

Abstract
Mitochondrial outer membrane permeabilization (MOMP) and release of mitochondrial intermembrane proteins like cytochrome c are critical steps in the control of apoptosis. Previous work has shown that MOMP depends on the functionally redundant multidomain proapoptotic proteins, Bak and Bax. Here we demonstrate that Bak and Bax are functionally non-redundant during Neisseria gonorrhoeae (Ngo)- and cisplatin-induced apoptosis. While the activation of Bak is caspase independent Bax activation needs Bak and active caspases. Silencing of either Bak or Bax resists both Ngo- and cisplatin- but not TNFα-induced apoptosis. Activation of Bak is required to release cytochrome c from the mitochondria; however, Bax is still required to activate effector caspases. Thus, both Bak and Bax are necessary to accomplish DNA damage and Ngo-induced apoptosis.
Keywords: apoptosis, Bak, Bax, DNA damage, Neisseria , mitochondria
Introduction
Apoptosis is a special type of programmed cell death that plays a crucial role in morphogenesis and tissue homeostasis. The major components of the apoptotic machinery are highly conserved during the course of evolution, underlying the fundamental importance of this phenomenon (Vaux and Korsmeyer, 1999). The key effector molecules of apoptosis are the caspases (cysteine aspartate specific proteases), which cleave hundreds of substrates to bring about the typical apoptotic morphology (Fischer et al, 2003). Apoptosis is activated by two major pathways namely the intrinsic or the mitochondria-mediated and the extrinsic or death receptor-mediated pathways. Depending on cell type the extrinsic pathway is also dependent on the mitochondrial pathway to induce apoptosis (Scaffidi et al, 1998). Mitochondria respond to several death stimuli by releasing proapoptogenic factors such as cytochrome c and Smac/DIABLO to the cytosol, which leads to the activation of caspases and apoptosis.
Bacterial pathogens profoundly modulate host cell apoptosis to achieve successful infection (Weinrauch and Zychlinsky, 1999). The plethora of bacterial proteins inducing apoptosis include the pore forming alpha toxin from Staphylococcus aureus, listeriolysin O from Listeria monocytogenes, VacA from Helicobacter pylori and PorB from Neisseria gonorrhoeae. Indeed, mitochondria have emerged as a target for modulating host cell apoptosis by bacterial and viral pathogens (Müller and Rudel, 2001). We have previously shown that PorB from Ngo translocates to host cell mitochondria to induce mitochondrial membrane potential (MMP) loss and cytochrome c release to effect apoptosis (Müller et al, 2000).
The genus Neisseria comprises of the human pathogenic species N. gonorrhoeae and N. meningitidis, which cause gonorrhea and meningitis, respectively. The initial contact with the host takes place at the epithelia, where a strong association of the pathogen to the cells is brought about by pili and Opa proteins (Naumann et al, 1999). Attachment to epithelia leads to the transfer of the outer membrane porin PorB to the host cell cytoplasmic membranes (Weel and van Putten, 1991; Rudel et al, 1996) and the mitochondria (Massari et al, 2000; Müller et al, 2000). Studies in yeast revealed that the import of PorB to mitochondria is dependent on the host mitochondrial import machinery and as in the bacterial outer membrane, translocated PorB can form trimers and multimers at the mitochondrial outer membrane (Müller et al, 2002). Although purified PorB induces membrane potential loss and cytochrome c release in intact cells and in isolated mitochondria, transfected PorB did not induce cytochrome c release despite mitochondrial localization and loss of membrane potential (Müller et al, 2002). This suggests that there may be a second signal required to induce cytochrome c release during infection (Müller et al, 2002).
The release of proapoptotic proteins from the mitochondria during apoptosis is primarily mediated by the mitochondrial outer membrane permeabilization (MOMP). The permeabilization of the mitochondrial outer membrane is mainly effected by the multimerization of pore-forming so-called ‘multidomain' proapoptotic family members Bax and Bak, which are activated by the members of the BH3 only family (Korsmeyer et al, 2000). Recent evidence has shown that Bax and Bak can function in a redundant fashion to release cytochrome c in response to various apoptosis inducers (Wei et al, 2001). Unlike Bax, Bak resides in the mitochondrial outer membrane and remains inactive by the direct interaction with antiapoptotic Bcl-2 family members like Mcl-1 or Bcl-XL. On the onset of apoptosis, Bak is activated either by displacement and/or degradation of Mcl-1 by the BH3 only members or p53 to cause cytochrome c release and apoptosis (Mihara et al, 2003; Chipuk et al, 2004; Leu et al, 2004).
