

Abstract
Metabolism is a vital cellular process, and its malfunction is a major contributor to human disease. Metabolic networks are complex and highly interconnected, and thus systems-level computational approaches are required to elucidate and understand metabolic genotype–phenotype relationships. We have manually reconstructed the global human metabolic network based on Build 35 of the genome annotation and a comprehensive evaluation of >50 years of legacy data (i.e., bibliomic data). Herein we describe the reconstruction process and demonstrate how the resulting genome-scale (or global) network can be used (i) for the discovery of missing information, (ii) for the formulation of an in silico model, and (iii) as a structured context for analyzing high-throughput biological data sets. Our comprehensive evaluation of the literature revealed many gaps in the current understanding of human metabolism that require future experimental investigation. Mathematical analysis of network structure elucidated the implications of intracellular compartmentalization and the potential use of correlated reaction sets for alternative drug target identification. Integrated analysis of high-throughput data sets within the context of the reconstruction enabled a global assessment of functional metabolic states. These results highlight some of the applications enabled by the reconstructed human metabolic network. The establishment of this network represents an important step toward genome-scale human systems biology.
Keywords: constraint based, metabolism, model, systems biology
An individual's metabolism is determined by one's genetics, environment, and nutrition. With the available human genome sequence and its annotation (1–3), we can hope to define the human body's complement of metabolic enzymes. In addition, numerous metabolic genes and enzymes have been individually studied for decades, resulting in a collective knowledge base, or “bibliome,” that includes reaction mechanisms and well characterized interactions. Manual component-by-component (bottom-up) reconstruction of genomic and bibliomic data leads to a biochemically, genetically, and genomically structured (BiGG) reconstruction (4) that can be mathematically represented as an in silico model for computing allowable network states under governing chemical and genetic constraints (5). The procedure for integrating these diverse data types to form a network reconstruction and predictive model is well established for microorganisms (4) and has recently been applied to mouse hybridomas (6). Such in silico models have enabled hypothesis-driven biology, including the prediction of the outcome of adaptive evolution (7–11) and the identification and discovery of candidates for missing metabolic functions that were subsequently experimentally verified (12). Because metabolic networks are more complex in mammals than in single-celled organisms, there is likely to be an even greater opportunity for the use of computational models to understand the basis of normal and abnormal cellular function.
Here we present the reconstruction of the global human metabolic map. Homo sapiens Recon 1 is a comprehensive literature-based genome-scale metabolic reconstruction that accounts for the functions of 1,496 ORFs, 2,004 proteins, 2,766 metabolites, and 3,311 metabolic and transport reactions. This network reconstruction was transformed into an in silico model of human metabolism and validated through the simulation of 288 known metabolic functions found in a variety of cell and tissue types. Recon 1 (i) enables the identification of gaps in our understanding of human metabolism, (ii) facilitates the computational interrogation of the overall properties of the human metabolic network, and (iii) provides context for analysis of “-omics” data sets. These examples are described in further detail herein.
Results and Discussion
Reconstruction and Validation of H. sapiens Recon 1.
A well annotated genome sequence is vital for bottom-up reconstruction because it enables the rapid identification of candidate network components (4) and the assembly of a preliminary network (13) that can be used as a starting point for manual curation [supporting information (SI) Fig. 5]. We used Enzyme Commission numbers (14) and Gene Ontologies (15) to identify an initial set of 1,865 human metabolic genes from the November 2004 annotations (Build 35) of Kyoto Encyclopedia of Genes and Genomes (KEGG) (16), National Center for Biotechnology Information's LocusLink (17) [now EntrezGene (18)], and the H-Invitational Database (19). These genes were mapped to a rudimentary network of 3,623 metabolic enzymes and 3,673 reactions from KEGG's LIGAND database and the compartmentalized yeast metabolic reconstruction (20). In addition to establishing initial network scope, LIGAND's pathway-based organizational structure also facilitated parallel network assembly. A team of researchers simultaneously curated network components by evaluation of >50 years of biological evidence from >1,500 primary literature articles, reviews, and biochemical textbooks. Strict quality control/quality assurance methods were used throughout the reconstruction (see Materials and Methods). Manual literature-based reconstruction ensured that the network components and their interactions were based on direct physical evidence and reflected the current knowledge of human metabolism.
