

Abstract
In humans, mutations in the X-linked MECP2 gene, are the cause of Rett syndrome (RTT), a neurodevelopmental disorder that affects mainly girls. MeCP2 binds to methylated CpGs and is thought to act as a transcriptional repressor. In male mice, deletion or targeted mutation of Mecp2 leads to lethality and causes a neuronal phenotype. Selective mutation of Mecp2 in postnatal neurons results in a similar, although delayed, phenotype, suggesting that the symptoms are caused by MeCP2 deficiency in postmitotic neurons. In agreement with this idea, expression of a Mecp2 transgene in postmitotic neurons of Mecp2-null mutant mice resulted in the phenotypical rescue of the symptoms. To assess whether postnatal activation of MeCP2 in mutant animals could also affect the progression of the disorder, we constructed a conditionally active Mecp2 “rescue transgene” that was activated between P0 and P30. The Mecp2 transgene was under the control of the CAGGS promoter and was activated by using brain specific Cre-mediated recombination. Our results indicate that postnatal, neuron-specific activation of MeCP2 as late as 2–4 weeks of age significantly prolonged the lifespan of mutant animals and delayed the onset of neurologic symptoms.
Keywords: Rett syndrome, CamKinase, Nestin, Tau, behavioral
Rett syndrome (RTT) is an X-linked neurological disorder that predominantly affects girls (1). First signs of illness include deceleration of head growth between 2–4 months of age. Affected individuals usually develop full-blown symptoms between the ages of 1 and 4 years. The disorder is characterized by social withdrawal, loss of motor skills, and gait ataxia. Many affected individuals develop stereotypic hand movement and autonomic dysfunctions such as irregular respiration. All RTT subjects have severe mental retardation.
Mutations in the X-linked MECP2 gene (methyl-CpG-binding protein 2) account for nearly 80% of RTT cases (2–5). To study the RTT etiology, we and others established mouse models by engineering Mecp2 gene deletions (6–8). Conditional mutations have shown that although MeCP2 is also expressed outside the nervous system, deficiency in neurons is responsible for the phenotype (7). Electrophysiological analyses demonstrated that MeCP2 deficiency shifts the balance between excitation and inhibition in cortical neurons (9), and, more recently, MeCP2 has been implicated in regulating RNA splicing (10). Male mutant mice develop normally until 4–5 weeks of age, when RTT-like symptoms, including reduced brain weight, decreased CA2 neuron size, hindlimb clasping, and impaired locomotor function, become manifest (6, 7). A recent study showed that postnatal loss of MeCP2 in the forebrain is sufficient to cause many of the behavioral aspects of RTT in mice (11).
Given the strong biochemical evidence that MeCP2 binds methylated CpGs and functions in establishing or maintaining a repressive chromatin state by recruiting histone deacetylases (12–14) and H3 K9 methylating enzymes (15), it is surprising that only few differences in gene expression were observed between WT and mutant (16–20). Among the identified targets of MeCP2 are glucocorticoid-regulated genes (19), Hairy 2 (21), and Bdnf. Bdnf (brain-derived-neurotrophic-factor), the first identified target gene, is repressed in resting neurons by binding of MeCP2 to the neuronal-specific promoter region. Resting neurons from Mecp2-null animals have slightly higher BDNF expression (22–24), whereas calcium influx triggers MeCP2 phosphorylation, leading to Bdnf activation (25). Recent in vivo data suggested a functional interaction between the two genes because overexpression of BDNF was shown to extend the lifespan, partially rescue locomotor defects, and reverse the electrophysiological deficits of MeCP2-deficient animals (24, 26). MeCP2 has also been implicated in the regulation of imprinted genes such as Ube3a (27), Gabrb3 (28), and Dlx5 (29), although some of these claims have been challenged (30). Thus, despite the great insights derived from those studies, it remains unclear whether RTT is the result of misregulation of few key genes, such as Bdnf, or whether it is caused by misregulation of multiple genes, each with an incremental contribution to the phenotype.
Because overt symptoms become manifest only at 1–2 years of age in RTT females and at 4–5 weeks of age in male Mecp2 mutant mice, it would be important to know whether the behavioral and physiological symptoms are due to abnormal brain development or to neuronal dysfunction caused by MeCP2 deficiency in mature neurons. If MeCP2 plays an ongoing critical role in mature CNS neurons and only prolonged deficiency leads to neuronal dysfunctions, it may be possible to restore these functions by reactivating MeCP2 or by affecting expression of other genes in adult neurons. Indeed, previous results have demonstrated that a Tau promoter-driven reactivation of a Mecp2 transgene in postmitotic neurons rescued the MeCP2-deficient phenotype in mice (31). These experiments did not address the possibility that activation of MeCP2 at later ages could affect the course of the disorder. Here, we use Cre-mediated recombination in Mecp2 −/y mice to show that activation of MeCP2 as late as 2–4 weeks of age can delay lethality and the onset of symptoms.
Results
Generation of the CAGGS Lox–Stop–Lox Mecp2 Transgene.