Here we show that the multidomain proapoptotic proteins Bak and Bax are activated sequentially during Ngo- and cisplatin-induced apoptosis. Bak activation precedes Bax activation to release cytochrome c from the mitochondria. Activation of Bax is Bak and caspase-9 dependent and is obligatory for effector caspase activation. Consistently, silencing of either Bak or Bax renders HeLa cells resistant to Ngo- and cisplatin- but not TNF/CHX-induced apoptosis, suggesting distinct roles for Bax and Bak during Ngo infection and DNA damage-induced apoptosis.
Results
Loss of mitochondrial membrane potential or cytochrome c release during Ngo infection is independent of caspases
To understand if Ngo infection-mediated loss of MMP is caspase-dependent, we checked if pretreatment of cells with zVAD-fmk (zVAD), a broad-spectrum caspase inhibitor, has any influence on Ngo-induced MMP loss. To perform these experiments, we first checked if pretreatment of cells with 50 μM of zVAD could inhibit caspase activity after induction of apoptosis with various inducers. As expected, 50 μM of zVAD could effectively inhibit Ngo-, cisplatin- and TNFα and cycloheximide (TNFα/CHX)-induced caspase 3/7 activity in HeLa cells (Supplementary Figure S1). These data suggested that zVAD is indeed a potent inhibitor of caspases in these cells irrespective of the inducer and length of apoptosis induction. HeLa cells infected with Ngo strain VPI or treated with TNFα/CHX underwent apoptosis in a zVAD-sensitive manner (Figure 1A). However, zVAD had no significant effect on infection-induced MMP loss as measured by TMRE staining (Figure 1B), indicating that MMP loss occurred in infected cells independent of caspase activity. To check if cytochrome c was still released from the mitochondria of the cells pretreated with zVAD upon Ngo infection, we performed immunofluorescence analysis of control, infected and infected zVAD-treated cells at 15 h postinfection, the time point at which most of the infected cells undergo apoptosis. Inhibition of caspases failed to prevent MMP loss or cytochrome c release (Figure 1C and D). Immunoblot analyses revealed that cytochrome c is released to the cytosol during Ngo infection in the presence of zVAD (Figure 1E and F). This suggests that during Ngo infection, cytochrome c release is a caspase-independent event.
Figure 1.
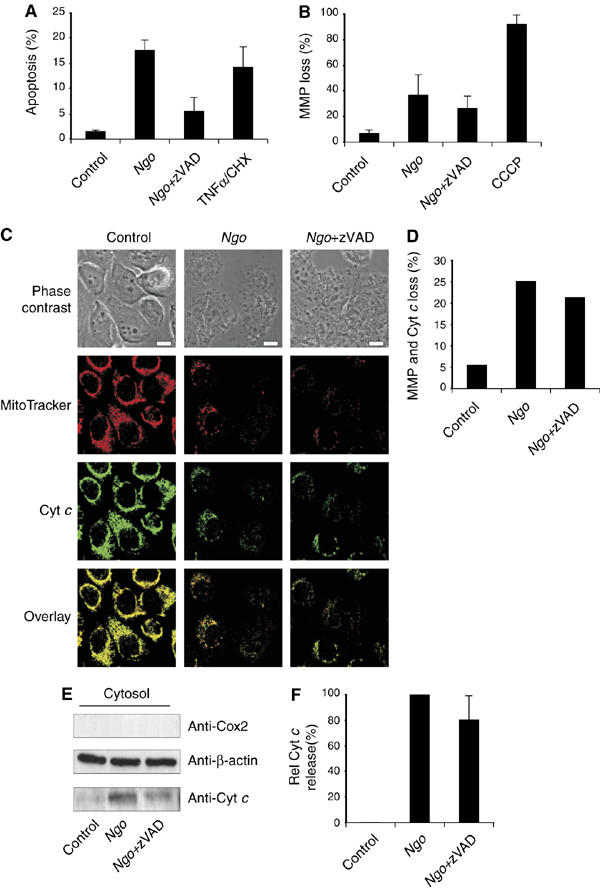
Loss of mitochondrial membrane potential (MMP) and cytochrome c release during Ngo infection are independent of caspase activation. (A) DNA fragmentation of HeLa cells infected in the presence or absence of broad-spectrum caspase inhibitor zVAD was measured by FACS analysis using sub-G assay. TNFα/CHX was used as a control. (B) Additionally the loss of MMP was measured using TMRE staining by FACS analysis. Cells treated with the protonophore CCCP were used as a control. (C) To visualize MMP-loss and cytochrome c release-Ngo-infected cells were stained with MitoTracker orange anti-cytochrome c antibody. Overlays and the corresponding phase contrast images are shown. Size bars represent 10 μm. (D) Cells with mitochondria that showed MMP loss and cytochrome c release were quantified using a confocal microscope. At least 150 cells per sample were counted from a representative experiment. (E) The amount of cytochrome c released to the cytosol was monitored in noninfected (control), infected (Ngo) and infected zVAD-treated (Ngo+zVAD) cells. Released cytochrome c was visualized using anti-cytochrome c antibody and anti-β-actin antibody was used as a loading control. Purity of the cytosol was checked using anti-Cox II antibody. (F) Relative amounts of released cytochrome c were quantified by a densitometric analysis. To determine the effect of zVAD, pretreatment uninfected samples were set as blank and cytosol of infected cells were set to 100%. Panels (A), (B) and (F) show data from three independent experiments and the error bars represent the ±s.d. of the mean.