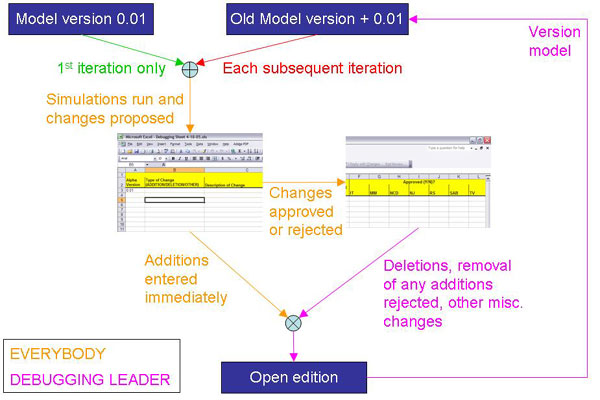
Bottom-up reconstructions can be represented mathematically, enabling the computational interrogation of their properties (4, 21). We validated the basic functionality of the human metabolic network by simulating 288 known metabolic functions in silico (SI Tables 2–4). Like genome sequence assembly and annotation, network reconstruction is an iterative process, and consequently several rounds of iterative gap analysis (i.e., targeted literature searches to identify metabolic reactions that were missed initially) and comprehensive revalidation were required to achieve a BiGG database and a high-quality network reconstruction (SI Fig. 6).
The result of five iterative rounds of reconstruction and validation is H. sapiens Recon 1 and the first human cellular process to be comprehensively modeled at this level of detail and accuracy (Table 1). It was almost entirely constructed from human-specific data and includes many reactions directly extracted from the literature that are not described in any chart or database. Furthermore, Recon 1 represents several hierarchical levels of detail, namely:
Carefully formulated metabolites and reactions, which account for known reaction stoichiometry, substrate/cofactor specificity, and directionality, as well as overall conservation of mass and charge-based metabolite ionization states at pH 7.2.
Full compartmentalization of metabolites in and their exchange between seven intracellular locations (cytoplasm, mitochondria, nucleus, endoplasmic reticulum, Golgi apparatus, lysosome, and peroxisome) and the extracellular environment.
Precise Boolean descriptions of gene–protein relationships such as alternatively spliced variants, protein complexes, and isozymes.
Confidence scores and literature references based on known biological evidence associated with each gene, protein, and reaction.
Table 1.
H. sapiens Recon 1 network statistics
| Component | Number |
|---|---|
| Genes | 1,496 |
| Transcripts* | 1,905 |
| Proteins | 2,004 |
| Complex-associated reactions* | 248 |
| Isozyme-associated reactions* | 946 |
| Intrasystem reactions | 3,311 |
| Metabolic | 2,233 |
| Transport† | 1,078 |
| Exchange reactions† | 432 |
| Compartment-specific metabolites | 2,712 |
| Cytoplasm | 995 |
| Extracellular space | 388 |
| Mitochondrion | 383 |
| Golgi apparatus | 279 |
| Endoplasmic reticulum | 231 |
| Lysosome | 207 |
| Peroxisome | 139 |
| Nucleus | 90 |
| Citations | 1,587 |
| Primary literature | 1,378 |
| Review articles | 188 |
| Textbooks | 21 |
| Validated metabolic functions | 288 |
| Knowledge gaps‡ | 356 |
*See Materials and Methods for definitions of transcripts, complexes, and isozymes.
†Transport reactions refer to intrasystem transport across a boundary (organellar and plasma membranes), whereas exchange reactions describe metabolite transport across the system boundary, e.g., into and out of the extracellular space from the surrounding medium.
‡Number of “dead-end” metabolites only produced or consumed.
The entire contents of H. sapiens Recon 1 is freely available in several formats [searchable database, metabolite and reaction lists, human-specific metabolic maps, stoichiometric matrix, and Systems Biology Markup Language (22)] at http://bigg.ucsd.edu and in SI Figs. 7–16 and SI Tables 5–10. We now describe three applications of Recon 1.
Quantitative Characterization of the Human Metabolic Bibliome Reveals an Uneven Knowledge Landscape.
Bottom-up reconstruction of Recon 1 required extensive manual surveys of the primary literature to evaluate biological evidence associated with each gene, protein, and reaction. Viewing confidence scores for these individual components at the system level reveals a global knowledge landscape with specific “peaks” and “valleys” in our understanding of human metabolism (Fig. 1). Three categories of metabolic pathways were identified based on the degree of characterization of their corresponding reactions.
Fig. 1.

Human metabolic knowledge landscape. Colors represent the percentage of reactions within a pathway that have a confidence score of 3 (biochemical or genetic evidence), 2 (physiological data or evidence from a nonhuman mammalian cell), 1 (modeling evidence), or 0 (unevaluated). Metabolic pathways (primarily defined by the Kyoto Encyclopedia of Genes and Genomes LIGAND database) were classified into three categories based on their level of characterization as detailed in the text.
Category I pathways are those where extensive primary literature is available. Chondroitin sulfate is a common component of proteoglycans, which are important in cell adhesion, signaling, and connective tissue composition. Catabolism of chondroitin sulfate chains (SI Fig. 15) is a typical example of a Category I pathway in which nearly all of the enzymes have been biochemically characterized and their corresponding genes have been identified. However, the initial steps of chondroitin sulfate catabolism in endosomes (CSBPASEly) and final degradation of the core tetrasaccharide linkage (LINKDEG2ly) are not well known (23).
