

Abstract
The DYT1 gene containing a trinucleotide deletion (ΔGAG) is linked to early-onset dystonia, a neurological movement disorder of involuntary muscle contractions. To understand Dyt1’s contribution to dystonia, we produced and analyzed Dyt1 knockdown (KD) mice that expressed a reduced level of torsinA protein encoded by Dyt1. Knockdown mice exhibited deficits in motor control and a decreased trend in dopamine with a significant reduction in 3,4-dihydroxyphenylacetic acid. These alterations are similar to those displayed by previously reported Dyt1 ΔGAG knockin heterozygous mice, suggesting that the partial loss of torsinA function contributes to the pathology of the disease.
Keywords: Dyt1, torsinA, dystonia, knockdown, Tor1A, Dyt1 ΔGAG
A trinucleotide deletion (ΔGAG) in the DYT1 gene is found in a majority of patients with Oppenheim’s early-onset dystonia, a neurological disorder of uncontrollable muscle contractions (Ozelius et al., 1997). DYT1 codes for torsinA protein, and the ΔGAG deletion removes a glutamic acid (ΔE) from the protein. The role of mutant torsinA in the development of dystonia is unknown, but possible functions of normal torsinA were reported to include its involvement in cytoskeletal dynamics, nuclear membrane formation, and neuroprotection (Kuner et al., 2003; Bragg et al., 2004; Gonzalez-Alegre & Paulson, 2004; Goodchild & Dauer, 2004; Shashidharan et al., 2004; Hewett et al., 2006; Kock et al., 2006). Also unknown is the nature of the genetic mutation in torsinA, an important aspect of the pathophysiology of dystonia that may affect the development of genetic-based therapeutics.
The ΔGAG mutation of DYT1 has been speculated to work through a toxic-gain-of-function mechanism by reports of protein aggregates caused by an overexpression of mutant torsinA in cultured cells and Drosophila (Hewett et al., 2000; Kustedjo et al., 2000; Koh et al., 2004). Also, in patient tissues and two mouse models, an overexpression transgenic and our Dyt1 ΔGAG knockin (Dyt1 ΔGAG) mouse lines, aggregates containing torsinA and ubiquitin were found primarily in the brainstem, even though torsinA is widely expressed throughout the brain (McNaught et al., 2004; Dang et al., 2005; Shashidharan et al., 2005). In contrast, other published findings have suggested that ΔGAG causes a loss of normal protein function (Torres et al., 2004; Goodchild et al., 2005). For example, nuclear envelope abnormalities were noted in neurons of Dyt1 knockout and homozygous Dyt1 ΔGAG mice, indicating that the mutant protein may be equivalent to the absence of the protein (Goodchild et al., 2005).
To further address the genetic mechanism of the ΔGAG mutation, we report behavioral and biochemical analyses of Dyt1 knockdown (KD) mice that displayed motor deficits similar to the behavioral phenotype previously shown in our Dyt1 ΔGAG mice that carry one mutant allele. This is the first report to demonstrate that reducing torsinA expression can lead to abnormal motor development and biochemical changes in the brain.
All experimental procedures in this report were carried out in compliance with the USPHS Guide for Care and Use of Laboratory Animals and approved by University of Illinois Institutional Animal Care and Use Committee.
Dyt1 KD mouse was generated in the process of making Dyt1 ΔGAG mice (Dang et al., 2005). The targeting vector used had the implicated GAG removed in exon 5 of Dyt1 and a STOP sequence (Lakso et al., 1992) containing a false translation signal, splice donor site and a poly(A) tail inserted into intron 4 (Fig. 1a). The Dyt1 KD could be generated using this construct because of the multiple sites of recombination that occurred in transfected stem cells. Twenty eight of 73 clones screened after transfection had homologously recombined the targeting vector as determined by Southern blot analysis (Dang et al., 2005). The targeted clones were further screened for the presence of ΔGAG by amplification of a 360-bp sequence spanning across ΔGAG of exon 5 that was then digested with EarI, which yielded two different digest patterns specific to the presence or absence of ΔGAG. From this screening, 25 of the 28 clones were found not to contain ΔGAG. In these clones, it appeared that an additional undesirable recombination occurred upstream of ΔGAG (Fig. 1b). Germline transmitted mice produced from these colonies were first named Dyt1 STOP. These mice were genotyped and distinguish from wild-type mice (WT) by PCR using the following two pairs of primers, 84, 5′-CGCTTGGGTGGAGAGGCTATTC-3′ and 85, 5′-GCAAGGTGAGATGACAGGAGATC-3′ and tcko1 and tcko2 (Dang et al., 2005).