Activation of Bax but not Bak needs active caspases during Ngo- and cisplatin-induced apoptosis
Release of cytochrome c is primarily effected by MOMP, which is mediated by the activation of proapoptotic Bcl-2 family members Bak and Bax. Upon activation they undergo conformational changes, which can be monitored by conformation-specific antibodies. In order to test whether Ngo activates Bak and Bax, infected cells were stained with antiactive Bak and antiactive Bax antiserum and analyzed by fluorescence activated cell sorting (FACS) (Figure 2A and B). These analyzes clearly showed that Bak and Bax are activated in infected cells as evidenced by a shift in FL-1 channel. Moreover, the results were further confirmed by immunoprecipitation experiments showing that infection-induced Bak and Bax activation were similarly efficient as TNFα/CHX treatment (Figure 2C).
Figure 2.
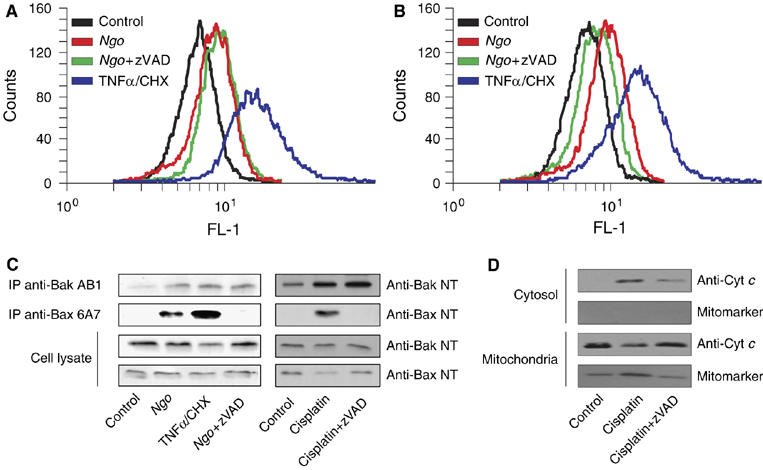
Activation of Bax but not Bak needs active caspases during Ngo and cisplatin induced apoptosis. HeLa cells infected with Ngo were stained for active Bak (A) and active Bax (B) using conformation-specific antibodies and quantified by FACS analysis. (C) The activation of Bax and Bak during infection and additionally cisplatin induced apoptosis was monitored in immunoprecipitation experiments using the same antibodies. Cells induced to apoptosis with TNFα/CHX were used as a positive control. Pretreatment of cells with zVAD blocked Bax but not Bak activation upon infection and cisplatin induction. (D) The release of cytochrome c was analyzed by subcellular fractionation. HeLa cells were induced to apoptosis with cisplatin in the presence or absence of zVAD for 20 h and subcellular fractions were prepared as mentioned in Materials and methods. The level of cytochrome c in various fractions was analyzed by immunoblotting. The purity of the fractions was monitored by the presence of the mitochondrial marker protein Cox II.