To investigate whether the phenotype in MeCP2-deficient mice could be affected by postnatal, brain-specific reactivation of MeCP2, we engineered a conditional rescue transgene. MeCP2 is present in two isoforms: e1 and e2 (32–34). These isoforms have a slightly different N-terminal region and are expressed at different levels but have been demonstrated to be functionally equivalent in mice because brain expression of the e2 isoform alone rescued the phenotype of Mecp2 −/y mice (31). We used the mouse Mecp2e2 cDNA placed downstream of a Stop cassette containing a neo-resistance gene and three polyadenylation signals (Fig. 1 A). The synthetic CAGGS promoter, a hybrid promoter–enhancer containing the β-actin enhancer and the CMV early promoter (35), was placed upstream of the Stop cassette. Because the Stop cassette was flanked by LoxP sites (36), Cre-mediated recombination would result in expression of MeCP2e2 from the CAGGS promoter. C10 ES cells were used to target the transgene to the modified Col1a1 locus by frt/Flpe recombinase-mediated site-specific integration (37), and the targeted ES cells were injected into blastocysts to generate CAGGS LoxP-Stop-LoxP (LSL) Mecp2 mice.
Fig. 1.

Schematic representation of the rescue construct. (A) Mecp2e2 cDNA (white box), CAGGS promoter (dark gray box), stop cassette (light gray box), LoxP sites (black boxes). The construct was targeted to the Col1a1 locus. Expression of Cre will loop out the stop cassette, allowing the expression of MeCP2e2. (B) Characteristics of the Cre lines used. (C) Western blot analysis of tissue from WT (lanes 1), transgenic (CAGGS LSL Mecp2; Mecp2 −/y, lanes 2), and Mecp2 −/y (KO) mice (lane 3). No MeCP2 signal was detected in brain, lung, and spleen of null animals containing the CAGGS LSL Mecp2 transgene. Anti-GAPDH was used as loading control. (D) Western blot of total brain extracts of 6-week-old animals. Lane 1, endogenous MeCP2 in WT brain. The higher, stronger band corresponds to MeCP2e1, and the lower, fainter band corresponds to the MeCP2e2. Lane 2: Mecp2 −/y (KO); CAGGS LSL Mecp2. Lanes 3 and 4: Nestin and Tau Cre rescued animals, respectively. Lanes 5 and 6: C93 and C159 rescued mice. (E and F) Western blot analysis of CAGGS MeCP2 expression in cortex (Co), hippocampus (Hi), and cerebellum (Ce). Tau and Nestin Cre activate the transgene in all areas (E, lanes 4–9), whereas C93 and C159 activate Mecp2 in cortex and hippocampus (F, lanes 4, 5, 7, and 8) but not in cerebellum (F, lanes 6 and 9). E and F, lanes 1–3 show endogenous MeCP2.
To test whether the LSL element repressed the transgene expression, CAGGS LSL Mecp2 transgenic mice were crossed with Mecp2+/− mice (7) to generate animals carrying the Mecp2-null allele as well as the conditional rescue construct. Western blot analysis of extracts from brain, lung, and spleen of transgenic mice (CAGGS LSL Mecp2; Mecp2 −/y) failed to detect MeCP2 protein (Fig. 1 C), indicating that the LSL cassette efficiently repressed expression of the transgene.
Brain-Specific Activation of the Rescue Transgene by Cre-Mediated Recombination.
We crossed the Mecp2+/−; CAGGS LSL Mecp2 mice with various Cre transgenic lines to test whether removal of the STOP cassette would result in activation of the CAGGS Mecp2 transgene. Four different lines were used that expressed Cre recombinase during embryogenesis (Nestin Cre and Tau Cre) or during postnatal life (CamKinaseII Cre 93 and 159). The nestin Cre line activates the recombinase at embryonic day (E)10 in precursors of neurons and glia (7, 38), whereas the Tau Cre line activates the enzyme in postmitotic neurons (31, 39). In the CamKinaseII Cre line 93 (C93) the recombinase is activated in the postnatal forebrain, hippocampus, midbrain, and brainstem, reaching maximum expression at around postnatal day (P)21 (40), whereas in the CamKinaseII Cre line 159 (C159), Cre starts to be expressed in forebrain only at around P15 and reaches maximal activity at ≈P30 (41). The main characteristics of the Cre lines used are summarized in Fig. 1 B.
To assess Mecp2 transgene expression, we performed Western blot analysis on total brain extracts from 6-week-old animals. As shown in Fig. 1 D (lane 1), WT mice expressed the two MeCP2 isoforms e1 and e2 at a ratio of 9 to 1 as reported (33, 34), whereas only the e2 form was expressed from the CAGGS Mecp2 transgene (lanes 3–6). Mice carrying the Nestin (lane 3) or the Tau transgene (lane 4) expressed MeCP2 at a similar level as WT mice, whereas C93 double-transgenic animals (lane 5) had much lower MeCP2 expression. No MeCP2 protein was detected in total brain extracts from mice carrying the C159 transgene (lane 6). We also performed Western blot analysis on specific subregions of the brain (Fig. 1 E and F). The level of MeCP2 in frontal cortex, hippocampus, and cerebellum of Nestin Cre and Tau Cre mice was similar to the level of MeCP2 in WT mice (Fig. 1 E). Total MeCP2 levels in animals carrying the C93 Cre or the C159 Cre were similar or reduced, respectively, as compared with WT mice, and no expression was found in the cerebellum (Fig. 1 F).