Fig. 1.
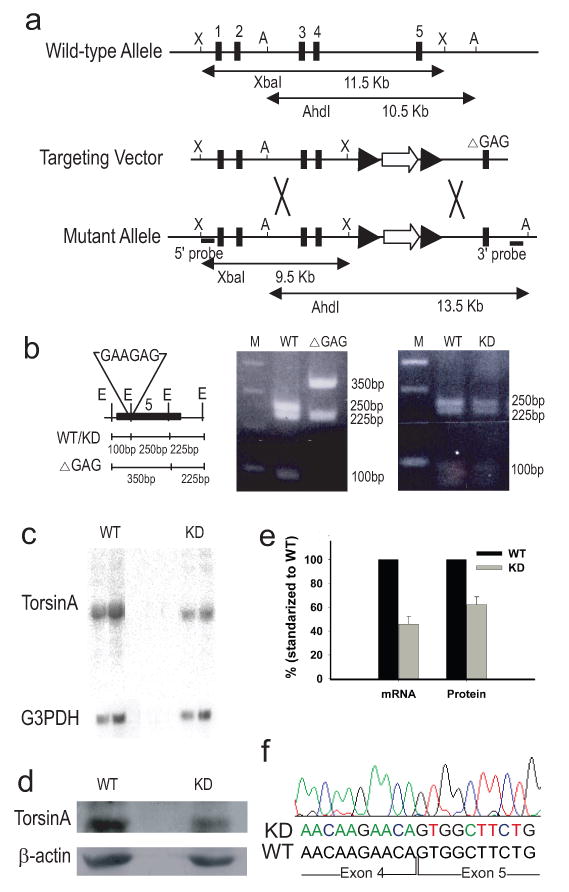
Generation of Dyt1 KD mice. (a) Targeting construct and the genomic organization of Dyt1 gene. Exon 5 in the targeting construct carried a GAG deletion, which was not homologously recombined into the modified allele of targeted ES cells due to a frequent recombination spot upstream of ΔGAG. The sizes and locations of the restriction fragments for the identification of targeted clones are indicated. Black rectangles with number: exons; black arrowheads: loxP sites; open arrow: PGKNeoSTOP cassette; X: XbaI site; A, AhdI site. Big X: possible sites of recombination. (b) Digestion of a 360-bp sequence spanning across ΔGAG of exon 5 with EarI gave two different digest patterns from sequences with and without ΔGAG. Dyt1 KD mice were generated from colonies that had the PGKNeoSTOP cassette, but did not have GAG. Black rectangle: exon 5; E: EarI; M: standard marker ladder. (c) Northern blot analysis of total RNA samples from brain tissues of WT and Dyt1 KD mice showed Dyt1 KD mice had 46% ± 6% (P < 0.0001, n=3 WT, 3 KD) of Dyt1 mRNA produced in WT mice. G3PDH quantity was used for loading control. (d) Western blot analysis of torsinA protein from brain tissues of WT and Dyt1 KD mice showed Dyt1 KD mice had 64% ± 5% (P = 0.009, n=3 WT, 3 KD) of torsinA protein expressed in WT mice. β-actin was used for loading control. (e) mRNA and protein levels of torsinA in KD mice relative to that of WT mice. (f) Trace and sequence of Dyt1 mRNA transcript in KD mice at the splice junction between exons 4 and 5 showed that KD mice have mRNA transcripts that are unaltered.