Since Bak and Bax may be activated by caspase-dependent or -independent pathways, we tested the involvement of caspases in the infection-induced Bak and Bax activation. HeLa cells were infected or pretreated with zVAD before infection and 15 h later active Bak or Bax were precipitated. Surprisingly, Bak, but not Bax was precipitated with conformation-specific antibodies in the presence of the pan-caspase inhibitor zVAD (Figure 2C). The caspase-independent and -dependent activation of Bak and Bax, respectively, was also confirmed by FACS analysis (Figure 2A). Consistent with the zVAD-dependent activation, translocation of Bax to mitochondria was impaired in the presence of zVAD upon Ngo infection (Supplementary Figure S2). The requirement of active caspases for Bax but not for Bak is an intriguing observation, as Bax and Bak have been shown to be activated in a similar manner. To check if the observed phenomenon is specific to Ngo-induced apoptosis, we tested if blocking caspases has any influence in the activation of Bax during cisplatin-induced apoptosis. Interestingly, pretreatment of cells with zVAD blocked Bax but not Bak activation during cisplatin-induced apoptosis (Figure 2C). As previously observed for infected cells, translocation of Bax to mitochondria was impaired in the presence of zVAD upon cisplatin treatment (Supplementary Figure S2). Finally, to confirm if the observed phenotype is not confined to a single-cell type, we have performed these experiments in a second epithelial cell line, HEp-2, derived from human larynx carcinoma. As expected, zVAD inhibited Bax but not Bak activation when treated with cisplatin as observed in HeLa cells (Supplementary Figure S3). These data showed that Bak and Bax are activated by zVAD-independent and -dependent mechanisms, respectively, upon Ngo-infection and cisplatin treatment in these cells.
As cytochrome c is released from the mitochondria despite zVAD treatment during Ngo infection, we checked if cytochrome c release during cisplatin induction is also independent of active caspases. Interestingly, cytochrome c is still released upon cisplatin treatment despite zVAD treatment (Figure 2D). These data suggested that cytochrome c is released in these cells even when caspases were inhibited during Ngo- and cisplatin-mediated apoptosis.
Bak is required for Ngo- and cisplatin-induced Bax activation
The differential requirement for caspases could either mean that Bak and Bax are activated simultaneously by different pathways or sequentially by the same initial stimulus. Time course experiments with Ngo revealed Bak being activated between 4 and 6 h postinfection and Bax between 6 and 8 h postinfection (Supplementary Figure S4) suggesting a sequential rather than a parallel activation of Bak and Bax. To investigate whether Bax activation depends on active Bak, we designed short-interfering RNAs (siRNAs) for transient silencing of bak and bax. In addition, expression vectors for short hairpin RNAs (shRNAs) were introduced into HeLa cells by lentiviral transduction to permanently silence the expression of Bak and Bax by RNA interference (RNAi). Both, siRNAs and shRNAs reduced the homologous mRNA by more than 80% (Supplementary Figure S5). Moreover, a clear reduction of the homologous, but not the heterologous protein indicated that silencing of one gene did not affect the amount of the other protein by the siRNAs and shRNAs (Figure 3A–C). HeLa cells depleted of Bak were subsequently infected with Ngo or treated with cisplatin and activation of Bax was monitored by immunoprecipitation with conformation-specific antibody. Ngo infection and cisplatin treatment failed to activate Bax in the absence of Bak (Figure 3D and E) indicating that Bak is required for the infection- and cisplatin-induced activation of Bax. Treatment of HeLa-shBak with TNFα/CHX induced a conformational change in Bax (Figure 3F) demonstrating that TNF-receptor-induced activation of Bax is independent of Bak in these cells.
Figure 3.
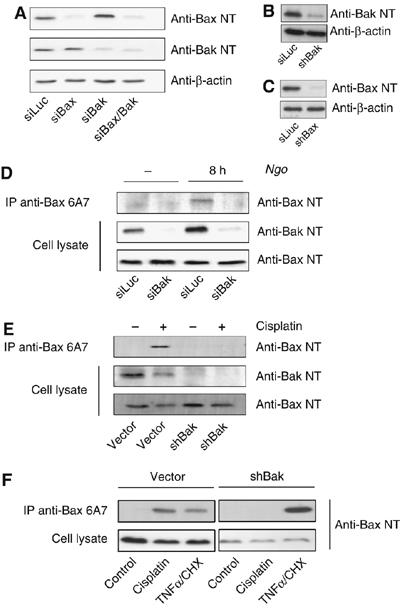
Bak is required for Ngo and cisplatin induced Bax activation. For further analysis Bak and Bax were silenced using RNAi. (A) A transient knockdown was established using siRNAs against Bax, Bak or Bax and Bak. After transfection the efficiency of gene silencing was monitored by immunoblot analysis. (B–C) Expression of Bak or Bax was permanently silenced by generating cell lines stably expressing shRNA directed against the Bak (shBak) or Bax (shBax) mRNA. The knockdown was validated by immunoblot. (D) Cells transfected with siRNA against Bak were infected for 8 h with Ngo and monitored for Bax activation by immunoprecipitating active Bax as shown before. No activation of Bax was detected in Bak-deficient cells. (E–F) In parallel, dependency of Bak- for Bax-activation was monitored upon cisplatin treatment. Consistently, Bax-activation was inhibited by siRNA- and shRNA-mediated knockdown of Bak, whereas TNFα treatment still activated Bax under the same conditions.