Category II pathways, such as glyoxylate metabolism (SI Fig. 16), have a roughly equal proportion of highly characterized enzymes and those with moderate biological evidence (see Materials and Methods). For instance, although the peroxisomal and mitochondrial degradation of glyoxylate to l-glycine (reactions AGTix and AGTim, respectively) has been extensively studied, the presence of the glycerate kinase reaction (GLYCK2) was inferred based on the observation that individuals with d-glycericaciduria (who lack the enzyme catalyzing this reaction) cannot further metabolize d-glycerate and excrete gram amounts of it in their urine (24).
Category III pathways exhibit a wide range of confidence scores and gene coverage. That some of these pathways have not been completely elucidated is somewhat surprising, and arguably these knowledge deficits may not have been comprehensively identified without a systems approach. For example, the mechanism that cycles the end products of vitamin C degradation back to the glycolytic pathway appear to be poorly understood (SI Fig. 17) despite evidence in human erythrocytes that it may be used as an energy source (25). Furthermore, although most of ubiquinone 10 biosynthesis was inferred from physiological evidence, there are a few well studied enzymes interspersed in the pathway (SI Fig. 18). A large number of intracellular transport reactions are also included in this category, indicating that as a whole they require considerably more investigation to elucidate precise mechanistic reactions. Thus, the reconstruction of H. sapiens Recon 1 has resulted in a comprehensive appraisal of our knowledge of human metabolism and has led to direct suggestions where further experimental studies are needed (SI Tables 9 and 10).
Singular Value Decomposition (SVD) of the Stoichiometric Matrix Highlights the Importance of Compartmentalization.
Network capabilities are constrained in part by the overall structure of the stoichiometric matrix (S), which can be analyzed by using computational approaches (21). SVD (26) can identify the most influential components of a network. We used SVD to calculate the effective dimensionality of the human network to assess the number of network components needed to account for a given percentage of its structure. The cumulative normalized singular value spectrum is shown in Fig. 2 and compared with that of previous reconstructions (20, 27). The human and yeast networks are considered with and without metabolite compartmentalization to evaluate its effect on network complexity. We observed that the compartmentalized networks have a significantly larger effective dimensionality, requiring a larger number of independent modes to fully describe their contents. For a functional metabolic model, this result may be interpreted as the expansion of nonredundant metabolic functionality rather than simply a division of linear metabolic pathways across various cellular compartments.
Fig. 2.

Normalized cumulative singular value spectra for H. sapiens, S. cerevisiae, and E. coli and dominant metabolite modes. (A) Compartmentalized networks have a greater effective dimensionality than their noncompartmentalized counterparts, requiring a larger number of singular values to completely reconstruct the network. Each spectrum shows the number of decomposed modes (x axis) required to reconstruct a given fraction (y axis) of the S matrix's content. (B) The first five modes of the human metabolite coupling matrix (38) highlight the importance of the production and exchange of energy equivalents and the potentially significant impact of osmotic regulation. c, cytoplasmic; e, extracellular; g, Golgi apparatus; m, mitochondrial.
SVD was further applied to demonstrate unbiased systemic links between the metabolites of Recon 1. To minimize any bias arising from differing stoichiometric coefficients and lumped reactions, the metabolite-coupling matrix M (28) was constructed and decomposed. The five most dominant modes of the SVD of M (Fig. 2) are interpreted as independent groups of interacting metabolites, ordered with monotonically decreasing importance. These have biologically meaningful interpretations as currency exchanges, including high-energy phosphate group transfer (modes 1 and 4), reducing equivalent exchange (mode 2), and sugar transfer (mode 3). Mode 5 is dominated by ions and water, linking biochemical and mechanical relationships in the cell by osmotic force balance. Interestingly, these modes are compartment-specific, highlighting known relationships between compartments and cellular functions. Collectively, these observations highlight some of the implications of compartmentalization in the human metabolic reconstruction and support the notion that compartments may function as independent reaction sets to achieve specific metabolic objectives (29). This finding reinforces the idea that intracellular compartmentalization has functional implications for various metabolites, including glutathione (30), amino acids (31–33), cholesterol and bile acids (34, 35), and sphingolipids (36).
Coupled Reaction Sets Suggest Potential Alternatives to Known Drug Targets.