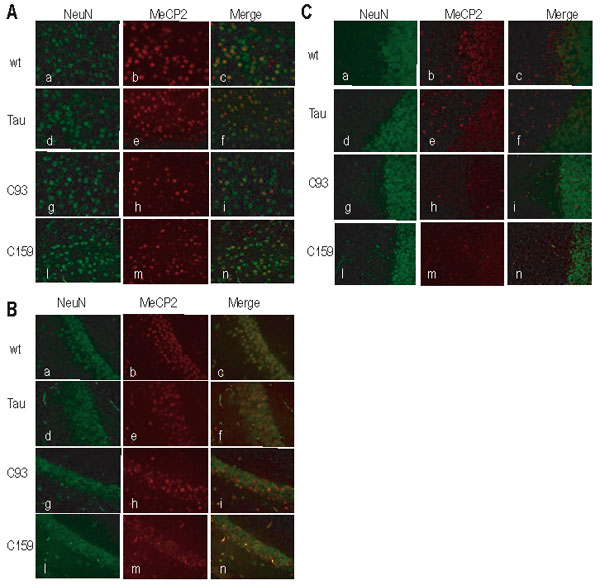
To evaluate MeCP2 expression in single cells, we performed immunohistochemical analysis on brain sections. Staining for the neuronal-specific nuclear protein (NeuN) and MeCP2 was used to determine the fraction of neurons that had activated the transgene in different brain regions. Fig. 2 D shows that CAGGS MeCP2 staining was nuclear and localized to heterochromatin-dense areas (Fig. 2 Aii, arrowheads), whereas NeuN showed both nuclear and cytoplasmic staining (Fig. 2 Ai). We quantified the number of neurons that expressed the Mecp2 transgene. As shown in Fig. 2 B–D, 70–90% of the neurons expressed CAGGS MeCP2 throughout the entire brain in Nestin Cre and Tau Cre animals. In contrast, CamKinaseII-driven Cre was activated only to specific areas of the forebrain: Cre93 and Cre159 expressed MeCP2 in ≈50% of frontal cortical neurons, C93 expressed MeCP2 in all regions of the hippocampus (CA1, CA2, CA3, DG), and expression in the C159 animals was restricted to the dentate gyrus. No MeCP2 protein was found in the cerebellum of these mice (representative images for all three regions are shown as supporting information (SI) Fig. 5). We also noticed some variability between different CAGGS MeCP2 expressing cells: the majority had WT levels of MeCP2, but ≈20% of the cells had higher or lower than endogenous MeCP2 signal (data not shown).
Fig. 2.

Expression of the CAGGS Mecp2 in different areas of the brain. (A) LV cortical neurons from a C93 rescued animal. CAGGS MeCP2 localizes in heterochromatin-rich areas of the nucleus (arrowheads in ii). (i) NeuN (neuronal nuclear-specific marker) in green. (ii) MeCP2 in red. (iii) Merge. (B–D) Percentage of neurons expressing CAGGS MeCP2. For each genotype (mice were at least 3 months old), we analyzed LV of the cortex (M1-S1) (B), hippocampal CA1/CA2 boundary (C), and cerebellum (D). Nestin (N) and Tau Cre (T) rescued animals expressed MeCP2 in 70–90% of neurons in all brain regions surveyed. C93 rescued mice had MeCP2 expression in 50% of the neurons in cortex and hippocampus. C159 rescued mice had maximum expression in the cortex (60% of neurons) but little or no expression elsewhere. No MeCP2-positive cells were found in the cerebellum of C93 and C159 rescued animals. In WT mice, 100% of neurons express MeCP2, whereas no expression was observed in Mecp2 −/y mice (data not shown). At least 100 cells per animal were included in the count. “n” is the number of animals analyzed for each genotype.
The results shown in Figs. 1 and 2 indicate that the CAGGS Mecp2 transgene was activated by all Cre transgenes, although the level and the anatomical distribution of MeCP2 expression differed among the four tested Cre lines. In the following discussion, the Mecp2-null mutant animals carrying the CAGGS Mecp2 transgene in combination with one of the different Cre transgenes will be referred to as “rescued” mice and the corresponding transgene combination as “rescue transgenes.”
Cre-Mediated Expression of MeCP2 Significantly Delays Lethality and Progression of Symptoms.
Mecp2-null mice develop symptoms between 4 and 6 weeks and die ≈10–12 weeks of age (7, 24). Fig. 3 A shows that Nestin Cre- and Tau Cre-mediated activation of Mecp2 increased the life span of Mecp2 mutants to >8 months. Also, a smaller but significant increase in life span by ≈4 weeks was observed when the Mecp2 transgene was activated by C93 and C159 Cre (Fig. 3 B).
