

Abstract
To effect movement, motoneurons must respond appropriately to motor commands. Their responsiveness to these inputs, or excitability, is regulated by neuromodulators. Possible sources of modulation include the abundant cholinergic “C boutons” that surround motoneuron somata. In the present study, recordings from motoneurons in spinal cord slices demonstrated that cholinergic activation of m2-type muscarinic receptors increases excitability by reducing the action potential afterhyperpolarization. Analyses of isolated spinal cord preparations in which fictive locomotion was elicited demonstrated that endogenous cholinergic inputs increase motoneuron excitability during locomotion. Anatomical data indicate that C boutons originate from a discrete group of interneurons lateral to the central canal, the medial partition neurons. These results highlight a unique component of spinal motor networks that is critical in ensuring that sufficient output is generated by motoneurons to drive motor behavior.
Keywords: afterhyperpolarization, C bouton, central pattern generator, muscarinic receptors, spinal cord
To generate movement, it is necessary for motoneurons (MNs) to integrate the inputs (motor commands) they receive and produce an output sufficient to effect muscular contraction. The relationship of input to output is determined by neuronal excitability, which in the case of MNs is known to be regulated by identified descending modulatory systems (1). Given that these descending systems are disrupted after spinal cord injury, strategies aimed at restoring movement need to address not only premotor circuits that provide motor commands but also any modulatory systems that ensure MNs are sufficiently excitable to respond to these commands. Spinal premotor circuits for locomotion can be activated after spinal transection (2) and provide one clear target for treatments designed to produce functional recovery. Should an intrinsic spinal modulatory system exist, this would be an important additional target for such strategies.
The somata and proximal dendrites of MNs are contacted by large cholinergic varicosities named “C boutons” (3–11). It has been known since 1972 (12) that C boutons originate from spinal cord neurons, but the location of these cells remains unknown (10). Although the C bouton synapse has been anatomically characterized and shown to be associated with postsynaptic type 2 muscarinic (m2) receptors (8–10), neither the physiological effects of m2 receptor activation on MNs nor the roles of C boutons in motor activity are known. In the absence of motor behavior, exogenous application of cholinergic agonists affects MN excitability via undefined mechanisms (13–17). We therefore studied the possibility that the intrinsic spinal neurons that give rise to the C boutons regulate MN excitability via activation of m2 receptors, and that this system is used during motor behavior.
Results
The Effects of Muscarinic Receptor Activation on Spinal MNs.
Because C boutons are closely associated with postsynaptic m2 receptors by the second postnatal week (8–10), we investigated the effects of muscarinic receptor activation on spinal MNs by using whole-cell patch-clamp recordings in spinal cord slices from postnatal day 9 (P9)–P15 mice.
Muscarine applied locally (100 μM, pressure-injected, micropipette tip diameter ≈2 μm) or via the perfusate (50 μM) induced varied subthreshold responses in MNs held at −60 mV in voltage clamp or at rest (−58 to −75 mV) in current clamp. These responses included inward currents (−30 to −500 pA, n = 10) or depolarizations (1–17 mV, n = 13), outward currents (10–60 pA, n = 11) or hyperpolarizations (1–2 mV, n = 6), and no effect (n = 2). There was no obvious relationship between the type of response and developmental stage or the passive membrane properties of MNs. The current–voltage relationships for inward (n = 2) and outward (n = 4) currents were established by using local application of muscarine (Fig. 1 A and B Right). These data revealed that both the inward and outward currents increased with depolarization and had reversal potentials (−95 ± 8 mV and −94 ± 5 mV, respectively) near the equilibrium potential for K+ as calculated for our solutions by using the Nernst equation (−98 mV). In addition, muscarine-induced inward currents were associated with a decrease in conductance (−10 ± 7%), whereas outward currents were associated with an increase in conductance (+3.7 ± 0.5%). Together, these data indicate that activation of muscarinic receptors on MNs can either enhance or reduce a resting K+ conductance. We also investigated whether muscarine-induced currents were mediated by m2 receptors. Muscarine-induced outward currents (Fig. 1B Left, n = 3), but not inward currents (Fig. 1A Left, n = 2) or depolarizations (n = 6), were blocked by the relatively selective m2 channel antagonist methoctramine (10 μM) (18, 19). These data indicate that only the small outward currents are likely to be activated by acetylcholine released from C boutons.
Fig. 1.

Muscarinic receptor activation increases spinal MN excitability. (A and B) Voltage-clamp traces (Left) and the current–voltage relationships (Right) of methoctramine-insensitive inward currents (A) and methoctramine-sensitive outward currents (B) induced by muscarine (100 μM, locally applied) in MNs held at −60 mV. The methoctramine was bath-applied (10 μM). (C) Increased MN firing in response to the same amplitude of current injection in the presence of muscarine (50 μM, bath-applied) compared with control. (D) Increased steady-state f–I slope for an MN in the presence of muscarine (50 μM, bath-applied). (E) Pooled data showing changes in the slopes of f–I plots in response to bath applications of muscarine (50 μM, n = 19), oxotremorine (100 μM, n = 6), or muscarine (50 μM) with methoctramine (10 μM, n = 8). #, significantly different to muscarine alone.