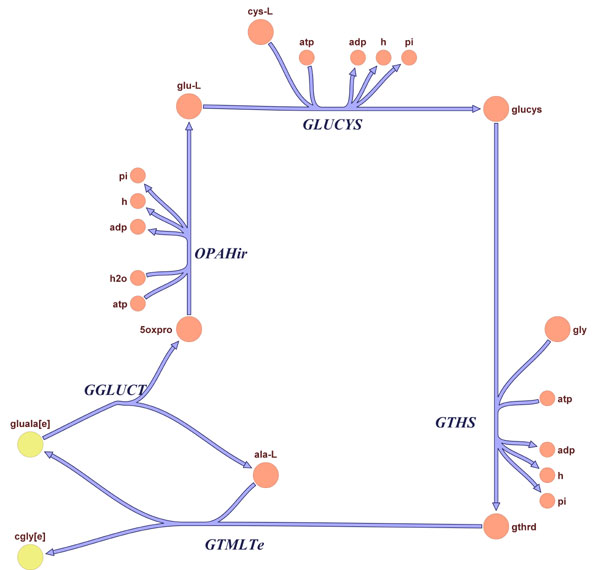
Recon 1 has also enabled in silico characterization of the known human metabolic map by using well established constraint-based methods that have been applied extensively to microbial metabolism (37). Coupled reaction sets consist of reactions that are active together in functional states of a network (29, 38). Thus, a flux through one reaction results in a directionally equivalent flux in other reactions in the set. Flux coupling analysis (39) was used to identify coupled reaction sets under aerobic glucose metabolic conditions. One of the largest of the >250 coupled reaction sets identified under these metabolic conditions involves two branches of cholesterol biosynthesis (Fig. 3A and SI Fig. 19). 3-Hydroxy-3-methylglutaryl-CoA reductase (Entrez Gene ID 3156), a primary metabolic target of the antilipidemic class of statin drugs, is in this coupled reaction set; other members of the set are thus identified as potential alternative drug targets for treating hyperlipidemia. It has been proposed that deficiencies in enzymes belonging to the same functionally coupled reaction set may have similar phenotypes, and examples of this have been reported for the human mitochondria (38). This hypothesis is supported by the example in Fig. 3B (SI Fig. 20), which depicts a coupled reaction set involved in the production and transport of glutathione. The Online Mendelian Inheritance in Man (OMIM) (40, 41) identification tags associated with the genes encoding glutathione synthetase (MIM no. 231900; Entrez Gene ID, 2937) and glutamate-cysteine ligase (OMIM no. 230450; Entrez Gene ID, 2729) indicate that both deficiencies result in hemolytic anemia. These examples demonstrate that in silico experiments with these models provide an analytical approach to studying the causes and consequences of disease states, which can potentially lead to insights into new drug treatment targets. The use of functionally grouped reactions, such as coupled (39) and correlated reaction sets (42), present a promising approach for the functional analysis of complex networks with applications in elucidating causal relationships in the diseases and potentially identifying new treatment strategies and drug targets (29).
Fig. 3.

Coupled reaction sets involving cholesterol biosynthesis and glutathione production and transport. (A) The cholesterol biosynthesis coupled set includes all reactions except those shaded in gray. Note that the groups of reactions are not all directly connected and could not be identified by visual inspection alone. (B) Reactions in the glutathione reaction set were mapped to disease associations by using Mendelian Inheritance in Man identification tags. Deficiencies in glutathione synthetase (GTHS) or glutamate-cysteine ligase (GLUCYS) both result in hemolytic anemia, supporting the notion that enzyme deficiencies in the same coupled set may have similar phenotypes. Interference with or decreases in GLUCYS activity is associated with an increased risk of myocardial infarctions (MI). A high-resolution version of this figure is available in SI Figs. 19 and 20.
Integrated Analysis of Gene Expression Data Reveals the Effects of Gastric Bypass Surgery on Skeletal Muscle Metabolism.
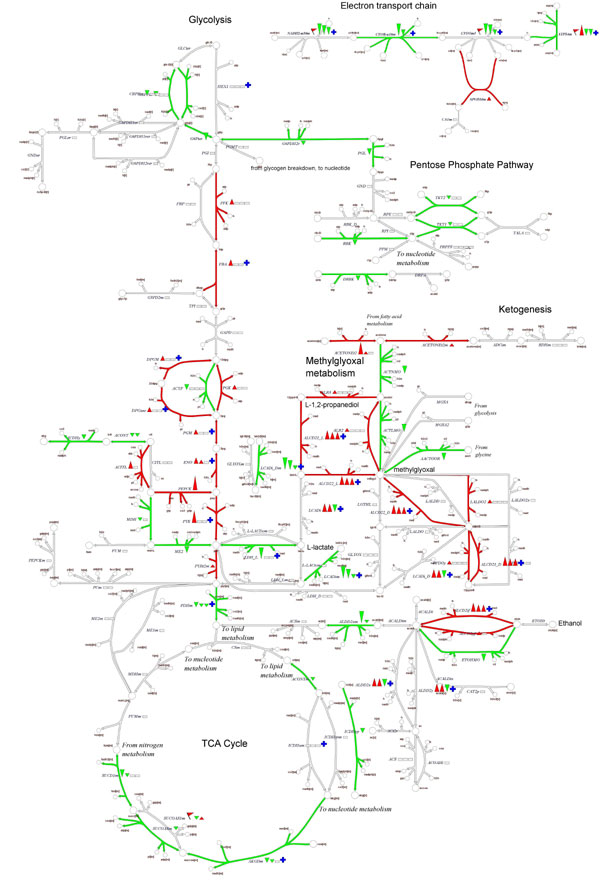
Recon 1 was used as a context for interpreting the effects of gastric bypass surgery on skeletal muscle metabolism. In this study, gene expression data were acquired from the same patients before and after bariatric surgery. Consequently, many genetic factors that might contribute to interindividual differences are not a concern in this analysis. Published gene expression measurements (43) were mapped to the reconstructed network by using gene–transcript–protein–reaction associations (see Materials and Methods) and visualized on maps of central metabolism and the electron transport chain (Fig. 4; SI Fig. 21). We observed a general trend of up-regulated anaerobic metabolism and down-regulated oxidative phosphorylation after surgery, with many genes in glycolysis, pentose phosphate pathway, methylglyoxal metabolism, and oxidative phosphorylation showing subtle but