Fig. 3.

Rescued animals have higher probability of survival than Mecp2 null littermates. Kaplan–Meier survival curves were generated by plotting the percentage of live mice (y axis) against number of days after birth (x axis). “n” indicates the number of animals analyzed. (A) Nestin (N) and Tau (T) Cre double-transgenic animals lived up to 280 days, whereas control mice died within 106 days after birth. (B) C93 and C159 mice lived up to 140 and 160 days, respectively (C159 vs. null: log rank test P = 0.0431; C93 vs. C159 P = 0.725). (C–E) Spontaneous nocturnal activity was measured by placing the animals in cages equipped with a movement detector (infrared beam). On the y axis, the number of beam interruptions over 10 h, each bar represents a different genotype. Animals were monitored for 5 weeks (C), during the symptomatic stage (5–15 weeks) (D), and later than 15 weeks (E). No significant difference between WT (black bar), mutant (white bar), and rescued mice was found in animals younger than 4 weeks of age. Between 5 and 15 weeks of age, the Mecp2 mutant animals became hypoactive, whereas rescued animals maintained normal levels of activity. At later age (E), the rescued animals showed lower activity that WT controls. Wt, KO, N, T, C93, and C159 are as in A and B.
Mecp2 mutant animals progressively lose motor coordination starting at 4 weeks and become severely lethargic. To assess whether activation of MeCP2 would affect the progressive behavioral alterations characteristic of the disorder, we tested nocturnal activity by placing animals in cages equipped with an infrared-beam movement detector. Fig. 3 C–E show the results of baseline activity of mice at different ages. As expected, there was no difference in nocturnal activity at 4–5 weeks in any of the genotypes (Fig. 3 C). At 6–15 weeks of age, Mecp2-null mice were significantly less active than WT mice (Fig. 3 D). Rescued mice carrying the Nestin Cre, the Tau Cre, or the C93 transgenes were as active as WT mice. C159 double-rescued mice were more active than Mecp2-null mutant mice but less active than the other genotypes. MeCP2 −/y animals (white bar) start to deteriorate and become uncoordinated and hypoactive, whereas all mutant mice carrying the rescue transgenes had nocturnal baseline activities very similar to WT level (black bar). No Mecp2-null mutant animals survived beyond 15 weeks of age and all other genotypes showed less activity than WT mice (Fig. 3 E).
These results suggest that Cre-mediated activation of the Mecp2 transgene significantly prolonged the life span and delayed behavioral deterioration. The extent of rescue was, however, directly correlated with the time and level of Cre transgene activation. Early activation of Cre in most neurons (Nestin Cre and Tau Cre) led to the most efficient rescue. Importantly, postnatal activation of MeCP2 between 1 and 2 weeks of age by the C93 Cre-driven transgene led to a significant rescue of symptoms, whereas later activation of Cre between 2 and 4 weeks, as in C159, was less efficient.
Physical Development and Brain Growth Are Normal in Rescued Animals.
In addition to motor dysfunction, Mecp2 mutant animals display decreased brain weight and smaller neurons and become obese with age (7). Fig. 4 A shows that body weight of mutant animals carrying any rescue transgene combination was indistinguishable from that of WT (black line) littermates, whereas MeCP2-deficient animals tended to gain weight (red line). Consistent with this finding, rescued animals showed no significant difference in brain weight when compared with WT littermates (Fig. 4 B). Finally, no significant decrease in neuronal cell size in hippocampus and cortex was seen in the C93 rescued animals, whereas Mecp2-null mutants have smaller sized neurons (7) (Fig. 4 C). Representative images from the hippocampi of these mice are shown in SI Fig. 6.
Fig. 4.

Rescued animals have normal physical development and brain weight. (A) Body weight in grams. We followed the weight of WT (black line) and rescued animals and found no significant difference, whereas null mutant animals (red line) became overweight starting at 5 weeks. By 8 weeks, they were significantly overweight compared with WT and rescued subjects. Nestin Cre*, purple line (n = 4); Tau Cre*, yellow line (n = 3); C93*, green line (n = 5); C159*, blue line (n = 5); WT, black line (n = 4); KO*, red line (n = 4) (*Mecp2 −/y; CAGGS LSL Mecp2). (B) Brain weights in grams. White circles represent WT, and red triangles represent Mecp2 null (*) brains. Green and yellow squares represent brains from C93 (*) rescued animals and Tau Cre* rescued animals, respectively. Paired t test WT vs. transgenic P = 0.000110 (using the first four time points); WT vs. C93 P = 0.0788. (C) Neurons of C93 double-transgenic animals have a normal nuclear size. Two-month-old C93 Cre double-transgenic (gray bar), control WT (black bar), and null transgenic (white bar) littermates were analyzed for the comparison. Images were taken from the primary motor cortex (layer V) at ×40 magnification, and measurements from at least 200 cells were used for each bar. The animals used for the analysis were 8 weeks old. WT, KO, C93 as described above.