Next, we examined the effects of muscarinic receptor activation on MN output. Regardless of the polarity of subthreshold responses, bath applications of muscarine (50 μM) were associated with an increase in the frequency of repetitive action potential firing evoked by the injection of square current pulses (Fig. 1C). To study the effects of muscarine on frequency–current (f–I) relationships relative to the resting membrane potential, subthreshold changes in potential were offset via injection of direct current, and a series of current steps of increasing magnitude were applied. Plots of steady-state firing frequency (mean frequency during the last 500 ms) versus injected current demonstrated a significant increase in the slope of f–I relationships from 86 ± 11 Hz nA−1 in control to 112 ± 14 Hz nA−1 in the presence of muscarine (37 ± 7% increase, n = 19; Fig. 1 D and E). Application of oxotremorine (100 μM), an m2-preferring muscarinic agonist (20), similarly increased the slope of f–I relationships (35 ± 8% increase, n = 6; Fig. 1E). Methoctramine alone (10 μM, n = 8) had no effect on f–I slopes and blocked muscarine-induced increases in the f–I slope (Fig. 1E; n = 8). These data indicate that activation of the muscarinic receptor subtype (m2) found at the C boutons results in increased gain of the input–output relation (i.e., increased excitability) of MNs.
Subsequent experiments investigated the mechanism by which m2 receptor activation increases MN excitability. Because increased f–I gain can result from a reduction in the postspike afterhyperpolarization (AHP), we investigated whether m2 receptor activation reduces the AHP in spinal MNs of mice. Action potentials were elicited by brief (10-ms) current pulses, and AHP amplitudes were measured as the difference between resting membrane voltage and peak hyperpolarization. Muscarine applied locally (100 μM, 1- to 5-sec duration) caused a rapid reduction in the amplitude of the AHP (n = 6; Fig. 2A and B). Bath applications of muscarine (50 μM) elicited longer-lasting reductions in AHP amplitude and thus were used to quantify the reduction: from 4.3 ± 0.7 mV to 1.9 ± 0.5 mV (n = 10; Fig. 2C). Reductions in the AHP induced by muscarine, applied locally (n = 5; Fig. 2 A and B) or via the perfusate (n = 8; Fig. 2C), were prevented by bath application of methoctramine (10 μM), indicating involvement of m2-type receptors.
Fig. 2.

Muscarinic receptor activation increases spinal MN excitability via a reduction in the AHP. (A) Single action potentials (truncated) and AHPs elicited in an MN showing the methoctramine-sensitive (10 μM, bath-applied) effects of muscarine (100 μM, locally applied). (B) Plots of AHP amplitude versus time relative to the local application of muscarine (100 μM) in control and in the presence of methoctramine (10 μM). (C) Average AHP amplitude in MNs in control conditions (n = 10), during bath application of muscarine (50 μM, n = 10) and during bath application of both muscarine (50 μM) and methoctramine (10 μM, n = 8). (D) Unchanged amplitude of calcium currents (measured at the end of voltage steps, −60 mV to a test potential of 0 mV, 500-ms duration, delivered every 30 sec) plotted versus time during bath application of muscarine (50 μM). CdCl2 (500 μM) eliminated the currents, confirming the specificity of our protocol for voltage-activated Ca2+ currents (n = 2). Traces at bottom show examples of currents elicited in control, muscarine, and CdCl2. (E and F) Steady-state f–I plots for one MN (E) and average MN f–I slopes (F; n = 6) in control, apamin (100 nM), and both apamin and muscarine (50 μM). #, significantly different to control.
The AHP is mediated by a Ca2+-dependent K+ (KCa) conductance (21). Because muscarine modulates voltage-activated Ca2+ currents in several types of neuron (22), we investigated whether the reduction in AHP is mediated by reduced Ca2+ influx. We found no evidence of muscarinic modulation of Ca2+ currents in mouse spinal MNs (Fig. 2D; n = 5). This result indicates that the m2 receptor-mediated reduction in amplitude of the AHP results from modulation of the KCa conductance rather than reduction in voltage-activated Ca2+ conductance.
Next, we determined whether modulation of KCa conductance is the primary mechanism underlying increased excitability of MNs in response to muscarine. We applied the SK-type KCa channel blocker apamin to abolish the AHP (21) and then tested whether the muscarinic-mediated increase in MN excitability also was blocked. As reported previously (23), applications of apamin (100 nM) to the perfusate increased MN firing rates and the slopes of f–I relationships (from 91 ± 26 Hz nA−1 to 197 ± 57 Hz nA−1, n = 6; Fig. 2 E and F). The subsequent addition of muscarine (50 μM) to the perfusate produced subthreshold responses in five of six MNs but no further increase in the slopes of f–I relationships (182 ± 41 Hz nA−1 with muscarine, n = 6; Fig. 2 E and F). Together, the above data indicate that activation of the m2 receptors, found at postsynaptic sites of C boutons, increases MN excitability by reducing a SK-type KCa conductance, which in turn decreases AHP amplitude.