Bak and Bax are required for infection and cisplatin-induced apoptosis
Previous studies with knockout mice revealed that Bak and Bax exert very similar functions in most of the stress-induced apoptotic pathways and can functionally substitute each other (Wei et al, 2001). To test whether Ngo induces apoptosis via either Bak or Bax, we silenced their expression and analyzed infection-induced apoptosis by TUNEL analysis. The fraction of apoptotic cells was reduced from 29% in the control transfected cells to 7% (P<0.001) in Bak-silenced and to 16% (P<0.005) in Bax-silenced cells (Figure 4A). Consistently, 27% of the infected cells transfected with control siRNA exhibited an increased caspase activity indicative of induction of apoptosis (Figure 4B). Surprisingly, the fraction of apoptotic cells was significantly reduced in cells with reduced Bak expression (11%; P<0.001) but also in siBax transfected cells (18%; P<0.001). To confirm these results, cleaved caspase-3 was detected by immunoblot analysis. Infection induced a strong activation of caspase-3 in control-transfected cells (Figure 4C) whereas cleaved caspases-3 was strongly reduced or not detectable in infected cells with reduced expression of Bax or Bak or both, respectively (Figure 4C). Similar results were obtained when the Bak- or Bax-silenced cells were induced to apoptosis with cisplatin. Depletion of either Bak or Bax reduced the apoptotic response and the activation of caspase-3 (Figure 4D and E). In contrast, neither Bak nor Bax were required for TNFα/CHX-induced apoptosis and effector caspase activation (Figure 4D and E) ruling out unspecific inhibition of caspase-3 activation due to experimental settings. Finally, HeLa cells depleted of both, Bax and Bak by siRNAs (Supplementary Figure S6A) resisted Ngo- and cisplatin-, but not TNFα/CHX-induced apoptosis (Figure 4C and Supplementary Figure S6B). These data suggested that Bak and Bax are functionally non-redundant and are both required for infection- and cisplatin-induced apoptosis.
Figure 4.
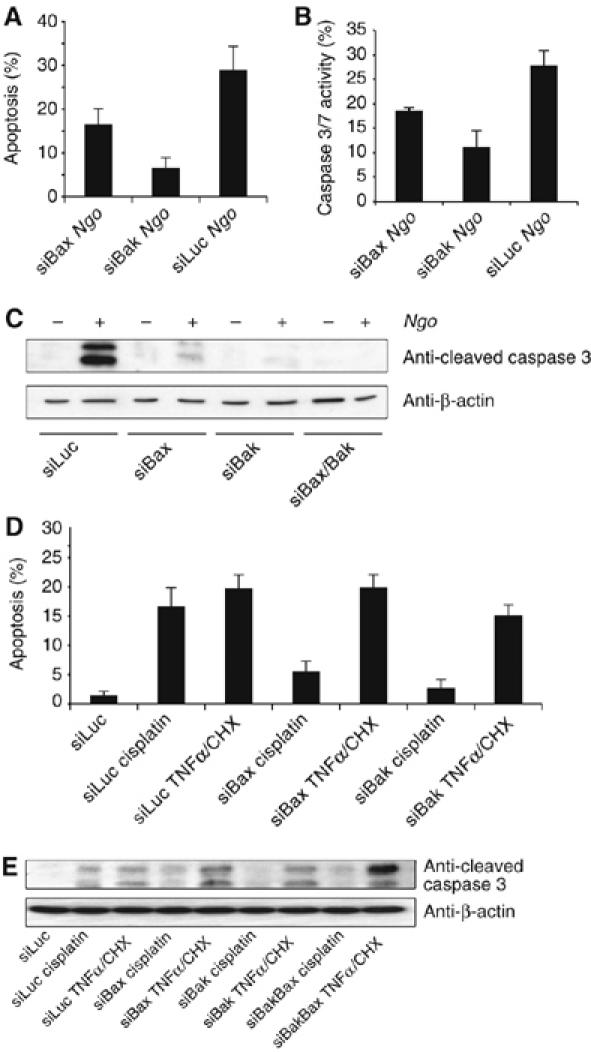
Bax and Bak are required for Ngo- and cisplatin-induced apoptosis. (A) Bak or Bax deficient cells were infected with Ngo and apoptosis was quantified by TUNEL analysis. (B) Additionally the effector caspase activity was measured by FACS analysis using the CaspAce assay kit. (C) The activation of effector caspase-3 upon Ngo infection was monitored by detecting the appearance of p19/17 fragments using cleaved caspase-3 antibody. Consistently, Bak but not Bax knockdown completely abrogated effector caspase activation and thereby apoptosis upon Ngo infection. (D) Cisplatin-induced apoptosis measured by Hoechst 33342 staining and enumeration of fragmented nuclei showed a clear dependency on Bak and to a lesser degree on Bax. TNFα, which induces apoptosis independent of Bak and Bax, was used as a control. (E) Additionally effector caspase activity was measured as described before. As for Ngo infection silencing of Bak inhibited the activation of effector caspases upon cisplatin treatment. (A), (B) and (D) show data from three independent experiments and the error bars represent the ±s.d. of the mean.