Discussion
An unresolved issue in RTT is whether the manifestation of overt symptoms can be affected by restoring MeCP2 expression in postnatal neurons. Previous results showed that Tau promoter-driven activation of MeCP2 in postmitotic mutant neurons prevents development of an obvious phenotype, consistent with the conclusion that RTT is not a developmental disorder but rather is caused by MeCP2 deficiency in mature neurons (31). However, these experiments did not resolve the important question of whether later reactivation of MeCP2 in postnatal life could affect the phenotype progression. Here, we show that induction of a Mecp2 transgene in postnatal mutant animals delayed onset of symptoms and time of death.
To achieve activation of MeCP2 in postnatal neurons, we constructed a conditionally active rescue transgene by placing a LoxP–Stop–LoxP cassette in front of the Mecp2e2 cDNA. MeCP2 was activated by crossing the conditional transgene into different Cre lines that induced recombination during early development (Nestin Cre and Tau Cre), at 2 weeks (C93) or between 2 and 4 weeks of age (C159). Immunohistochemistry indicated that the Nestin and Tau Cre transgenes activated Mecp2 in all regions of the brain, whereas expression of the CamKinaseII Cre transgenes was restricted to a smaller fraction of cells in a given region and was not detected in the cerebellum.
Mecp2-null animals are characterized by severe motor impairment and lethargy beginning at ≈4 weeks of age and death at 8–12 weeks of age. These phenotypes were significantly delayed when the Mecp2 rescue transgenes were activated. The extent of the rescue seemed to depend on the timing of Mecp2 activation and on the fraction of neurons that had undergone Cre-mediated recombination. Thus, Nestin Cre- and Tau Cre-mediated MeCP2 activation extended the life span of mutant mice to 8 months, whereas the C93 and C159 transgenes extended life span by ≈4 weeks. A significant improvement of basal nocturnal activity was found in mutant animals carrying the rescue transgenes. Finally, anatomical alterations typical for RTT, such as a decreased brain weight and neuronal cell size, were not seen in rescued animals.
The most important conclusion from these results is that reactivation of MeCP2 in mutant animals even as late as 2–4 weeks of age can result in a significant amelioration of RTT-like symptoms. However, full restoration of the phenotype was not achieved with any of the rescue transgenes, even when the transgene was activated early in development in progenitor cells (Nestin Cre) or at a time when neurons become postmitotic (Tau Cre). Inappropriate expression of MeCP2 probably contributed to this partial rescue. The immunohistochemical data indicated that temporal and spatial expression of the transgene varied considerably depending on the Cre line used, with 80% of cells expressing MeCP2 in brains of the Nestin and Tau rescued mice and only 10% expressing MeCP2 in the brains of C159 animals. Insufficient or no activation may have occurred in thalamus, cerebellum, brainstem, and medulla (CamKinaseII Cre lines) that may be important for the RTT phenotype. For example the cerebellum plays an important role in the integration of sensory perception and motor output, and deficiencies in the medullar respiratory network could be the cause of fatal breathing irregularities in Mecp2-null mice (42). Lack of expression of Mecp2 in these areas may have contributed to the reduced motor activity and viability of the CamKinaseII Cre rescued animals. In addition, CAGGS promoter-driven Mecp2 expression may have been inappropriate, (too low or too high) in a given neuron, compared with WT levels. Overexpression of MeCP2 has been shown to have detrimental effects on CNS function both in humans and mice (31, 43–45). We note that expression of MeCP2 from the Tau locus (31) fully rescued the phenotype, yet expression of Cre from the same locus was not sufficient to achieve complete rescue in our system. Cre-mediated excision is a stochastic event that does not happen at the same time in all cells because it depends on both the level of Cre enzyme and the availability of the flanking LoxP sites. This may have created mosaicism within the neuronal population. Direct expression from the Tau locus may have resulted in a higher fraction of neurons expressing appropriate levels of MeCP2, emphasizing the limitations of the Cre system.
A major unresolved issue in the pathogenesis of RTT is whether the disorder is caused by dysfunction of postnatal neurons at a time when symptoms become apparent or whether it is a prenatal developmental disorder with postnatal phenotypic manifestation in the CNS. Our results are consistent with the notion that the physical deterioration of null mice can be rescued by reactivation of MeCP2, even at an age when overt symptoms are about to commence. This finding implies that newborn RTT individuals might not have irreversible neuronal damage and that pathological alterations of neurons may occur only later in life. If correct, this presumption would encourage developing therapeutic strategies aimed at affecting the course of the RTT by reversing gene-expression abnormalities caused by MeCP2 deficiency. Although it would be difficult to reverse a mutation in the Mecp2 gene, it may be possible to modulate the activity of downstream genes that have been shown to impact the phenotype progression, such as Bdnf (24). Identification of such downstream genes should further advance our understanding of the molecular basis of RTT.
Methods
Generation of the Conditional Mecp2 LSL Transgene.