The Role of Cholinergic Inputs to MNs During Locomotion.
To investigate whether spinal cholinergic inputs modulate MN excitability during motor behavior, we used a postnatal (P6–P9) mouse isolated spinal cord preparation (Fig. 3B) that elicits locomotor-related activity in response to application of 5-hydroxytryptamine (5-HT; 10 μM), NMDA (5 μM), and dopamine (25–50 μM) (24). During ventral root recordings, cholinergic drugs were applied locally via pressure injection (1- to 10-sec duration, tip diameter ≈10–15 μm) to the motor pool on one side of the spinal cord at the level of the second lumbar segment. Local applications were used specifically to investigate the effects on motor output and avoid effects on rhythm-generating networks. Application of methoctramine (100 μM or 1 mM) caused a rapid, reversible reduction in the amplitude of locomotor-related bursts recorded from the ipsilateral ventral root (Fig. 3A). Calculations based on 10 bursts before and 10 bursts immediately after drug applications demonstrated that 100 μM methoctramine reduced burst amplitude by 18 ± 4%, and 1 mM methoctramine reduced burst amplitude by 43 ± 5% (Fig. 3 A and C; n = 4). Application of the general muscarinic receptor antagonist atropine (1 mM, 3–10 sec) also reduced burst amplitude (27 ± 4%, n = 5; data not shown). Simultaneous analysis of ipsilateral and contralateral nerve roots showed that methoctramine (1 mM, 1–5 sec) only reduced burst amplitude on the side of drug injection (Fig. 3 D and E; 31 ± 3% reduction, n = 8). Methoctramine had no effect on burst frequency (Fig. 3F) or left–right alternation (Fig. 3D), indicating that local applications had no significant effect on rhythm-generating networks.
Fig. 3.

Endogenous cholinergic activity increases MN output during fictive locomotion. (A) Local methoctramine application (100 μM and 1 mM, 10-sec duration) decreased locomotor output as seen in rectified/integrated L2 ventral root recordings. (B) Schematic demonstrating positioning of the drug injection pipette through a slit made in the pia matter above the motor column. (C) Average reduction by methoctramine (100 μM and 1 mM) of locomotor-related burst amplitude reported relative to control (n = 4). (D) Local methoctramine application (1 mM, 5-sec duration) during fictive locomotion did not affect contralateral activity. (E and F) Average amplitude and frequency of locomotor-related bursts recorded before and during methoctramine application (1 mM, 1- to 5-sec duration, n = 8). (G) Rectified/integrated (Upper) and raw (Lower) traces of locomotor output recorded from the L2 root during the local application of physostigmine (1 mM, 5-sec duration). ∗, significantly different to 100 μM methoctramine. #, significantly different to control.
We also investigated the effect of enhancing endogenous cholinergic actions on MNs by locally applying the cholinesterase inhibitor physostigmine. Application of physostigmine (1 mM) caused a delayed (up to ≈1 min) and long-lasting increase in the tonic activity recorded from the ipsilateral ventral root in all preparations (n = 5, Fig. 3G Lower, raw trace). In two of five preparations, there also was a clear increase in the amplitude of the phasic locomotor-related bursts (Fig. 3G Upper, integrated trace).
Together, these data indicate that cholinergic inputs to MNs are active during drug-induced fictive locomotion. By activating m2 receptors, which are found at synapses formed by C boutons, cholinergic inputs increase MN excitability, thus ensuring that appropriate output is generated during motor behavior.
The Source of Cholinergic Inputs to MNs.
