal leucine zipper and, thereby, DNA binding (6). To address this hypothesis, rC/EBPβ/LAP was phosphorylated and cross-linked with glutaraldehyde. As illustrated in Fig. 4A, dimer was generated within 30 sec of exposure to the cross-linking agent and was markedly facilitated by dual phosphorylation. At longer times of exposure to glutaraldehyde, rC/EBPβ/LAP formed multimers as well as dimers, which was also increased by phosphorylation. These results suggested that phosphorylation promoted the interaction between rC/EBPβ/LAP monomers, supporting the occurrence of a conformational change. Given that rC/EBPβ/LAP exists in equilibrium between monomeric and dimeric forms, phosphorylation appeared to shift the equilibrium toward dimer, thereby promoting binding to the cognate C/EBP regulatory element. Because the use of glutaraldehyde precluded DNA binding analysis, we sought another approach to investigate the role of dimerization in DNA binding (see next paragraph).
Fig. 4.

Effect of phosphorylation and dimerization on the DNA binding activity of rC/EBPβ/LAP. (A) rC/EBPβ/LAP was phosphorylated or not with MAPK and GSK3β and then treated with 0.05% glutaraldehyde for the indicated times. Proteins were resolved by SDS/10% PAGE and immunoblotted with anti-C/EBPβ N-terminal antibody (see Fig. 1C Lower). (B) rC/EBPβ/LAP (1 ng) doubly phosphorylated or not with MAPK and GSK3β was mildly oxidized with 5 mM GSSG for the times indicated, the reaction terminated with 20 mM iodoacetamide, and the reaction mixture subjected to nonreducing SDS/PAGE (50 ng) or EMSA (5 ng). (C) Unphosphorylated (N) or doubly phosphorylated (P) rC/EBPβ/LAP was incubated with 5 mM GSSG at room temperature for 30 min and then treated with 1 unit of thrombin. Samples were resolved by nonreducing SDS/12% PAGE gel and immunoblotted with anti-C/EBPβ C-terminal antibody. The arrow on the right identifies the 34-kDa (17-kDa/17-kDa) dimer of the doubly phosphorylated C-terminal thrombin cleavage products of the 56-kDa homodimer (upper arrow on left) or residual uncross-linked monomer (lower arrow on left).
Further evidence for a phosphorylation-induced conformational change/dimerization derives from studies in which rC/EBPβ/LAP was exposed to mild oxidizing conditions with oxidized glutathione (GSSG). It is of interest that C/EBPβ/LAP possesses five sulfhydryl groups (−SH), one of which is C-terminal and abuts the leucine zipper dimerization domain. Thus, this –SH group pair in the C/EBPβ/LAP homodimer might be expected to readily undergo oxidation to form an interchain disulfide bond that would stabilize the leucine zipper dimer and create a DNA binding pocket. Thus, we tested the effect of GSSG-induced oxidation of rC/EBPβ/LAP on DNA binding activity before and after dual phosphorylation. When rC/EBPβ/LAP was exposed to GSSG, both the un- and dual-phosphorylated proteins were converted to dimers (Fig. 4B). Of interest, however, only doubly phosphorylated rC/EBPβ/LAP exhibited DNA binding activity on dimerization. Moreover, phosphorylation status alone appeared to have little effect on dimerization caused by GSSG. The close correlation between dimerization and DNA binding activity of GSSG-treated C/EBPβ/LAP suggests that dimerization is necessary, but without phosphorylation is not sufficient to induce a conformation that will bind DNA. Rather, dual phosphorylation is necessary to induce the “active” conformation. Moreover, comparison of the extent of dimerization of unphosphorylated versus phosphorylated rC/EBPβ/LAP suggests that dual phosphorylation increases the extent of dimerization (Fig. 4B Upper).