We cloned a NcoI/SpeI Mecp2e2 cDNA pA fragment from (31) into pBluscript. The CAGGS promoter (35) was inserted into the SalI-EcoRI site upstream of Mecp2pA. The LoxP–Stop–LoxP neo-resistance cassette was cloned into the EcoRI site between the CAGGS and the Mecp2pA (36). The whole CAGGS LSL Mecp2 pA fragment was released from pBluscript with a SalI digest, blunted, and inserted into the EcoRV site of pKATGfrt plasmid (37). We targeted the Cola1 locus in C10 cells (37). Hygromycin-resistant ES clones were analyzed by Southern blot (37). Positively targeted clones were injected into C57BL/6-DBA2 blastocysts. Chimeric offspring were bred to C57BL/6 mice and screened for germ-line transmission.
Mating Schemes and Genotyping.
Male mice homozygous for the LSL transgene were mated to Mecp2+/− females from the germ-line null colony (7) to obtain LSL/+; Mecp2+/− females that were then mated to males carrying the Cre transgene. Genotyping for the Mecp2 locus and for the Cre was done as published (7, 40). Genotyping for the Col1a1 locus was done as in ref. 24.
Isolation of Frontal Cortex, Hippocampus, and Cerebellum.
For forebrain samples, the forebrain was separated from the mid- and hindbrain by a coronal cut along the rostral border of the superior colliculi. For hippocampus samples: the cerebellum was removed, and the brains were cut midsagittally; under a dissecting microscope, the anterior border of the hippocampus was identified as the fimbria hippocampi; dorsally, the hippocampal white matter was carefully separated from overlying cerebral cortex along the alveus; and, finally, the subiculum was bisected to release the hippocampal gray matter. For brain weight analysis, brains were freshly isolated and weighed.
Immunoblot Analysis.
Organs were harvested and snap-frozen in liquid nitrogen. Tissues were homogenized with a Polytron in a lysis buffer containing 125 mM Tris and 1% SDS (pH 6.8) supplemented with a proteinase-inhibitor mix (Roche, Indianapolis, IN). Protein concentrations were determined by BCA (Pierce, Rockford, IL). Sample buffer was added to a final concentration of 12.5% glycerol and 0.25% 2-mercaptoethanol. A total of 50 μg of protein was loaded on 4–12% Nu-Page gel, probed with an anti-MeCP2 rabbit polyclonal antibody (cat. no. 07-013; Upstate Biotechnology, Lake Placid, NY) or anti-GAPDH rabbit polyclonal (Abcam, Cambridge, MA) and visualized by using ECL (Amersham Pharmacia, Piscataway, NJ).
Immunohistochemistry.
Brains were perfused with 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4), postfixed in 4% paraformaldehyde overnight at 4°C, washed in PBS, processed by TISSUE-TEK VIP machine (Miles Scientific, Naperville, IL), and imbedded in paraffin. Five-micrometer serial sections were deparaffinized and treated with 10 μg/ml proteinase K in 50 mM Tris/5 mM EDTA for 10 min at room temperature. Slides were blocked in 5% normal goat serum and incubated with anti-MeCP2 polyclonal (23) and mouse anti-NeuN (Chemicon, Temecula, CA) at a dilution of 1:200. The primary antibodies were detected with a rhodamine-conjugated goat anti-rabbit IgG or with FITC-conjugated donkey anti-mouse IgG (Jackson ImmunoResearch, West Grove, PA), both at a dilution of 1:500. Sections were finally rinsed in PBS DAPI and mounted in Vectashield (Vector Laboratories, Burlingame, CA). Images were taken with a fluorescence microscope (Leica, Bannockburn, IL) at ×40 magnification.
Nuclear Size Measurements.
For nuclear size measurement, brains were perfused and imbedded in paraffin as described above. Four-micrometer serial coronal sections were prepared and stained as described above. Digital pictures from the hippocampus (CA1–CA2) and somatosensory cortex LV were taken, and the nuclear area (arbitrary unit) of the neuron was determined by tracing the outline of the nucleus in the DAPI channel, by using OpenLab. Neurons were identified by NeuN staining, and the results are expressed as the fraction of NeuN-positive cells that showed MeCP2 signal. Images were collected from at least two animals (8 weeks old) per genotype; 200 cells per animal were counted.
Spontaneous Activity Measurements.
Motor activity was measured by using an infrared beam-activated movement-monitoring chamber (Opto-Varimax-MiniA; Columbus Instruments, Columbus, OH). For each experiment, a mouse was placed in the chamber at least 3 h before recordings started. Movement was monitored during the normal 12-h dark cycle (7 p.m. to 7 a.m.). A total of seven WT mice, four transgenic controls, four NestinCre, four Tau Cre, seven CamkII93 Cre, and five CamkII159 Cre double-transgenic mice were used. One dark cycle per animal per time point was collected.
Supplementary Material
Acknowledgments
We thank of Jessica Dausman, Ruth Flannery, and Dong Dong Fu for excellent technical assistance; George Bell for statistical analysis; and Grant Welstead, Qiang Chang, and Heinz Linhart for critical reading of the manuscript. This work was supported by a predoctoral fellowship from the Boehringer Ingelheim Foundation (to E.G.), the Rett Syndrome Research Foundation, and National Institutes of Health Grants R01-HD045022, R37-CA084198, and R01-CA087869.