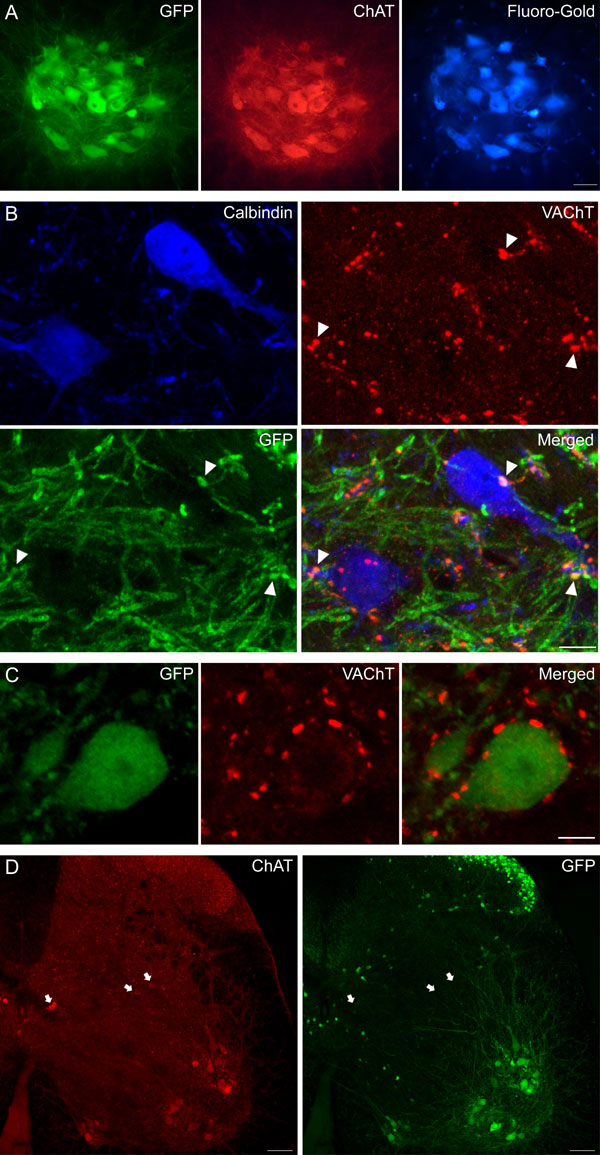
Having characterized the physiological roles of cholinergic inputs to MNs, we investigated which neurons in the spinal cord (12) give rise to the C boutons. Initially, we analyzed candidate populations of cholinergic neurons by using mice in which expression of enhanced GFP is controlled by the choline acetyltransferase (ChAT) promoter (25) [see supporting information (SI) Fig. 5 and SI Methods]. In the lumbar spinal cords of these animals, GFP expression was observed in all MNs and in their synaptic contacts on Renshaw cells. In contrast, C boutons and certain populations of ChAT+ interneurons (in dorsal horn and laminae VII and X) were GFP−. These data demonstrate that C boutons do not originate from MNs, which is consistent with previous reports (7), and leave the populations of GFP− cholinergic interneurons as possible sources.
In previous studies (R.H., A.J.T., D. J. Maxwell, unpublished work), we had noted that some cholinergic interneurons in the offspring of Dbx1-cre recombinase mice (26) crossed with a ROSA26-floxed-YFP reporter line (27) expressed yellow fluorescent protein (YFP). In the double-transgenic offspring, YFP is seen in all cells in which the promoter for Dbx1 is expressed at any point in development. We found that C boutons in these animals were YFP+ (Fig. 4 A–D), indicating that the cells of origin of these terminals must express YFP.
Fig. 4.

C boutons likely originate from interneurons located lateral to the central canal (lamina X/medial lamina VII). (A–D) High-magnification confocal images of a single optical section through an MN (M) from a Dbx1-YFP mouse. Staining for ChAT, YFP, and NOS indicates that C boutons (arrowheads) are YFP+ but lack NOS, whereas some other ChAT+ terminals are NOS+ (arrow). (E) A single optical section through the region surrounding the central canal (CC) from a Dbx1-YFP mouse. Asterisks mark ChAT+/YFP+/NOS− neurons. (Inset) Strong ChAT immunoreactivity in these cells. (F) Plots of all ChAT+ neurons (apart from those in the motor nuclei) from 10 60-μm upper lumbar spinal cord sections of a Dbx1-YFP mouse (gray matter outlined). Cells have been distinguished based on the presence or absence of YFP and NOS. (Scale bars: A–D, 5 μm; E, 20 μm.)
We therefore used immunohistochemistry to identify the ChAT+/YFP+ neurons in the spinal cord that could give rise to C boutons. To refine our search, we also used an antibody against the neuronal form of nitric oxide synthase (NOS), because we previously have shown that many cholinergic neurons in rat spinal cord contain NOS (28). In a set of five 60-μm sections (n = 2 animals), we analyzed 913 ChAT+ boutons on the somata or proximal dendrites of MNs and found that 901 (99%) of these were YFP+/NOS−, 10 were YFP−/NOS−, 1 was YFP−/NOS+, and 1 was YFP+/NOS+ (Fig. 4 A–D). We therefore used triple immunolabeling to reveal neurons with the same immunohistochemical profile as the C boutons: ChAT+/YFP+/NOS− (Fig. 4E). In 10 60-μm sections from the upper lumbar spinal cord of one Dbx1-YFP mouse, we identified 937 ChAT-immunoreactive cells that were not in the motor nuclei and found that 96 (10.2%) of these were YFP+/NOS−. These neurons, which presumably include the cells of origin of the C boutons, had a very restricted distribution (Fig. 4F). Most (76) were lateral to the central canal (lamina X and the medial part of lamina VII), whereas 12 were located dorsal to the central canal, 11 were in the ventromedial part of lamina VII, and one was in lamina V. The YFP+/NOS− cells that lay lateral to the central canal showed strong ChAT immunoreactivity compared with most of the cholinergic neurons that immediately surrounded the canal (data not shown). In another mouse, we examined sections from caudal thoracic spinal cord and observed an almost identical distribution of ChAT+/YFP+/NOS− neurons. Together, these results strongly suggest that the C boutons originate from a population of cells that are located in lamina X and the most medial part of lamina VII.