Because C/EBPβ/LAP possesses five cysteines, numerous combinations of disulfide bonds between the two monomeric molecules are possible. To minimize the number of possible combinations of cysteines involved, we took advantage of the fact that C/EBPβ/LAP can be digested with thrombin to generate N- and C-terminal halves of molecules that have two and three cysteines, respectively (Fig. 1C). Thus, if two C/EBPβ/LAP molecules are connected through any pair of cysteinyl–SH residues, the resulting 17 kDa from the C-terminal segment would produce a 34-kDa dimer with nonreducing SDS/PAGE (see Fig. 4D). Indeed, digestion of phosphorylated and GSSG-oxidized rC/EBPβ/LAP with thrombin produced a 34-kDa band that was not observed in unphosphorylated rC/EBPβ/LAP (Fig. 4C). This result indicates that only three cysteinyl–SHs (see Fig. 4D Lower) could have given rise to the S–S cross-links in the full-length native molecule that produced the 34-kDa dimer.
Effect of Mutating Cysteine Residues on DNA Binding Activity.
We focused our efforts on the three cysteine residues (Cys143, Cys201, and Cys296) located in the C-terminal segment after thrombin cleavage. The most obvious candidate for S–S bond formation in a dimer is C296 because this SH group is perfectly juxtaposed next to the leucine zipper at the C terminus. Each of the three cysteines in the C-terminal segment of rC/EBPβ/LAP was mutated to serine (C143S, C201S, and C296S), expressed and purified along with Wt as described in Fig. 1. To ascertain which of these cysteines is important in GSSG-induced disulfide bond formation, and thereby DNA binding activity, EMSA was conducted before and after phosphorylation. The mutant and Wt rC/EBPβ/LAP proteins were equally phosphorylated by MAPK and GSK3β (Fig. 5A). When subjected to oxidation with GSSG, the dimerized rC/EBPβ/LAPs were still observed on the nonreducing SDS/PAGE presumably because of disulfide bond formation through the remaining cysteine residues (data not shown). However, the DNA binding activity was markedly reduced by the C143S and C296S mutations (Fig. 5A), indicating that the cysteine residues that bracket the DNA binding domain are responsible for the oxidation-induced increase of DNA binding activity. This assertion was verified by the finding that the double Cys143/296 mutant exhibited even lower DNA binding activity after dual phosphorylation and GSSG-induced oxidation (Fig. 5B). Taken together, these findings indicate that phosphorylation of rC/EBPβ/LAP by MAPK and GSK3β produces a conformational change that favors dimerization. Although it is not known whether disulfide bond formation occurs in intact cells and is required for DNA binding in vivo, oxidation of rC/EBPβ/LAP in vitro, together with dual phosphorylation, promotes even stronger DNA binding.
Fig. 5.

Effect of cysteine mutations (Cys143, Cys201, Cys296 singly mutated and Cys143, Cys296 doubly mutated) in rC/EBPβ/LAP on DNA-binding activity. The Cys to Ser mutant recombinant proteins and Wt proteins were prepared and purified to homogeneity. Identical amounts of the proteins were subjected to protein staining, phosphorylation with [γ-32P] or unlabeled ATP, and EMSA. (A) Effect of single Cys mutations in rC/EBPβ/LAP. (B) Effect of double Cys mutations in rC/EBPβ/LAP.
Model.
A model (shown in Fig. 6) is proposed that accounts for the experimental results presented here and previously (6). We visualize that C/EBPβ/LAP is phosphorylated by MAPK immediately (≤2 h) after induction of differentiation, followed much later (≈14 h) by phosphorylation by GSK3β at which point a conformational change occurs that renders the leucine zipper of monomeric C/EBPβ/LAP accessible for dimerization. Dimerization would be expected to bring together the basic regions of the two monomers to create a “scissors-like” DNA binding pocket just above the coiled-coil leucine zipper (Fig. 6) as proposed by McKnight (16). The effect of dimerization on DNA binding is supported by the fact that mild oxidation by GSSG creates a disulfide-linked dimer, which, when disrupted by mutation of Cys143 and Cys296 (Fig. 5B), markedly decreases DNA binding (Fig. 6). In the cell where disulfide links would not be expected because of a reducing environment, C/EBPβ/LAP would exist in a monomer–dimer equilibrium that would be shifted to “active” dimer by dual phosphorylation. It should be noted that under reducing conditions in vitro [i.e., in the presence of 1 mM dithiothreitol (DTT)], despite the disruption of disulfide bond formation, DNA binding occurs but only after dual phosphorylation (Fig. 2A). The monomer–dimer equilibrium would be further shifted to dimer when DNA becomes locked into the basic DNA binding domain by the scissors grip proposed by McKnight (16).