Abbreviation
- RTT
Rett syndrome.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/cgi/content/full/0610593104/DC1.
References
- 1.Hagberg B, Hagberg G. Eur Child Adolesc Psychiatry. 1997;6(Suppl 1):5–7. [PubMed] [Google Scholar]
- 2.Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 3.Van den Veyver IB, Zoghbi HY. Curr Opin Genet Dev. 2000;10:275–279. doi: 10.1016/s0959-437x(00)00083-6. [DOI] [PubMed] [Google Scholar]
- 4.Wan M, Lee SS, Zhang X, Houwink-Manville I, Song HR, Amir RE, Budden S, Naidu S, Pereira JL, Lo IF, et al. Am J Hum Genet. 1999;65:1520–1529. doi: 10.1086/302690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xiang F, Buervenich S, Nicolao P, Bailey ME, Zhang Z, Anvret M. J Med Genet. 2000;37:250–255. doi: 10.1136/jmg.37.4.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guy J, Hendrich B, Holmes M, Martin JE, Bird A. Nat Genet. 2001;27:322–326. doi: 10.1038/85899. [DOI] [PubMed] [Google Scholar]
- 7.Chen RZ, Akbarian S, Tudor M, Jaenisch R. Nat Genet. 2001;27:327–331. doi: 10.1038/85906. [DOI] [PubMed] [Google Scholar]
- 8.Shahbazian M, Young J, Yuva-Paylor L, Spencer C, Antalffy B, Noebels J, Armstrong D, Paylor R, Zoghbi H. Neuron. 2002;35:243–254. doi: 10.1016/s0896-6273(02)00768-7. [DOI] [PubMed] [Google Scholar]
- 9.Dani VS, Chang Q, Maffei A, Turrigiano GG, Jaenisch R, Nelson SB. Proc Natl Acad Sci USA. 2005;102:12560–12565. doi: 10.1073/pnas.0506071102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Young JI, Hong EP, Castle JC, Crespo-Barreto J, Bowman AB, Rose MF, Kang D, Richman R, Johnson JM, Berget S, et al. Proc Natl Acad Sci USA. 2005;102:17551–17558. doi: 10.1073/pnas.0507856102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gemelli T, Berton O, Nelson ED, Perrotti LI, Jaenisch R, Monteggia LM. Biol Psychiatry. 2006;59:468–476. doi: 10.1016/j.biopsych.2005.07.025. [DOI] [PubMed] [Google Scholar]
- 12.Lewis JD, Meehan RR, Henzel WJ, Maurer-Fogy I, Jeppesen P, Klein F, Bird A. Cell. 1992;69:905–914. doi: 10.1016/0092-8674(92)90610-o. [DOI] [PubMed] [Google Scholar]
- 13.Nan X, Campoy FJ, Bird A. Cell. 1997;88:471–481. doi: 10.1016/s0092-8674(00)81887-5. [DOI] [PubMed] [Google Scholar]
- 14.Klose RJ, Bird AP. Trends Biochem Sci. 2006;31:89–97. doi: 10.1016/j.tibs.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 15.Fuks F, Hurd PJ, Wolf D, Nan X, Bird AP, Kouzarides T. J Biol Chem. 2003;278:4035–4040. doi: 10.1074/jbc.M210256200. [DOI] [PubMed] [Google Scholar]
- 16.Traynor J, Agarwal P, Lazzeroni L, Francke U. BMC Med Genet. 2002;3:12. doi: 10.1186/1471-2350-3-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tudor M, Akbarian S, Chen RZ, Jaenisch R. Proc Natl Acad Sci USA. 2002;99:15536–15541. doi: 10.1073/pnas.242566899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matarazzo V, Ronnett GV. Proc Natl Acad Sci USA. 2004;101:7763–7768. doi: 10.1073/pnas.0307083101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nuber UA, Kriaucionis S, Roloff TC, Guy J, Selfridge J, Steinhoff C, Schulz R, Lipkowitz B, Ropers HH, Holmes MC, et al. Hum Mol Genet. 2005;14:2247–2256. doi: 10.1093/hmg/ddi229. [DOI] [PubMed] [Google Scholar]
- 20.Ballestar E, Ropero S, Alaminos M, Armstrong J, Setien F, Agrelo R, Fraga MF, Herranz M, Avila S, Pineda M, et al. Hum Genet. 2005;116:91–104. doi: 10.1007/s00439-004-1200-0. [DOI] [PubMed] [Google Scholar]
- 21.Vetter ML. Dev Cell. 2003;5:359–360. doi: 10.1016/s1534-5807(03)00267-3. [DOI] [PubMed] [Google Scholar]
- 22.Martinowich K, Hattori D, Wu H, Fouse S, He F, Hu Y, Fan G, Sun YE. Science. 2003;302:890–893. doi: 10.1126/science.1090842. [DOI] [PubMed] [Google Scholar]
- 23.Chen WG, Chang Q, Lin Y, Meissner A, West AE, Griffith EC, Jaenisch R, Greenberg ME. Science. 2003;302:885–889. doi: 10.1126/science.1086446. [DOI] [PubMed] [Google Scholar]