Discussion
In the present study, we demonstrate that activation of the m2 receptor, which is found at synapses formed by C boutons, leads to an increase in excitability of MNs via a reduction in amplitude of the AHP. In addition, we show that cholinergic inputs to MNs act via m2 receptors to increase MN excitability during rhythmic motor behavior. This function ensures that MNs fire at sufficient rates to effect appropriate muscle contraction while keeping excitatory input to MNs at a minimum.
Activation of postsynaptic muscarinic receptors has a wide range of actions on neuronal ion channels (22). Our data are consistent in part with previous findings in salamander MNs (15) and rat hypoglossal MNs (17), which showed a muscarine-induced reduction in amplitude of the AHP. Although this reduction in the AHP (which led to increased MN excitability) was consistent, the subthreshold effects of muscarine were varied. Because subthreshold depolarizations were not blocked by methoctramine, which reduced the ventral root output during fictive locomotion, it is clear that they did not contribute significantly to the increased excitability during rhythmic activity.
Our data indicating that cholinergic inputs to MNs are active during locomotion and that they increase MN excitability via m2 receptor-mediated effects on the AHP are consistent with previous studies that demonstrated that the AHP of spinal MNs is a target for modulation during motor behavior. This finding was first demonstrated in the decerebrate cat in which, during fictive locomotion, the amplitude of the AHP in spinal MNs was reduced (29). Similar reductions in AHP amplitude have been reported in MN recordings from isolated neonatal rat spinal cords during fictive locomotion (30), indicating that spinal mechanisms are involved.
Although there are other cholinergic inputs to spinal MNs besides C boutons (6, 31), several lines of evidence suggest that C boutons are responsible for the increased MN excitability during locomotor-related activity. First, being on or close to MN somata, C boutons are well positioned to act as modulators of the intrinsic properties, such as the AHP, that regulate neuronal output. Second, we have shown that the increase in excitability is mediated by m2 receptors, which are found exclusively postsynaptic to C boutons (8–10). Although m2 receptor knockout animals showed no obvious change in motor coordination (32), this finding does not preclude a role for m2 receptor-mediated increase in MN excitability. For example, in the absence of m2 receptors, there may be compensatory modulation (e.g., from descending serotonergic systems) or increased drive from last-order interneurons to MNs. Third, anatomical evidence concerning expression of ion channels at C bouton synapses correlates well with the physiological data in the present study. This evidence includes the presence of discrete clusters of small conductance KCa channels in close apposition to C boutons (R. E. Fyffe, personal communication).
Several groups of cholinergic interneurons have been identified in the spinal cord (33). In addition to autonomic neurons (which are present at thoraco-lumbar and sacral levels), there are scattered neurons in the dorsal horn, cells that surround the central canal (central canal cluster cells), and a population of neurons that occupy a region extending from lamina X to the lateral edge of the gray matter. The latter show strong ChAT immunoreactivity and were named partition cells because they are located between dorsal and ventral horns. We found that the YFP+/NOS− neurons, which likely give rise to the C boutons, are lateral to the central canal and show strong ChAT immunoreactivity. This profile identifies them as belonging to the medial group of partition neurons (33). Interestingly, YFP was not present in all of the strongly ChAT+ neurons in this region, suggesting that the C boutons originate from a specific subpopulation of medial partition cells that express Dbx1 at some stage during their development.
The results of the present study indicate that this subpopulation of medial partition neurons is responsible for increasing MN excitability during fictive locomotion in the mouse. This finding is supported by experiments in the cat in which Fos expression revealed that many cholinergic neurons, but particularly those located “in the medial portion of lamina VII close to lamina X,” are active during fictive locomotion (34). The distribution of these active cells is very similar to that of the YFP+/NOS− cells seen in the present study [compare our Fig. 4F with figure 7 of Huang et al. (34)].
The experiments presented here indicate that C boutons are important in regulating MN output. Interestingly, previous studies suggest that they also may be involved in certain pathological conditions. For example, after sacral spinal cord transection, C boutons transiently disappear from sacrocaudal MNs; the time course of their loss and reappearance mirrors the time course of spinal shock (35, 36). In addition, there is a loss of cholinergic terminals (37) and muscarinic binding sites (38) on human MNs in sporadic amyotrophic lateral sclerosis (ALS). Whether this loss of C boutons is involved in the pathogenesis of the disease, and whether it contributes to the symptomatic weakness of the disease, however, is not clear.
C boutons also are relevant to strategies aimed at restoring motor function after spinal cord injury. One clear goal of such strategies is to activate spinal rhythm-generating networks. However, it will also be important to ensure that MNs respond to the motor commands that they receive. Because the C bouton system is present after chronic spinal cord injury (35) and regulates MN excitability, it is a potential target for strategies aimed at regaining motor function after injury.
Materials and Methods
All procedures were approved by the Dalhousie University Animal Care Committee or the Ethical Review Process Applications Panel of the University of Glasgow and conformed to the guidelines of the Canadian Council of Animal Care or the U.K. Animals (Scientific Procedures) Act 1986.
Electrophysiology.
In vitro whole spinal cord preparation.