Bak-dependent release of cytochrome c during Ngo- and cisplatin-induced apoptosis
The requirement of both Bak and Bax during cisplatin- and Ngo-induced apoptosis suggested that both Bax and Bak have distinct roles during stress-induced apoptosis. One of the key events during stress-induced apoptosis is the MOMP and the release of cytochrome c to trigger the formation of the apoptosome. As cytochrome c is still released in the absence of active Bax (zVAD treated cells), we hypothesized that the release of cytochrome c is primarily influenced by Bak in these cells. To test this hypothesis, we made use of the HeLa cell lines stably expressing shRNAs to silence Bak and Bax expression (see Figure 3B and C). Consistently both shBax- and shBak-expressing HeLa cells were profoundly resistant to cisplatin-induced apoptosis (Supplementary Figure S7A and B) reconfirming the distinct roles for Bak and Bax during stress-induced apoptosis. In addition, we have tested if shBax and shBak cells were resistant to other inducers of the intrinsic pathway of apoptosis. Interestingly, both shBak and shBax cells resisted brefeldin A-, staurosporine- and doxorubicin-induced apoptosis (Supplementary Figure S8).
To test for the specific role of Bak and Bax, control and shBak- and shBax-expressing cells were infected with Ngo or treated with cisplatin and the release of cytochrome c was monitored by immunofluorescence analysis (Figure 5A and B). To our surprise, cytochrome c release was blocked in the shBak but not in the shBax cells when infected with Ngo or induced with cisplatin suggesting the specific role of Bak in releasing cytochrome c during Ngo- and cisplatin-induced apoptosis (Figure 5A and B). To further confirm these observations, subcellular fractionation experiments were performed and the amounts of released cytochrome c was monitored by immunoblot analysis. Consistent with the immunofluorescence data, release of cytochrome c is abrogated in the shBak-cells but not in shBax-expressing cells upon Ngo infection or cisplatin treatment (Figure 5C and D). These data suggested that Bak is specifically required to release cytochrome c from the mitochondria of these cells during Ngo- and cisplatin-induced apoptosis.
Figure 5.
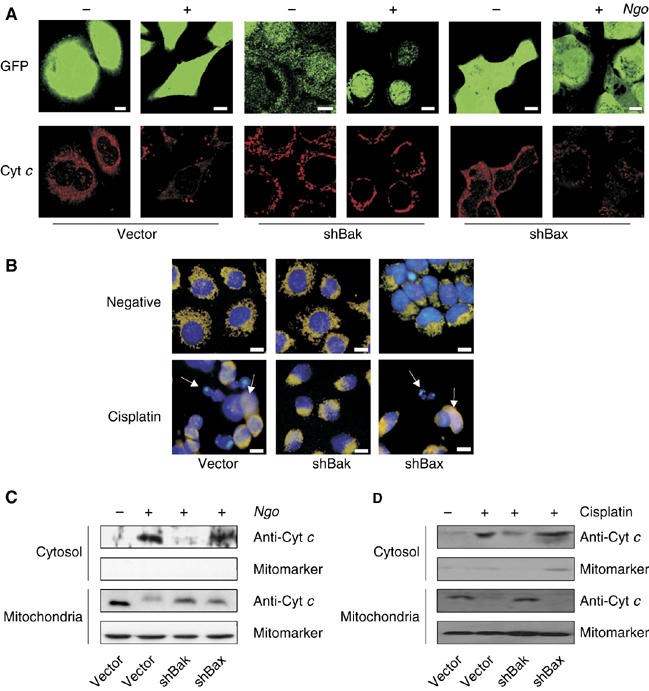
Bak-dependent release of cytochrome c during Ngo- and cisplatin-induced apoptosis. (A) HeLa cells permanently expressing shBak or shBax were infected and cytochrome c release upon infection was visualized by immunofluorescence using anti-cytochrome c antibody. The GFP-expression derived from the vector used for shRNA transcription. (B) Cisplatin-induced cytochrome c release was monitored by co-staining the cells with anticytochrome c antibody and Hoechst 33342. Arrows indicate cells showing a release of cytochrome c. (C–D) Additionally the release of cytochrome c in shBak and shBax cells upon infection and cisplatin treatment was monitored by the use of anticytochrome c antibody in cytosolic extracts upon subcellular fractionation. ShBak cells showed no release of Cytochrome c, whereas shBax and controls released Cytochrome c upon cisplatin treatment or Ngo infection.