To quantify the expression of Dyt1 transcript, total brain RNA was extracted from three pairs of adult Dyt1 KD and WT mice using RNeasy Protect Kit (Qiagen). Northern blot analysis was performed using two radioactive DNA probes, one specific for exon 5 and the other for exons 1 to 4. Blots were analyzed using a Fuji Phosphor Imager BAS1000. The membranes were then reprobed with a G3PDH cDNA probe (Promega). Quantification of Dyt1 mRNA transcript was performed with standardization to G3PDH.
To quantify the expression of torsinA protein, Western blot analysis was performed using whole brain protein that was extracted from 3 pairs of adult Dyt1 KD and WT mice with the following lysate buffer: 100 mM Tris-HCl (pH 8.0), 1% SDS, 150 mM NaCl and protease inhibitor cocktail (Sigma Aldrich). Protein extract was separated on a 13% SDS-PAGE acrylamide gel. The transferred membrane was blocked with nonfat milk, incubated at 4°C overnight in primary torsinA antibody, DM2A8 (Hewett et al., 2004), and processed with an ECL detection analysis system (GE Healthcare). Membranes with reprobed with β-actin antibody (Chemicon). Image J (NIH) was used to quantify torsinA protein standardized to β-actin quantity.
To confirm that the gene cassette inserted between exons 4 and 5 of Dyt1 did not alter the expressed Dyt1 mRNA transcript in KD mice by either prematurely terminating transcription or interfering with splicing of the exons, Dyt1 mRNA in KD mice was sequenced. RT-PCR was performed using Qiagen One-Step RT-PCR kit and primers specific to sequences in exons 1 and 5.
All behavioral tests were performed by investigators blind to the genotypes of the mice. The test group comprised of 12 WT females, 13 Dyt1 KD females, 8 WT males, and 8 Dyt1 KD males at 6 months of age. Mice were observed on the rotarod and beam-walking tests. The first test measures gross motor coordination and balance while the second measures finer changes in motor control. For the rotarod test, an Economex accelerating rotarod (Columbus Instruments) was used as previously described (Dang et al., 2005). Briefly, the apparatus has an initial speed of 4 rpm and gradually accelerated at a rate of 0.2 rpm/sec. The latency to fall was measured with a cutoff time of 2 min. Mice were tested for three trials on each day for two days. The beam-walking test was performed also as previously described (Dang et al., 2005). Animals were trained to traverse a medium square beam (14 mm wide) and then tested on the training beam and three additional beams (round: 17 mm and 10 mm diameter, square: 7 mm wide). Beam transversal time and number of hindpaw slips for each of the two trials per beam were measured.
Mice were also tested for spontaneous activity in the open-field apparatus (AccuScan Instruments) for 15 min using DigiPro software (Dang et al., 2005). Statistical analysis was performed using SAS/STAT and SAS/Analyst software (Versions 8.2 and 9.1) mainly with ANOVA or by logistic regression (GENMOD) as previously described with significance assigned at the P ≤ 0.05 level (Dang et al., 2005).
For striatal tissue dopamine (DA) and metabolite measurements, striata were dissected from Dyt1 KD and WT male mice of 8 to 9 months old (n = 8 KD, 14 WT) and measured by HPLC as previously described (Dang et al., 2005). One-way ANOVA was used to analyze these measurements as well as the protein and RNA measurements. Means and standard errors were obtained using Tukey’s HSD method.
Since the main rate limiting step of DA turnover is catalyzed by monoamine oxidase B (MAO-B), we measured the activity of MAO-B using MAO-Glo Assay kit (Promega). Striata were dissected from 4 WT and 3 KD mouse brains and homogenized for 1 min in nine volumes of ice-cold 100mM HEPES-NaOH (pH7.5), 5% glycerol solution. Aliquot of the homogenate corresponding to 50 ng of wet tissue was used for each MAO-B reaction for one hour. The luminescent signal was measured for 12 seconds by a Monolight 2010 luminometer (Analytical Luminescence Laboratory). The experiment was done in duplicate. MAO-B activity was expressed as RLU/hour/ng of tissue wet weight.