The preparation of the in vitro isolated whole spinal cord was similar to that described in ref. 24. Briefly, Swiss–Webster mice (P6–P9) were anesthetized with ketamine (500 mg/kg), and their spinal cords were isolated from the mid-cervical to upper sacral segments and secured in a continually perfused recording chamber. Glass suction electrodes were attached to ventral roots for recording. Signals were amplified, filtered (30–3,000 Hz), rectified, integrated, and acquired at 1 kHz by using a Digidata 1322A A/D board and AxoScope software. Offline analysis was performed using Clampfit software (Molecular Devices, Union City, CA).
Spinal cord slice preparation.
Experiments were performed on spinal cord slices obtained from P9–P15 Swiss–Webster or C57BL/6 mice. One to 3 days before experimentation animals received i.p. injections of Fluoro-Gold (0.04 mg/g; Fluorochrome Inc., Denver, CO) to retrogradely label MNs (39). Spinal cord slices were prepared as described in ref. 23. Fluoro-Gold-positive MNs were visualized with epifluorescence and infrared differential interference contrast microscopy by using a Leica DMLFSA upright microscope. Signals recorded by using whole-cell patch-clamp techniques were amplified and filtered (4 kHz low-pass Bessel filter) with a MultiClamp 700B amplifier and acquired at ≥10 kHz by using a Digidata 1322A A/D board and pClamp software (Molecular Devices). Series resistance compensation (≥60%) was used during all voltage-clamp recordings. All data are reported as mean ± SE. Differences in means were compared by using Student's t test. Values of P < 0.05 were considered significant.
Solutions and drugs.
The recording solution contained 127 mM NaCl, 3 mM KCl, 2 mM CaCl2, 1 mM MgSO4, 26 mM NaHCO3, 1.25 mM NaH2PO4, and 10 mM d-glucose (equilibrated with 95% O2/5% CO2). The standard pipette solution for whole-cell patch-clamp recordings contained 140 mM potassium methane sulfonate, 10 mM NaCl, 1 mM CaCl2, 10 mM Hepes, 1 mM EGTA, 3 mM ATP-Mg, and 0.4 mM GTP (pH 7.2–7.3 adjusted with KOH, osmolarity adjusted to ≈300 mosM with sucrose).
For experiments investigating Ca2+ currents, external and pipette solutions were designed to eliminate Na+ and K+ currents (40, 41). The external solution contained 115 mM NaCl, 3 mM KCl, 30 mM tetraethyl ammonium chloride (TEA-Cl), 10 mM Hepes, 1 mM MgCl2, 2 mM CaCl2, 10 mM d-glucose, 4 mM 4-aminopyridine (4-AP), and 0.5 μM tetrodotoxin (TTX) (gassed with 100% O2, pH 7.35 adjusted with NaOH, osmolarity ≈305 mosM). The pipette solution contained 100 mM Cs methane sulfonate, 30 mM TEA-Cl, 10 mM NaCl, 1 mM CaCl2, 10 mM Hepes, 1 mM EGTA, 3 mM ATP-Mg, and 0.4 mM GTP (pH 7.2–7.3 adjusted with KOH, osmolarity adjusted to ≈295 mosM with sucrose).
Drug application was by means of either addition to the perfusate or local pressure injection (PMI-100 Pressure Micro Injector; Dagan Corporation, Minneapolis, MN) via a pipette visually inserted into the motor pool. As previously demonstrated, local applications required much higher drug concentrations (10–100×) than bath applications because of the very small volume being injected into a large bath volume and because of the requirement for sufficient diffusion of drugs into the tissue (42, 43).
Anatomy.
The double-transgenic offspring of Dbx1-cre recombinase mice (26) crossed with a ROSA26-stop-YFP reporter line (27) were anesthetized and perfused with 4% formaldehyde. Transverse 60-μm spinal cord sections were cut with a Vibratome, incubated for 72 h in rabbit anti-GFP (which recognizes YFP; 1:4,000; Abcam, Cambridge, U.K.) and goat anti-ChAT (1:2,000; Chemicon, Billerica, MA), and then incubated for 24 h in Alexa 488 donkey anti-rabbit IgG (1:500; Invitrogen, Carlsbad, CA) and biotinylated donkey anti-goat IgG (1:500; Jackson Immunoresearch, West Grove, PA). ChAT was revealed with tyramide signal amplification (Perkin-Elmer Life Sciences, Boston, MA). Sections were incubated for 48 h in sheep anti-neuronal NOS (1:2,000; provided by P. C. Emson) and 24 h in Cy5-donkey anti-goat IgG (1:100; Jackson Immunoresearch).
Confocal images were acquired with a Bio-Rad (Hercules, CA) Radiance 2100 confocal microscope. Ten sections from each of two Dbx1-YFP mice were scanned through their full thickness. Locations of all ChAT-positive neurons outside motor nuclei were plotted with Neurolucida (MicroBrightField, Williston, VT), and the presence or absence of immunostaining for YFP and NOS was noted.