Caspase-9 is required to activate Bax
Release of cytochrome c to the cytosol triggers the formation of apoptosome where caspase-9 is recruited for activation. As activation of Bax is caspase dependent, we tested if the silencing of caspase-9 can inhibit Bax activation. Therefore, we used siRNAs to silence the expression of caspase-9 in HeLa cells and checked for the activation of Bax upon Ngo infection. Indeed, Ngo infection activated only Bak but not Bax in caspase-9 silenced cells (Figure 6A). Bax activation was also abrogated upon Ngo infection when caspase-9 activity was inhibited by the caspase-9-specific chemical inhibitor LEHD-fmk (Figure 6B). As expected cisplatin treatment also failed to induce the activation of Bax in cells depleted of caspase-9 (Figure 6C), suggesting a role of caspase-9 in Bax activation. To further confirm the requirement of the apoptosome in activating Bax, we investigated the influence of Apaf-1, the adaptor protein responsible for the formation of the apoptosome complex activating caspase-9. HeLa cells depleted of Apaf-1 by RNAi were induced to apoptosis with Ngo or cisplatin. The efficiency of the knockdown was monitored by real-time PCR and immunoblot analysis (Supplementary Figure S9A and B). In line with our previous observations, loss of Apaf-1 caused resistance for both, Ngo- and cisplatin-induced apoptosis (data not shown). Apoptosis induction failed to activate Bax but still activated Bak in cells depleted of Apaf-1 (Figure 6D and E) suggesting that apoptosome formation and activation of caspase-9 are obligatory for the activation of Bax during Ngo- and cisplatin-mediated apoptosis in these cells.
Figure 6.
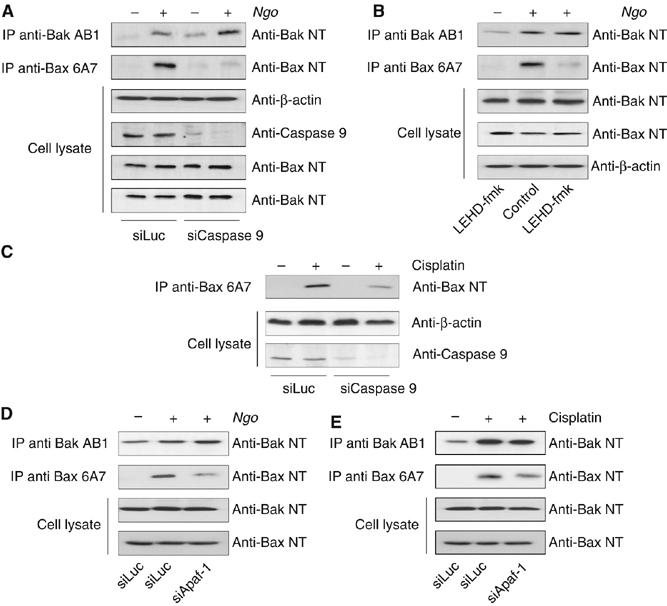
Caspase-9 is required to activate Bax. (A) Caspase-9 expression was silenced in infected or non-infected cells using siRNA and active Bak or Bax was precipitated as indicated. Knockdown of caspase-9 was validated using anticaspase-9 antibody. An influence of caspase-9 knockdown on the expression levels of Bak and Bax was excluded by additional analysis of the cell lysates using anti-Bak NT and anti-Bax NT antibodies. (B) Infected and non-infected cells were pretreated with the caspase-9 inhibitor LEHD-fmk and active Bax and Bak were precipitated as indicated. (C) The effect of caspase-9 knockdown on Bax activation upon cisplatin treatment was analysed as described before. The activation of Bax upon infection and cisplatin treatment was dependent on caspase-9. (D, E) Apaf-1 is required for the activation of Bax. Apaf-1 expression was silenced with siRNAs and active Bax and Bak were immunoprecipitated as indicated.