Fig. 6.

Effect of phosphorylation- and dimerization-induced conformational changes on the DNA binding activity of C/EBPβ/LAP. Visualized are the effects of (i) the phosphorylation by MAPK and GSK3β on the conformation and dimerization of C/EBPβ/LAP monomer, and (ii) the GSSG-induced Cys-oxidation/dimerization on the monomer–dimer equilibrium of doubly phosphorylated C/EBPβ/LAP. The size of the DNA symbol is intended to indicate the tightness of DNA binding.
Although it is generally assumed but not proven that the nuclear environment is poised in a reducing state, it would be of interest to ascertain whether changes in redox state occur in the nucleus physiologically and whether such changes could affect disulfide-linked dimerization of C/EBPβ/LAP.
Materials and Methods
Purification and in Vitro Phosphorylation of rC/EBPβ/LAP.
The cDNA encoding C/EBPβ/LAP was cloned into pGEX-6P vector (GE Healthcare, Piscataway, NJ) and transformed into BL21(DE3)pLysS (Novagen, Madison, WI). Mutations described were introduced by using the QuikChange site-directed mutagenesis kit (Invitrogen, Carlsbad, CA). The GST-fusion protein was expressed, purified with glutathione agarose column, cleaved with PreScrisson Protease (GE Healthcare), and applied directly to the column. The released recombinant protein was then further purified by CM-Sepharose ion-exchange chromatography (GE Healthcare). rC/EBPβ/LAP was eluted with 250∼300 mM NaCl and stored at −80°C until use. Where indicated, the protein was treated with 1 unit of thrombin (GE Healthcare). For phosphorylation 500 ng of Wt or mutant rC/EBPβ was incubated with activated MAPK and/or GSK3β in 50 mM Hepes (pH 7.6), 10 mM MgCl2, 0.5 mM ATP, and 1 mM DTT at 30°C for 30 min. Where indicated, 5 μCi of [γ-32P]ATP was included.
Cell Culture, Induction of Differentiation, and Nuclear Extract Preparation.
Differentiation of postconfluent 3T3-L1 preadipocytes (on day 0) was as described (17). Nuclear extracts were prepared with NUN buffer (1, 18) containing 1 mM DTT for EMSA. For cell-free transcription assay, nuclear extracts were prepared from 2-day postconfluent undifferentiated preadipocytes or from adipocyte 2 days after induction of differentiation (day 2) by a modification (13) of the protocol of Dignam (19). Nuclear extracts were dialyzed in 25 mM Hepes (pH 7.6), 0.1 mM EDTA, 40 mM KCl, 10% glycerol, and 1 mM DTT and had a final protein concentration of 3∼4 mg/ml.
EMSA.
EMSA was performed essentially as described (1). Briefly, labeled probe was incubated with rC/EBPβ/LAP (1∼10 ng) or nuclear extract (5 μg) in a reaction mixture containing 10 mM Hepes (pH 7.6), 1 mM EDTA, 7% glycerol, 0.2 (for recombinant protein) or 1.0 (for crude nuclear extract) μg of poly(dIdC), 5 μg of BSA, 100 mM NaCl, and 1 mM DTT. In oxidation-induced binding experiments, DTT was omitted and 5 mM oxidized GSSG [prepared in 10 mM Hepes (pH 7.6)] was added. The C/EBP-binding sequences of adipocyte-specific genes were the following: C/EBPα (4), GGCGT TGCGC CACGA TCTCT CT; aP2/422 (20), CCAAA GTTGA GAAAT TTCTA TTAA; SCD1 (21), AGGGG GCTGA GGAAA TACTG AACA; PPARγ (22), ACTGC AATTT TAAAA AGCAA TCAA; and GLUT4 (23), TCAAT TCTTT CAGAA ATTTC GCAG. C/EBP-binding sequences are italic.