Pups of the first 22 litters of Dyt1 STOP had the following normal Mendelian genotype ratios: 48 WT, 84 heterozygotes, and 39 homozygotes. Although the Dyt1 knockout genotype is lethal (Goodchild et al., 2005), our homozygous Dyt1 STOP mice survived into adulthood, indicating that termination of transcription with the STOP sequence was incomplete. To determine the degree of transcription termination, Dyt1 expression of homozygotes was measured. Northern blot analysis using two different probes approximated Dyt1 mRNA in homozygous Dyt1 STOP mice to be 54% less than the level seen in WT mice (Fig. 1c and e). It is important to note that no additional RNA band was detected using either probe, which showed the absence of additional hybrid RNA molecules that could produce hybrid proteins containing any portion of torsinA. Nonsense-mediated mRNA decay has been well documented in mammalian cells (Frischmeyer, 1999). Western blot analysis showed that homozygous Dyt1 STOP mice expressed approximately 36% less protein than the level expressed in WT mice (Fig. 1d and e). For this reason, we called this mouse line Dyt1 KD and proceeded to characterize their phenotype. Sequencing of RT-PCR product from Dyt1 mRNA showed that KD mice had Dyt1 mRNA that was identical to that of WT mice. All four splice sites were preserved and the sequence of exon 5 was not missing from the transcript (Fig. 1f).
Motor behavioral tests showed that both the WT and Dyt1 KD mice of the 6-month-old group learned to walk on the rotarod and improved significantly over the course of six trials [F(5,200) = 16.99, P < 0.001]. There was no significant difference in latency to falling between WT and Dyt1 KD mice [F(1,38) = 0.12, P = 0.73; Figure 2a] across all trials.
Fig. 2.
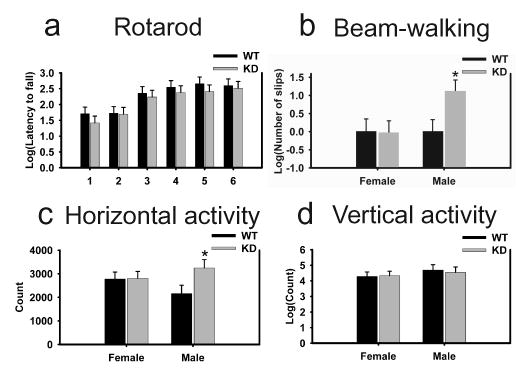
Motor performance of Dyt1 KD mice on rotarod, beam-walking and open-field tests. (a) Dyt1 KD mice performed comparable to WT mice on the accelerated rotarod test. 1–6: trial number. (b) Male Dyt1 KD mice showed a significantly larger number of slips as they crossed the beam with an almost 200% increase in relative number of slips (WT mice=1 slip). (c) Horizontal activity was significantly higher in male Dyt1 KD mice than WT controls. (d) No difference was observed in vertical activity of Dyt1 KD versus WT mice.*P ≤ 0.05.
In the beam-walking test, there was no significant difference between WT and Dyt1 KD mice in their latency to crossing [F(1,36) = 0.12, P = 0.73; data not shown]. Analysis of the beam-walking slips showed a significant interaction between genotype and sex (DF=1, χ2=6.00, P = 0.014, GENMOD). Although there was no significant difference between female WT and Dyt1 KD mice (P = 0.98), male Dyt1 KD mice displayed a significant increase of nearly 218% more slips than their WT littermates (P = 0.016, parameter estimate = e1.15 ± 0.48, Fig. 2b).
Spontaneous movement levels of mice were measured in the open-field apparatus. The statistical analysis of horizontal activity showed a genotype and sex interaction that approached significance [F(1,36) = 2.67, P = 0.11]. Pairwise comparison showed a significant difference between Dyt1 KD and WT male mice [P = 0.037, Fig. 2c], with Dyt1 KD more hyperactive than WT mice. Vertical activity between Dyt1 KD and WT mice did not differ [F(1,36) = 0.03, P = 0.87, Fig. 2d].