Supplementary Material
Acknowledgments
We are grateful to Dr. P. C. Emson (Babraham Institute, Babraham, Cambridge, U.K.) for the gift of antibody against neuronal NOS; Dr. M. E. Hatten (The Rockefeller University, New York, NY) and the GENSAT Project for the gift of the ChAT-GFP mice; Drs. A. Pierani (Centre National de la Recherche Scientifique, Paris, France) and T. M. Jessell (Columbia University, New York, NY) for the spinal cords from Dbx1-cre × ROSA26-stop-YFP mice; Prof. D. J. Maxwell for helpful discussion; Dr. L. Jordan for comments on an earlier version of the manuscript; and Mr. R. Kerr, Ms A. Alcos, and Ms. H. Zhang for superb technical assistance. This work was supported by the Canadian Institutes of Health Research, the Nova Scotia Health Research Foundation, the Human Frontier Science Program, and the Wellcome Trust. G.B.M. was supported by New Zealand Foundation for Research Science and Technology Postdoctoral Fellowship DALH0201.
Abbreviations
- MN
motoneuron
- m2
type 2 muscarinic
- Pn
postnatal day n
- f–I
frequency–current
- AHP
afterhyperpolarization
- KCa
Ca2+-dependent K+
- ChAT
choline acetyltransferase
- YFP
yellow fluorescent protein
- NOS
nitric oxide synthase.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/cgi/content/full/0611134104/DC1.
References
- 1.Brownstone RM. Prog Neurobiol. 2006;78:156–172. doi: 10.1016/j.pneurobio.2006.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rossignol S. Curr Opin Neurobiol. 2000;10:708–716. doi: 10.1016/s0959-4388(00)00151-3. [DOI] [PubMed] [Google Scholar]
- 3.Conradi S, Skoglund S. Acta Physiol Scand Suppl. 1969;333:5–52. [PubMed] [Google Scholar]
- 4.Nagy JI, Yamamoto T, Jordan LM. Synapse. 1993;15:17–32. doi: 10.1002/syn.890150103. [DOI] [PubMed] [Google Scholar]
- 5.Li W, Ochalski PA, Brimijoin S, Jordan LM, Nagy JI. Neuroscience. 1995;65:879–891. doi: 10.1016/0306-4522(94)00511-3. [DOI] [PubMed] [Google Scholar]
- 6.Arvidsson U, Riedl M, Elde R, Meister B. J Comp Neurol. 1997;378:454–467. [PubMed] [Google Scholar]
- 7.Hellstrom J, Arvidsson U, Elde R, Cullheim S, Meister B. J Comp Neurol. 1999;411:578–590. [PubMed] [Google Scholar]
- 8.Wilson JM, Rempel J, Brownstone RM. J Comp Neurol. 2004;474:13–23. doi: 10.1002/cne.20089. [DOI] [PubMed] [Google Scholar]
- 9.Muennich EA, Fyffe RE. J Physiol. 2004;554:673–685. doi: 10.1113/jphysiol.2003.056192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hellstrom J, Oliveira AL, Meister B, Cullheim S. J Comp Neurol. 2003;460:476–486. doi: 10.1002/cne.10648. [DOI] [PubMed] [Google Scholar]
- 11.Welton J, Stewart W, Kerr R, Maxwell DJ. Brain Res. 1999;817:215–219. doi: 10.1016/s0006-8993(98)01208-6. [DOI] [PubMed] [Google Scholar]
- 12.McLaughlin BJ. J Comp Neurol. 1972;144:475–500. doi: 10.1002/cne.901440406. [DOI] [PubMed] [Google Scholar]
- 13.Alaburda A, Perrier JF, Hounsgaard J. J Physiol. 2002;540:875–881. doi: 10.1113/jphysiol.2001.015982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hornby TG, McDonagh JC, Reinking RM, Stuart DG. J Neurophysiol. 2002;88:86–97. doi: 10.1152/jn.2002.88.1.86. [DOI] [PubMed] [Google Scholar]
- 15.Chevallier S, Nagy F, Cabelguen JM. J Physiol. 2006;570:525–540. doi: 10.1113/jphysiol.2005.098970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zieglgansberger W, Reiter C. Neuropharmacology. 1974;13:519–527. doi: 10.1016/0028-3908(74)90141-5. [DOI] [PubMed] [Google Scholar]
- 17.Lape R, Nistri A. J Neurophysiol. 2000;83:2987–2995. doi: 10.1152/jn.2000.83.5.2987. [DOI] [PubMed] [Google Scholar]
- 18.Melchiorre C, Cassinelli A, Quaglia W. J Med Chem. 1987;30:201–204. doi: 10.1021/jm00384a034. [DOI] [PubMed] [Google Scholar]
- 19.Hulme EC, Birdsall NJ, Buckley NJ. Annu Rev Pharmacol Toxicol. 1990;30:633–673. doi: 10.1146/annurev.pa.30.040190.003221. [DOI] [PubMed] [Google Scholar]