Cell-Free Transcription.
Cell-free transcription was carried out as described (13, 24) with modification. Briefly, 40 μg of nuclear extract and/or 40 ng of rC/EBPβ/LAP were incubated with template DNA (experimental promoter and control; viral promoter: 500 ng and 50 ng, respectively) on ice for 10 min after which transcription was initiated by adding 0.6 mM each of ATP, GTP, CTP, and UTP in 40 μl containing 10 mM Hepes (pH 7.6), 3% glycerol, 25.5 mM KCl, 6 mM MgCl2, 1 mM DTT, and 10 units of RNasin (Promega, Madison, WI). After a 1-h incubation at 37°C, templates were digested by 20 units of RNase-free DNase I (Roche Diagnostics, Mannheim, Germany) for 30 min at 37°C and 370 μl containing 20 mM Tris·HCl (pH 7.5), 5 mM EDTA, 1% SDS, 250 mM NaCl, 5 μg of tRNA, and 20 μg of proteinase K was added. RNA was recovered by phenol/chloroform extraction, ethanol precipitation, and centrifugation. The washed pellet was reverse-transcribed by using random hexamers and subjected to PCR in the presence of 2 μCi of [α-32P]dCTP for 15 cycles to detect 150 bp of luciferase gene product. Template DNAs were promoters of C/EBPα (−205 bp) (4), 422/aP2 (−858 bp) (13), and SCD1 (−363 bp) (13) fused to the luciferase gene. An SV40 viral promoter construct was used for internal standardization. Transcription products were detected by using real-time PCR.
Glutaraldehyde Cross-Linking or Oxidation of C/EBPβ, Nonreducing SDS/PAGE, and Western Blotting.
After phosphorylation, rC/EBPβ was cross-linked in the presence of 0.05% glutaraldehyde. Briefly, 50 ng of protein was mixed with glutaraldehyde and incubated at room temperature for the indicated time. The reaction was stopped with SDS-containing loading buffer and subjected to SDS/12% PAGE. Similarly, rC/EBPβ was treated with GSSG followed by 20 mM iodoacetamide. Samples were run in 10% NuPAGE (Invitrogen) without reducing agent. Gels were transferred onto nitrocellulose membrane and immunoblotted with the appropriate antibodies.
Acknowledgments
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases/National Institutes of Health Grants DK38418 and DK66627 (to M.D.L.). Q.-Q.T. was supported by Program for Outstanding Medical Academic Leaders Grant B-LJ06032 and National Key Basic Research Project Grant 2006CB943700.
Abbreviations
- C/EBP
CCAAT enhancer binding protein
- LAP
liver activating protein
- LIP
liver inhibiting protein
- MAPK
mitogen-activated protein kinase
- GSK
glycogen synthase kinase β
- DTT
dithiothreitol
- GSSG
oxidized glutathione
- EMSA
electrophoretic mobility shift assay
- Wt
wild type.
Footnotes
The authors declare no conflict of interest.
¶As quantified by comparative EMSA experiments, 1 ng of pure rC/EBPβ/LAP is equivalent to the amount of C/EBPβ/LAP present in 5 μg of nuclear extract protein from 3T3-L1 cells 24 h after the induction of differentiation.