We also measured levels of striatal DA and DA metabolites, 3,4-dihydroxyphenylacetic acid (DOPAC) and homovanillic acid (HVA). Dyt1 KD mice had lower trends without significant differences in quantities of DA (P = 0.25) and HVA (P = 0.45; Figure 3). A significant reduction, however, was seen in DOPAC levels of Dyt1 KD versus WT mice (P = 0.04). Also, no significant difference was detected in the ratios of DOPAC to DA (P = 0.28) and HVA to DA (P = 0.48). Measurement of MAO-B activity to determine if the lower DOPAC with due to lower MAO activity showed no significant difference between that of WT and KD mice (mean +/− standard deviation, CT: 981+/−133, KD: 945+/−99, student’s t-test: P = 0.72).
Fig. 3.
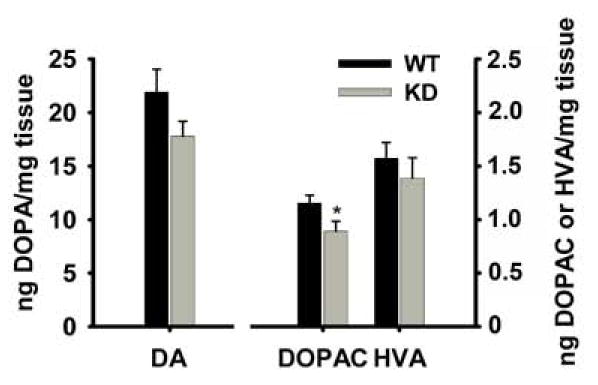
HPLC analysis of dopamine and metabolite levels showed a neurochemical change in KD mouse striatal tissues. Striatal DOPAC quantities were significantly lower in KD mice, but DA and DOPAC quantities were not significantly different between the two groups. *P < 0.05.
The motor deficit observed in Dyt1 KD mice showed that a reduction in torsinA can disrupt normal motor development, which replicates rather closely the disruption caused by the ΔGAG mutation of Dyt1 ΔGAG mice. Similar to the Dyt1 ΔGAG mice, Dyt1 KD mice showed no deficits in rotarod performance while displaying a significant increase in beam-walking slips. Furthermore, the hyperactivity, as indicated by heightened horizontal activity levels, seen previously in Dyt1 ΔGAG mice was also present in Dyt1 KD mice. The similarities between the two mouse lines also extend to the age of onset of beam-walking slips. Like Dyt1 ΔGAG mice, younger Dyt1 KD mice of approximately 3 months of age exhibited only a trend of, but not significant, increase in the number of slips during the beam-walking tests (data not shown), while in older mice the difference was significant. In addition, the appearance of motor abnormalities in males and not in females KD mimicked the sex-dependent deficit previously noted in Dyt1 ΔGAG mice.
In Dyt1 KD mice, DOPAC was significantly lower compared to WT while in Dyt1 ΔGAG mice, HVA was significantly lower. The reduced metabolites and the difference in type of metabolite that appeared lower in KD and Dyt1 ΔGAG mice may not be as consequential to the pathology as the overall trend of decrease in DA and metabolites seen in both lines of mice, considering that MAO activity is unchanged in KD mice. A shift of decreased striatal DA has been previously reported in dystonic patients and a transgenic Dyt1 mouse model (Furukawa et al., 2000; Shashidharan et al., 2005). Overall, an altered neurochemistry of the dopaminergic system seems to be a well documented part of early-onset dystonia’s pathology, and it is present in our KD mice as well.