- 20.Murakami Y, Matsumoto K, Ohta H, Watanabe H. Gen Pharmacol. 1996;27:833–836. doi: 10.1016/0306-3623(95)02084-5. [DOI] [PubMed] [Google Scholar]
- 21.Zhang L, Krnjevic K. Neurosci Lett. 1987;74:58–62. doi: 10.1016/0304-3940(87)90051-6. [DOI] [PubMed] [Google Scholar]
- 22.Caulfield MP, Robbins J, Higashida H, Brown DA. Prog Brain Res. 1993;98:293–301. doi: 10.1016/s0079-6123(08)62411-5. [DOI] [PubMed] [Google Scholar]
- 23.Miles GB, Dai Y, Brownstone RM. J Physiol. 2005;566:519–532. doi: 10.1113/jphysiol.2005.086033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiang Z, Carlin KP, Brownstone RM. Brain Res. 1999;816:493–499. doi: 10.1016/s0006-8993(98)01199-8. [DOI] [PubMed] [Google Scholar]
- 25.Gong S, Zheng C, Doughty ML, Losos K, Didkovsky N, Schambra UB, Nowak NJ, Joyner A, Leblanc G, Hatten ME, Heintz N. Nature. 2003;425:917–925. doi: 10.1038/nature02033. [DOI] [PubMed] [Google Scholar]
- 26.Bielle F, Griveau A, Narboux-Neme N, Vigneau S, Sigrist M, Arber S, Wassef M, Pierani A. Nat Neurosci. 2005;8:1002–1012. doi: 10.1038/nn1511. [DOI] [PubMed] [Google Scholar]
- 27.Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, Costantini F. BMC Dev Biol. 2001;1:4. doi: 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spike RC, Todd AJ, Johnston HM. J Comp Neurol. 1993;335:320–333. doi: 10.1002/cne.903350303. [DOI] [PubMed] [Google Scholar]
- 29.Brownstone RM, Jordan LM, Kriellaars DJ, Noga BR, Shefchyk SJ. Exp Brain Res. 1992;90:441–455. doi: 10.1007/BF00230927. [DOI] [PubMed] [Google Scholar]
- 30.Schmidt BJ. Exp Brain Res. 1994;99:214–222. doi: 10.1007/BF00239588. [DOI] [PubMed] [Google Scholar]
- 31.Nishimaru H, Restrepo CE, Ryge J, Yanagawa Y, Kiehn O. Proc Natl Acad Sci USA. 2005;102:5245–5249. doi: 10.1073/pnas.0501331102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gomeza J, Shannon H, Kostenis E, Felder C, Zhang L, Brodkin J, Grinberg A, Sheng H, Wess J. Proc Natl Acad Sci USA. 1999;96:1692–1697. doi: 10.1073/pnas.96.4.1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barber RP, Phelps PE, Houser CR, Crawford GD, Salvaterra PM, Vaughn JE. J Comp Neurol. 1984;229:329–346. doi: 10.1002/cne.902290305. [DOI] [PubMed] [Google Scholar]
- 34.Huang A, Noga BR, Carr PA, Fedirchuk B, Jordan LM. J Neurophysiol. 2000;83:3537–3547. doi: 10.1152/jn.2000.83.6.3537. [DOI] [PubMed] [Google Scholar]
- 35.Kitzman P. Exp Neurol. 2006;197:407–419. doi: 10.1016/j.expneurol.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 36.Bennett DJ, Gorassini M, Fouad K, Sanelli L, Han Y, Cheng J. J Neurotrauma. 1999;16:69–84. doi: 10.1089/neu.1999.16.69. [DOI] [PubMed] [Google Scholar]
- 37.Nagao M, Misawa H, Kato S, Hirai S. J Neuropathol Exp Neurol. 1998;57:329–333. doi: 10.1097/00005072-199804000-00004. [DOI] [PubMed] [Google Scholar]
- 38.Berger ML, Veitl M, Malessa S, Sluga E, Hornykiewicz O. J Neurol Sci. 1992;108:114–117. doi: 10.1016/0022-510x(92)90196-r. [DOI] [PubMed] [Google Scholar]
- 39.Leong SK, Ling EA. J Neurosci Methods. 1990;32:15–23. doi: 10.1016/0165-0270(90)90067-p. [DOI] [PubMed] [Google Scholar]
- 40.Miles GB, Lipski J, Lorier AR, Laslo P, Funk GD. Eur J Neurosci. 2004;20:903–913. doi: 10.1111/j.1460-9568.2004.03550.x. [DOI] [PubMed] [Google Scholar]
- 41.Carlin KP, Jiang Z, Brownstone RM. Eur J Neurosci. 2000;12:1624–1634. doi: 10.1046/j.1460-9568.2000.00050.x. [DOI] [PubMed] [Google Scholar]
- 42.Le Ray D, Brocard F, Dubuc R. J Neurophysiol. 2004;92:926–938. doi: 10.1152/jn.01025.2003. [DOI] [PubMed] [Google Scholar]
- 43.Liu G, Feldman JL, Smith JC. J Neurophysiol. 1990;64:423–436. doi: 10.1152/jn.1990.64.2.423. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}