consistent overall patterns of expression change (SI Tables 11–15). The relative decrease in mitochondrial bioenergetics is also evident in terms of the smaller number of mitochondrial reactions in the down-regulated network (SI Fig. 22) and is consistent with gene expression changes observed in the skeletal muscle of rhesus monkeys subjected to long-term caloric restriction (44). Comparison of gene expression data in the context of Recon 1 suggests that 1 year after surgery, patients may still be feeling the effects of calorie restriction even after weight stabilization. Thus, Recon 1 represents a versatile and effective integration tool, enabling visualization and analysis of genome-scale context in the context of a highly curated metabolic network.
Fig. 4.

Integrated analysis of gene expression data from gastric bypass patients before surgery and 1 year afterward. Expression measurements were to reactions in the global human metabolic network and then visualized on Recon 1's comprehensive collection of human metabolic maps. Reactions are color-coded based on their corresponding gene expression changes (green, down-regulated; red, up-regulated; white, no data available or reaction level conflict). Arrows next to reaction abbreviations indicate the magnitude of expression changes on a log10 scale (gray boxes indicate no data available). A high-resolution version of this figure is available in SI Fig. 21.
Conclusions
Reconstruction of the global (or genome-scale) human metabolic network in a standardized, quality-controlled, bottom-up manner is presented. H. sapiens Recon 1 is a BiGG reconstruction and represents a milestone in human systems biology. It is a mathematically structured database that enables systematic studies of the human metabolism and its properties. The reconstruction process required comprehensive review of the published human metabolic knowledge base (i.e., bibliomic data), and it led to a global quantitative assessment of network confidence that has highlighted specific areas of limited or poor understanding, such as intracellular transport of metabolites, which need further experimental investigation. The formulation of an in silico model from the reconstruction and initial analysis of the network structure highlighted the importance of intracellular compartmentalization. Further analyses demonstrated the potential utility of the model as a tool for discovery and for the analysis and interpretation of high-throughput data. These capabilities will likely be critical in elucidating underlying mechanisms of disease and identifying treatment strategies by developing cell-, tissue-, and context-specific models and building additional layers of complexity (such as gene regulation) into the framework.
Genome-scale microbial metabolic reconstructions have been widely used to successfully perform systems analysis to the point that models resulting from these reconstructions have become tools for hypothesis driven biological discovery (4). We expect that this global human metabolic reconstruction will not only become a prototype for other mammalian reconstructions but will hopefully also enable significant dimensions in the study of in human systems biology, some of which we have described herein. The future promise for individualized medicine and treatment will need a context to integrate and analyze data, and models resulting from these reconstructions can play a significant role in fulfilling this need. However, the development of cell-type or context-specific models will require the integration of various types of data, including transcriptomic, proteomic, fluxomic, and metabolomic measurements. Recon 1 provides the context for integration and analysis of these data into predictive models. For example, the developing field of nutrigenomics requires significant data integration and analysis to elucidate the influence of the diet on an organism's transcriptome, proteome, and metabolome (45). Achieving these ambitious goals will require top–down data sets in conjunction with quantitative bottom-up reconstructions such as H. sapiens Recon 1.
Materials and Methods
Reconstruction Procedure.