Our KD mouse model showed that a reduced level of normal torsinA caused deficits similar to that seen in knockin mice containing the mutant protein. The presence of mutant torsinA does not appear to be required for the development of the described behavioral and biochemical alterations. While this is support for a loss-of-function genetic mechanism, it must be noted that these findings do not exclude the possibility of a potential dominant-negative function of the ΔGAG mutation. The presence of mutant torsinA has in fact been shown to reduce the overall level of torsinA (Goodchild et al., 2005), suggesting that either the mutant protein is unstable by itself or it may interact also with normal torsinA to destabilize it, the former pointing to a loss-of-function and the latter to a dominant-negative mechanism (Breakefield et al., 2001). While this issue of dominant negative versus loss of function has yet to be elucidated, our findings narrow the search for the genetic mechanism of the GAG mutation by indicating that a toxic gain-of-function is not a likely candidate.
Acknowledgments
We thank Jianyong Li and Shinichi Mitsui for their technical assistance. We also thank Vijaya Ramesh and Jeff Hewett for the torsinA antibody and technical advice. This work was supported by funds from the Dystonia Medical Research Foundation, Bachmann-Strauss Dystonia & Parkinson Foundation, Inc., NIH/NINDS NS047692, and the Lucille P. Markey Charitable Trust.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bragg DC, Camp SM, Kaufman CA, Wilbur JD, Boston H, Schuback DE, Hanson PI, Sena-Esteves M, Breakefield XO. Perinuclear biogenesis of mutant torsin-A inclusions in cultured cells infected with tetracycline-regulated herpes simplex virus type 1 amplicon vectors. Neuroscience. 2004;125:651–661. doi: 10.1016/j.neuroscience.2004.01.053. [DOI] [PubMed] [Google Scholar]
- Breakefield XO, Kamm C, Hanson PI. TorsinA: movement at many levels. Neuron. 2001;31:9–12. doi: 10.1016/s0896-6273(01)00350-6. [DOI] [PubMed] [Google Scholar]
- Dang MT, Yokoi F, McNaught KS, Jengelley TA, Jackson T, Li J, Li Y. Generation and characterization of Dyt1 DeltaGAG knock-in mouse as a model for early-onset dystonia. Exp Neurol. 2005;196:452–463. doi: 10.1016/j.expneurol.2005.08.025. [DOI] [PubMed] [Google Scholar]
- Frischmeyer P, Dietz HC. Nonsense-mediated mRNA decay in health and disease. Hum Mol Genet. 1999;8:1893–1900. doi: 10.1093/hmg/8.10.1893. [DOI] [PubMed] [Google Scholar]
- Furukawa Y, Hornykiewicz O, Fahn S, Kish SJ. Striatal dopamine in early-onset primary torsion dystonia with the DYT1 mutation. Neurology. 2000;54:1193–1195. doi: 10.1212/wnl.54.5.1193. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Alegre P, Paulson HL. Aberrant cellular behavior of mutant torsinA implicates nuclear envelope dysfunction in DYT1 dystonia. J Neurosci. 2004;24:2593–2601. doi: 10.1523/JNEUROSCI.4461-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodchild RE, Dauer WT. Mislocalization to the nuclear envelope: an effect of the dystonia-causing torsinA mutation. Proc Natl Acad Sci USA. 2004;101:847–852. doi: 10.1073/pnas.0304375101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodchild RE, Kim CE, Dauer WT. Loss of the dystonia-associated protein torsinA selectively disrupts the neuronal nuclear envelope. Neuron. 2005;48:923–932. doi: 10.1016/j.neuron.2005.11.010. [DOI] [PubMed] [Google Scholar]