- 24.Chang Q, Khare G, Dani V, Nelson S, Jaenisch R. Neuron. 2006;49:341–348. doi: 10.1016/j.neuron.2005.12.027. [DOI] [PubMed] [Google Scholar]
- 25.Zhou Z, Hong EJ, Cohen S, Zhao WN, Ho HY, Schmidt L, Chen WG, Lin Y, Savner E, Griffith EC, et al. Neuron. 2006;52:255–269. doi: 10.1016/j.neuron.2006.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun YE, Wu H. Neuron. 2006;49:321–323. doi: 10.1016/j.neuron.2006.01.014. [DOI] [PubMed] [Google Scholar]
- 27.Makedonski K, Abuhatzira L, Kaufman Y, Razin A, Shemer R. Hum Mol Genet. 2005;14:1049–1058. doi: 10.1093/hmg/ddi097. [DOI] [PubMed] [Google Scholar]
- 28.Samaco RC, Hogart A, LaSalle JM. Hum Mol Genet. 2005;14:483–492. doi: 10.1093/hmg/ddi045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Horike S, Cai S, Miyano M, Cheng JF, Kohwi-Shigematsu T. Nat Genet. 2005;37:31–40. doi: 10.1038/ng1491. [DOI] [PubMed] [Google Scholar]
- 30.Jordan C, Francke U. Hum Mol Genet. 2006;15:2210–2215. doi: 10.1093/hmg/ddl146. [DOI] [PubMed] [Google Scholar]
- 31.Luikenhuis S, Giacometti E, Beard CF, Jaenisch R. Proc Natl Acad Sci USA. 2004;101:6033–6038. doi: 10.1073/pnas.0401626101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Quenard A, Yilmaz S, Fontaine H, Bienvenu T, Moncla A, des Portes V, Rivier F, Mathieu M, Raux G, Jonveaux P, et al. Eur J Med Genet. 2006;49:313–322. doi: 10.1016/j.ejmg.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 33.Mnatzakanian GN, Lohi H, Munteanu I, Alfred SE, Yamada T, MacLeod PJ, Jones JR, Scherer SW, Schanen NC, Friez MJ, et al. Nat Genet. 2004;36:339–341. doi: 10.1038/ng1327. [DOI] [PubMed] [Google Scholar]
- 34.Kriaucionis S, Bird A. Nucleic Acids Res. 2004;32:1818–1823. doi: 10.1093/nar/gkh349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Niwa H, Yamamura K, Miyazaki J. Gene. 1991;108:193–199. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- 36.Soriano P. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- 37.Beard C, Hochedlinger K, Plath K, Wutz A, Jaenisch R. Genesis. 2006;44:23–28. doi: 10.1002/gene.20180. [DOI] [PubMed] [Google Scholar]
- 38.Dubois NC, Hofmann D, Kaloulis K, Bishop JM, Trumpp A. Genesis. 2006;44:355–360. doi: 10.1002/dvg.20226. [DOI] [PubMed] [Google Scholar]
- 39.Tucker KL, Meyer M, Barde YA. Nat Neurosci. 2001;4:29–37. doi: 10.1038/82868. [DOI] [PubMed] [Google Scholar]
- 40.Fan G, Beard C, Chen RZ, Csankovszki G, Sun Y, Siniaia M, Biniszkiewicz D, Bates B, Lee PP, Kuhn R, et al. J Neurosci. 2001;21:788–797. doi: 10.1523/JNEUROSCI.21-03-00788.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Minichiello L, Korte M, Wolfer D, Kuhn R, Unsicker K, Cestari V, Rossi-Arnaud C, Lipp HP, Bonhoeffer T, Klein R. Neuron. 1999;24:401–414. doi: 10.1016/s0896-6273(00)80853-3. [DOI] [PubMed] [Google Scholar]
- 42.Viemari JC, Roux JC, Tryba AK, Saywell V, Burnet H, Pena F, Zanella S, Bevengut M, Barthelemy-Requin M, Herzing LB, et al. J Neurosci. 2005;25:11521–11530. doi: 10.1523/JNEUROSCI.4373-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Van Esch H, Bauters M, Ignatius J, Jansen M, Raynaud M, Hollanders K, Lugtenberg D, Bienvenu T, Jensen LR, Gecz J, et al. Am J Hum Genet. 2005;77:442–453. doi: 10.1086/444549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Meins M, Lehmann J, Gerresheim F, Herchenbach J, Hagedorn M, Hameister K, Epplen JT. J Med Genet. 2005;42:e12. doi: 10.1136/jmg.2004.023804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Collins AL, Levenson JM, Vilaythong AP, Richman R, Armstrong DL, Noebels JL, David Sweatt J, Zoghbi HY. Hum Mol Genet. 2004;13:2679–2689. doi: 10.1093/hmg/ddh282. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}