An initial component list was assembled as described in the text. This list was then divided into eight metabolic subsets (amino acids, carbohydrates, energy, glycans, lipids, nucleotides, secondary metabolites/xenobiotics, vitamins, and cofactors) for independent curation by a team of researchers. Putative gene assignments were verified based on evidence collected from genome annotation databases, namely EntrezGene (18), Gene Cards (46), and the scientific literature. Alternative transcripts were identified based on known RefSeq (17) mRNA transcripts for each locus. Substrate and cofactor preferences were identified from the literature and BRENDA (47). Metabolite formula and charge were calculated based on their ionization state at pH 7.2, which for simplicity was presumed to be constant across all compartments. Reaction directionality was determined from thermodynamic data or inferred from legacy data and textbooks. Compartmentalization was determined from protein localization data, sequence targeting signals, and indirect physiological evidence. If these data were unavailable, reactions were modeled as cytoplasmic. The intermembrane space of double-membrane organelles was also modeled as cytoplasmic. Gene–transcript–protein–reaction relationships (5, 6) were manually identified from the literature and formulated as Boolean logic statements. Isozymes (an “or” relationship) were defined as distinct proteins that catalyze the same substrate- and compartment-specific reaction and could arise from one gene due to alternative splicing or could be encoded by independent genes. Cases in which a reaction depended on the presence of more than one gene/protein (an “and” relationship, e.g., proteins with multiple subunits/chains or complexes composed of multiple enzymes) were classified as protein complexes. Confidence scores were assigned based on biological evidence associated with each reaction. Evidence from classical biochemical or genetic experiments, such as gene cloning and protein characterization, was given the highest confidence score (3). Midlevel scores (2) were assigned to reactions based on physiological data or biochemical/genetic evidence from a nonhuman mammalian cell (typically mouse, rat, or rabbit). Reactions with the lowest confidence score (1) were included solely based on in silico modeling because, during the process of model validation, they were deemed mandatory for a particular metabolic function. Transport reactions were entirely reconstructed based on literature reports and biochemistry textbooks because the current annotation of transporters is not sufficiently specific with regard to substrates and mechanisms.
Functional Validation and Gap Analysis.
The reconstruction was assembled in SimPheny (Genomatica, San Diego, CA), and the stoichiometric matrix was formulated as described (48). Exchange reactions (SI Table 6) were added to enable uptake and secretion of extracellular metabolites for the purpose of simulations. Functional validation was performed by using flux balance analysis (39), allowing recycled cofactor pairs to enter and leave the system as needed (SI Tables 2–4). Comprehensive gap analysis of the stoichiometric matrix was performed after each round of functional validation. Every “dead-end” metabolite that could not be produced or consumed was manually reexamined by returning to the literature to identify possible reactions describing its degradation, production, or transport. A final round of gap analysis was performed upon completion of H. sapiens Recon 1, and a description of unresolved gaps is provided in SI Table 9.
Network Analysis.
The singular value spectra (26) were computed for H. sapiens Recon 1, Saccharomyces cerevisiae iND750 (20), and Escherichia coli iJR904 (48) as the normalized cumulative sum of the singular values by using Matlab Ver. 6.5 (MathWorks, Natick, MA). The stoichiometric matrix was not altered or scaled before SVD. The human and yeast networks were decompartmentalized by reassigning all intracellular metabolites to the cytoplasm. Modes were defined as the largest elements (each >0.25×max column value) of the columns of U (identical to the columns of V), where UΣVT = ^S^ST and Ŝ is the binary form of S. Noncompartmentalized versions of the human and yeast model were created by summing up the rows of the S matrix corresponding to the same metabolite in the different intracellular compartments.
Coupled reaction sets were calculated with a bilinear optimization algorithm described by Burgard et al. (39) by using LINDO r6.1 (Lindo Systems, Chicago, IL) called from within Matlab Ver. 6.5 for aerobic glucose uptake conditions. Briefly, for all of the fluxes in the network under specified input conditions, if two fluxes always respond in the same manner (both increase or both decrease) to any perturbation or alternative flux distribution, they are said to be coupled.
Gene Expression Analysis.
Gene expression data for the gastric bypass study were downloaded from Gene Expression Omnibus (GEO) [GSE5109 (49)]. Expression signals were normalized by the average value for each chip. Log10 ratios of post/pregastric surgery probe expression signals were calculated for all three patients in the gastric bypass data set and mapped to Entrez Gene and RefSeq mRNA identifications (18) in H. sapiens Recon 1 based on database identifiers in the Affymetrix (Santa Clara, CA) U133A Plus 2.0 annotation file (50). Probes whose expression ratio was qualitatively inconsistent across all three patients (i.e., not all up or all down) or that conflicted at the gene level were removed from the data set. Average gene expression ratios were then mapped to the reaction network by using Recon 1's gene–transcript–protein–reaction associations.
Supplementary Material
Acknowledgments
We thank Dr. Markus Herrgård for performing the flux coupling computations, Mr. Jan Schellenberger for developing the BiGG Database, and Dr. Evelyn Travnik and Mr. Thomas Conrad for assistance in preparing the supporting information. This work was supported by Training Grants from the National Institutes of Health.
Abbreviations
- BiGG
biochemically, genetically, and genomically structured
- SVD
singular value decomposition.
Footnotes
Conflict of interest statement: N.C.D., S.A.B., N.J., I.T., M.L.M., T.D.V., R.S., and B.Ø.P. and the University of California at San Diego disclose potential financial conflict of interest related to U.S. Patent Application (Pub. No. 20040029149), published on February 12, 2004.
This article contains supporting information online at www.pnas.org/cgi/content/full/0610772104/DC1.