- Hewett J, Gonzalez-Agosti C, Slater D, Ziefer P, Li S, Bergeron D, Jacoby DJ, Ozelius LJ, Ramesh V, Breakefield XO. Mutant torsinA, responsible for early-onset torsion dystonia, forms membrane inclusions in cultured neural cells. Hum Mol Genet. 2000;9:1403–1413. doi: 10.1093/hmg/9.9.1403. [DOI] [PubMed] [Google Scholar]
- Hewett JW, Kamm C, Boston H, Beauchamp R, Naismith T, Ozelius L, Hanson PI, Breakefield XO, Ramesh V. TorsinB--perinuclear location and association with torsinA. J Neurochem. 2004;89:1186–1194. doi: 10.1111/j.1471-4159.2004.02404.x. [DOI] [PubMed] [Google Scholar]
- Hewett JW, Zeng J, Niland BP, Bragg DC, Breakefield XO. Dystonia-causing mutant torsinA inhibits cell adhesion and neurite extension through interference with cytoskeletal dynamics. Neurobiol Dis. 2006;22:98–111. doi: 10.1016/j.nbd.2005.10.012. [DOI] [PubMed] [Google Scholar]
- Kock N, Naismith TV, Boston HE, Ozelius LJ, Corey DP, Breakefield XO, Hanson PI. Effects of genetic variations in the dystonia protein torsinA: identification of polymorphism at residue 216 as protein modifier. Hum Mol Genet. 2006;15:1355–1364. doi: 10.1093/hmg/ddl055. [DOI] [PubMed] [Google Scholar]
- Koh YH, Rehfeld K, Ganetzky B. A Drosophila model of early onset torsion dystonia suggests impairment in TGF-beta signaling. Hum Mol Genet. 2004;13:2019–2030. doi: 10.1093/hmg/ddh208. [DOI] [PubMed] [Google Scholar]
- Kuner R, Teismann P, Trutzel A, Naim J, Richter A, Schmidt N, von Ahsen O, Bach A, Ferger B, Schneider A. TorsinA protects against oxidative stress in COS-1 and PC12 cells. Neurosci Lett. 2003;350:153–156. doi: 10.1016/s0304-3940(03)00904-2. [DOI] [PubMed] [Google Scholar]
- Kustedjo K, Bracey MH, Cravatt BF. Torsin A and its torsion dystonia-associated mutant forms are lumenal glycoproteins that exhibit distinct subcellular localizations. J Biol Chem. 2000;275:27933–27939. doi: 10.1074/jbc.M910025199. [DOI] [PubMed] [Google Scholar]
- Lakso M, Sauer B, Mosinger B, Jr, Lee EJ, Manning RW, Yu SH, Mulder KL, Westphal H. Targeted oncogene activation by site-specific recombination in transgenic mice. Proc Natl Acad Sci USA. 1992;89:6232–6236. doi: 10.1073/pnas.89.14.6232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNaught KS, Kapustin A, Jackson T, Jengelley TA, Jnobaptiste R, Shashidharan P, Perl DP, Pasik P, Olanow CW. Brainstem pathology in DYT1 primary torsion dystonia. Ann Neurol. 2004;56:540–547. doi: 10.1002/ana.20225. [DOI] [PubMed] [Google Scholar]
- Ozelius LJ, Hewett JW, Page CE, Bressman SB, Kramer PL, Shalish C, de Leon D, Brin MF, Raymond D, Corey DP, Fahn S, Risch NJ, Buckler AJ, Gusella JF, Breakefield XO. The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein. Nat Genet. 1997;17:40–48. doi: 10.1038/ng0997-40. [DOI] [PubMed] [Google Scholar]
- Shashidharan P, Paris N, Sandu D, Karthikeyan L, McNaught KS, Walker RH, Olanow CW. Overexpression of torsinA in PC12 cells protects against toxicity. J Neurochem. 2004;88:1019–1025. doi: 10.1046/j.1471-4159.2003.02233.x. [DOI] [PubMed] [Google Scholar]
- Shashidharan P, Sandu D, Potla U, Armata IA, Walker RH, McNaught KS, Weisz D, Sreenath T, Brin MF, Olanow CW. Transgenic mouse model of early-onset DYT1 dystonia. Hum Mol Genet. 2005;14:125–133. doi: 10.1093/hmg/ddi012. [DOI] [PubMed] [Google Scholar]
- Torres GE, Sweeney AL, Beaulieu JM, Shashidharan P, Caron MG. Effect of torsinA on membrane proteins reveals a loss of function and a dominant-negative phenotype of the dystonia-associated DeltaE-torsinA mutant. Proc Natl Acad Sci USA. 2004;101:15650–15655. doi: 10.1073/pnas.0308088101. [DOI] [PMC free article] [PubMed] [Google Scholar]