References
- 1.Tang Q-Q, Lane MD. Genes Dev. 1999;13:2231–2241. doi: 10.1101/gad.13.17.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tang Q-Q, Otto TC, Lane MD. Proc Natl Acad Sci USA. 2003;100:44–49. doi: 10.1073/pnas.0137044100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tang Q-Q, Otto TC, Lane MD. Proc Natl Acad Sci USA. 2003;100:850–855. doi: 10.1073/pnas.0337434100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Christy RJ, Kaestner KH, Geiman DE, Lane MD. Proc Natl Acad Sci USA. 1991;88:2593–2597. doi: 10.1073/pnas.88.6.2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.MacDougald OA, Lane MD. Annu Rev Biochem. 1995;64:345–373. doi: 10.1146/annurev.bi.64.070195.002021. [DOI] [PubMed] [Google Scholar]
- 6.Tang Q-Q, Grønborg M, Huang H, Kim JW, Otto TC, Pandey A, Lane MD. Proc Natl Acad Sci USA. 2005;102:9766–9771. doi: 10.1073/pnas.0503891102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Otto TC, Lane MD. Crit Rev Biochem Mol Biol. 2005;40:229–242. doi: 10.1080/10409230591008189. [DOI] [PubMed] [Google Scholar]
- 8.An MR, Hsieh CC, Reisner PD, Rabek JP, Scott SG, Kuninger DT, Papaconstantinou J. Mol Cell Biol. 1996;16:2295–2306. doi: 10.1128/mcb.16.5.2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cao Z, Umek RM, McKnight SL. Genes Dev. 1991;5:1538–1552. doi: 10.1101/gad.5.9.1538. [DOI] [PubMed] [Google Scholar]
- 10.Lane MD, Jiang M-S, Tang Q-Q. Pennington Center Nutrition Series: Nutrition, Genetics and Obesity. 1999;9:459–476. [Google Scholar]
- 11.Tang Q-Q, Lane MD. Proc Natl Acad Sci USA. 2000;97:12446–12450. doi: 10.1073/pnas.220425597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Williams SC, Cantwell CA, Johnson PF. Genes Dev. 1991;5:1553–1567. doi: 10.1101/gad.5.9.1553. [DOI] [PubMed] [Google Scholar]
- 13.Cheneval D, Christy RJ, Geiman D, Cornelius P, Lane MD. Proc Natl Acad Sci USA. 1991;88:8465–8469. doi: 10.1073/pnas.88.19.8465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Landschulz KT, Jump DB, MacDougald OA, Lane MD. Biochem Biophys Res Commun. 1994;200:763–768. doi: 10.1006/bbrc.1994.1516. [DOI] [PubMed] [Google Scholar]
- 15.Shuman JD, Vinson CR, McKnight SL. Science. 1990;249:771–774. doi: 10.1126/science.2202050. [DOI] [PubMed] [Google Scholar]
- 16.Vinson CR, Sigler PB, McKnight SL. Science. 1989;246:911–916. doi: 10.1126/science.2683088. [DOI] [PubMed] [Google Scholar]
- 17.Student AK, Hsu RY, Lane MD. J Biol Chem. 1980;255:4745–4750. [PubMed] [Google Scholar]
- 18.Lavery DJ, Schibler U. Genes Dev. 1993;7:1871–1884. doi: 10.1101/gad.7.10.1871. [DOI] [PubMed] [Google Scholar]
- 19.Dignam JD, Martin PL, Shastry BS, Roeder RG. Methods Enzymol. 1983;101:582–598. doi: 10.1016/0076-6879(83)01039-3. [DOI] [PubMed] [Google Scholar]
- 20.Cook JS, Lucas JJ, Sibley E, Bolanowski MA, Christy RJ, Kelly TJ, Lane MD. Proc Natl Acad Sci USA. 1988;85:2949–2953. doi: 10.1073/pnas.85.9.2949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ntambi JM, Buhrow SA, Kaestner KH, Christy RJ, Sibley E, Kelly TJ, Lane MD. J Biol Chem. 1988;263:17291–17300. [PubMed] [Google Scholar]
- 22.Zhu Y, Qi C, Korenberg JR, Chen XN, Noya D, Rao MS, Reddy JK. Proc Natl Acad Sci USA. 1995;92:7921–7925. doi: 10.1073/pnas.92.17.7921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaestner KH, Christy RJ, Lane MD. Proc Natl Acad Sci USA. 1990;87:251–255. doi: 10.1073/pnas.87.1.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gorski K, Carneiro M, Schibler U. Cell. 1986;47:767–776. doi: 10.1016/0092-8674(86)90519-2. [DOI] [PubMed] [Google Scholar]