References
- 1.International Human Gene Sequencing Consortium. Nature. 2004;431:931–945. [Google Scholar]
- 2.Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, et al. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- 3.McPherson JD, Marra M, Hillier L, Waterston RH, Chinwalla A, Wallis J, Sekhon M, Wylie K, Mardis ER, Wilson RK, et al. Nature. 2001;409:934–941. doi: 10.1038/35057157. [DOI] [PubMed] [Google Scholar]
- 4.Reed JL, Famili I, Thiele I, Palsson BO. Nat Rev Genet. 2006;7:130–141. doi: 10.1038/nrg1769. [DOI] [PubMed] [Google Scholar]
- 5.Price ND, Reed JL, Palsson B. Nat Rev Microbiol. 2004;2:886–897. doi: 10.1038/nrmicro1023. [DOI] [PubMed] [Google Scholar]
- 6.Sheikh K, Forster J, Nielsen LK. Biotechnol Prog. 2005;21:112–121. doi: 10.1021/bp0498138. [DOI] [PubMed] [Google Scholar]
- 7.Fong SS, Burgard AP, Herring CD, Knight EM, Blattner FR, Maranas CD, Palsson BØ. Biotechnol Bioeng. 2005;91:643–648. doi: 10.1002/bit.20542. [DOI] [PubMed] [Google Scholar]
- 8.Fong SS, Joyce AR, Palsson BØ. Genome Res. 2005;15:1365–1372. doi: 10.1101/gr.3832305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fong SS, Marciniak JY, Palsson BØ. J Bacteriol. 2003;185:6400–6408. doi: 10.1128/JB.185.21.6400-6408.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hua Q, Joyce AR, Fong SS, Palsson BØ. Biotechnol Bioeng. 2006;95:992–1002. doi: 10.1002/bit.21073. [DOI] [PubMed] [Google Scholar]
- 11.Ibarra RU, Edwards JS, Palsson BØ. Nature. 2002;420:186–189. doi: 10.1038/nature01149. [DOI] [PubMed] [Google Scholar]
- 12.Reed JR, Patel TR, Chen KH, Joyce AR, Applebee MK, Herring CD, Bui OT, Knight EK, Fong SS, Palsson BØ. Proc Natl Acad Sci USA. 2006;103:17480–17484. doi: 10.1073/pnas.0603364103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Romero P, Wagg J, Green ML, Kaiser D, Krummenacker M, Karp PD. Genome Biol. 2005;6:R2. doi: 10.1186/gb-2004-6-1-r2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.International Union of Biochemistry and Molecular Biology Nomenclature Committee. Enzyme Nomenclature 1992: IUB Recommendations of the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology on the Nomenclature and Classification of Enzymes. San Diego: Academic; 1992. [Google Scholar]
- 15.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kanehisa M, Goto S, Hattori M, Aoki-Kinoshita KF, Itoh M, Kawashima S, Katayama T, Araki M, Hirakawa M. Nucleic Acids Res. 2006;34:D354–D357. doi: 10.1093/nar/gkj102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pruitt KD, Maglott DR. Nucleic Acids Res. 2001;29:137–140. doi: 10.1093/nar/29.1.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maglott D, Ostell J, Pruitt KD, Tatusova T. Nucleic Acids Res. 2005;33:D54–D58. doi: 10.1093/nar/gki031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Imanishi T, Itoh T, Suzuki Y, O'Donovan C, Fukuchi S, Koyanagi KO, Barrero RA, Tamura T, Yamaguchi-Kabata Y, Tanino M, et al. PLoS Biol. 2004;2:e162. doi: 10.1371/journal.pbio.0020162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duarte NC, Herrgard MJ, Palsson BØ. Genome Res. 2004;14:1298–1309. doi: 10.1101/gr.2250904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Palsson BØ. Systems Biology: Determining the Capabilities of Reconstructed Networks. Cambridge, UK: Cambridge Univ Press; 2006. [Google Scholar]
- 22.Hucka M, Finney A, Sauro HM, Bolouri H, Doyle JC, Kitano H, Arkin AP, Bornstein BJ, Bray D, Cornish-Bowden A, et al. Bioinformatics. 2003;19:524–531. doi: 10.1093/bioinformatics/btg015. [DOI] [PubMed] [Google Scholar]
- 23.Winchester BG. Subcell Biochem. 1996;27:191–238. doi: 10.1007/978-1-4615-5833-0_7. [DOI] [PubMed] [Google Scholar]
- 24.Poore RE, Hurst CH, Assimos DG, Holmes RP. Am J Physiol. 1997;272:C289–C294. doi: 10.1152/ajpcell.1997.272.1.C289. [DOI] [PubMed] [Google Scholar]
- 25.Banhegyi G, Braun L, Csala M, Puskas F, Mandl J. Free Radic Biol Med. 1997;23:793–803. doi: 10.1016/s0891-5849(97)00062-2. [DOI] [PubMed] [Google Scholar]
- 26.Famili I, Palsson BØ. J Theor Biol. 2003;224:87–96. doi: 10.1016/s0022-5193(03)00146-2. [DOI] [PubMed] [Google Scholar]
- 27.Reed JL, Vo TD, Schilling CH, Palsson BØ. Genome Biol. 2003;4:R54. doi: 10.1186/gb-2003-4-9-r54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Becker SA, Price ND, Palsson BØ. BMC Bioinformatics. 2006;7:111. doi: 10.1186/1471-2105-7-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Papin JA, Reed JL, Palsson BØ. Trends Biochem Sci. 2004;29:641–647. doi: 10.1016/j.tibs.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 30.Smith CV, Jones DP, Guenthner TM, Lash LH, Lauterburg BH. Toxicol Appl Pharmacol. 1996;140:1–12. doi: 10.1006/taap.1996.0191. [DOI] [PubMed] [Google Scholar]
- 31.Gruetter R. Neurochem Int. 2002;41:143–154. doi: 10.1016/s0197-0186(02)00034-7. [DOI] [PubMed] [Google Scholar]
- 32.Sonnewald U, Hertz L, Schousboe A. J Cereb Blood Flow Metab. 1998;18:231–237. doi: 10.1097/00004647-199803000-00001. [DOI] [PubMed] [Google Scholar]
- 33.Sonnewald U, Schousboe A, Qu H, Waagepetersen HS. Neurochem Int. 2004;45:305–310. doi: 10.1016/j.neuint.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 34.Krisans SK. Ann NY Acad Sci. 1996;804:142–164. doi: 10.1111/j.1749-6632.1996.tb18614.x. [DOI] [PubMed] [Google Scholar]
- 35.Solaas K, Ulvestad A, Soreide O, Kase BF. J Lipid Res. 2000;41:1154–1162. [PubMed] [Google Scholar]
- 36.Kolter T, Sandhoff K. Biochim Biophys Acta. 2007;1758:2057–2079. doi: 10.1016/j.bbamem.2006.05.027. [DOI] [PubMed] [Google Scholar]
- 37.Trawick JD, Schilling CH. Biochem Pharmacol. 2006;71:1026–1035. doi: 10.1016/j.bcp.2005.10.049. [DOI] [PubMed] [Google Scholar]
- 38.Jamshidi N, Palsson BØ. Mol Syst Biol. 2006;2:38. doi: 10.1038/msb4100077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Burgard AP, Nikolaev EV, Schilling CH, Maranas CD. Genome Res. 2004;14:301–312. doi: 10.1101/gr.1926504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.OMIM. Online Mendelian Inheritance in Man. Baltimore: McKusick–Nathans Institute for Genetic Medicine, Johns Hopkins University, National Center for Biotechnology Information, National Library of Medicine; 2006. [Google Scholar]
- 41.Hamosh A, Scott AF, Amberger JS, Bocchini CA, McKusick VA. Nucleic Acids Res. 2005;33:D514–D517. doi: 10.1093/nar/gki033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thiele I, Price ND, Vo TD, Palsson BØ. J Biol Chem. 2005;280:11683–11695. doi: 10.1074/jbc.M409072200. [DOI] [PubMed] [Google Scholar]
- 43.Park JJ, Berggren JR, Hulver MW, Houmard JA, Hoffman EP. Physiol Genomics. 2007;27:114–121. doi: 10.1152/physiolgenomics.00045.2006. [DOI] [PubMed] [Google Scholar]
- 44.Kayo T, Allison DB, Weindruch R, Prolla TA. Proc Natl Acad Sci USA. 2001;98:5093–5098. doi: 10.1073/pnas.081061898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Muller M, Kersten S. Nat Rev Genet. 2003;4:315–322. doi: 10.1038/nrg1047. [DOI] [PubMed] [Google Scholar]
- 46.Safran M, Solomon I, Shmueli O, Lapidot M, Shen-Orr S, Adato A, Ben-Dor U, Esterman N, Rosen N, Peter I, et al. Bioinformatics. 2002;18:1542–1543. doi: 10.1093/bioinformatics/18.11.1542. [DOI] [PubMed] [Google Scholar]
- 47.Schomburg I, Chang A, Ebeling C, Gremse M, Heldt C, Huhn G, Schomburg D. Nucleic Acids Res. 2004;32:D431–D433. doi: 10.1093/nar/gkh081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Feist AM, Scholten JCM, Palsson BØ, Brockman FJ, Ideker T. Mol Syst Biol. 2006;2:E1–E14. doi: 10.1038/msb4100046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Edgar R, Domrachev M, Lash AE. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu G, Loraine AE, Shigeta R, Cline M, Cheng J, Valmeekam V, Sun S, Kulp D, Siani-Rose MA. Nucleic Acids Res. 2003;31:82–86. doi: 10.1093/nar/gkg121. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}