Discussion
Mitochondria play an important role during apoptosis by releasing several proapoptotic proteins like cytochrome c and Smac/DIABLO to the cytosol to activate caspases. Release of cytochrome c is primarily accomplished by the permeabilization of mitochondrial outer membrane by activated, proapoptotic Bcl-2 family members Bax and Bak. Previous studies with knockout MEFs revealed that Bax and Bak are functionally redundant and can substitute each other (Wei et al, 2001). An unexpected finding of the infection-induced and also the DNA damage-induced apoptosis described here was the distinct roles for Bak and Bax. This is in contrast to many apoptosis-inducing drugs activating apoptotic pathways for which Bak and Bax were functionally redundant (Wei et al, 2001). Interestingly, we found that Ngo infection and cisplatin treatment leads to the engagement of both, caspase-independent and caspase-dependent pathways resulting in the activation of Bak and Bax, respectively. We observed that Bak not only preceded, but was also required for Bax activation in accordance with their function in the same signaling cascade, but in a hierarchical manner.
Although we currently do not know how Bak and Bax are activated during infection, a role for the BH3 only proteins is likely. Proapoptotic activity of BH3 only proteins is controlled by transcriptional and post-translational mechanisms (Bouillet and Strasser, 2002). Control of Bad and Bik, for example, requires phosphorylation and dephosphorylation (Zha et al, 1996; Verma et al, 2001) whereas Bid activation involves cleavage by caspases or other proteases (Li et al, 1998; Luo et al, 1998; Deng et al, 2003). It is thus possible that the initial activation of Bak requires signals elicited early during infection or as a consequence of increasing bacterial load similar to treatment of cells with cisplatin. These signals cause the displacement of Mcl-1, a member of the Bcl-2 family inhibiting apoptosis, from Bak (Willis et al, 2005). Consistently, Mcl-1 co-precipitated with Bak in non-infected, but not in infected cells (Supplementary Figure S9), suggesting an efficient displacement of Mcl-1 from Bak upon infection. Once Bak is active, cytochrome c is released and Bax is activated in a caspase-9-dependent fashion to activate effector caspases. Recent observations with the caspase-3 and -7 double knockout fibroblasts also revealed a caspase-dependent activation of Bax and release of cytochrome c from the mitochondria (Lakhani et al, 2006).
The mechanism by which cytochrome c is released from the mitochondria has been intensively debated (Green and Kroemer, 2004). Two models have been proposed and experimental evidence exists for both. In the first model, a to-date undefined PT complex of the mitochondrial membranes is activated and leads to the loss of MMP. As a consequence, mitochondria swell causing the rupture of the outer membrane, release of cytochrome c and consequently apoptosis. However, the mitochondria of apoptotic cells remain not swollen in most of the cases including Ngo-induced apoptosis (data not shown). In the second model, MOMP is accomplished by formation of supra-molecular openings in the outer mitochondrial membrane promoted by activated Bax and cardiolipin in a manner inhibited by Bcl-XL (Kuwana et al, 2002). During Ngo- and cisplatin-induced apoptosis, activation of Bak is obligatory for cytochrome c release as silencing of BAK abrogated cytochrome c release and apoptosis. However, release of cytochrome c alone is not enough to drive the cells to apoptosis as cells silenced for the expression of Bax are also resistant to apoptosis. This suggests that Bax maybe involved either directly or indirectly in releasing other proapoptotic factors from the mitochondria, which may promote effector caspase activation.
Although Bak is required for the release of cytochrome c, we detect no role of Bak in Ngo-induced MMP loss (data not shown). Depending on the inducer of apoptosis, MMP loss may precede or follow caspase activation. In fact, active caspases cleave mitochondrial substrates like the p75 subunit of the electron transport chain complex I causing loss of MMP (Ricci et al, 2004). During Ngo-induced apoptosis, MMP loss occurs independent of Bak and caspases (Figure 1 and data not shown). This suggests that MMP loss is probably due to the targeting of PorB and the perforation of the inner mitochondrial membrane as loss of MMP is still evident in infected cells pretreated with zVAD (Figure 1B–D).
Induction of apoptosis may either support the infection or serve as a host defense strategy to eliminate the infected cell. Most importantly, pathogens often evolved unique ways to modulate the apoptotic program in host cells. Although these natural apoptosis pathways may have high therapeutic relevance, they are by far less well investigated than mechanisms underlying artificial, For example drug-induced apoptosis. Here we describe the molecular basis of naturally induced apoptosis by Ngo infection.
Materials and methods
Human cell culture and infection
HeLa cells (human cervix carcinoma), HEp-2 (human larynx carcinoma cell line) were grown in RPMI 1640 (Gibco) supplemented with 10% heat inactivated FCS in the presence of 5% CO2. Cells were seeded 24 h before infection, washed several times with RPMI without supplements. Infections were routinely performed at an multiplicity of infection (MOI) of 1 for 15 h. For inhibition of caspases, cells were preincubated with 50 μM zVAD-fmk (Bachem) for 15 min before infection/induction and throughout the respective experiment. 293T cells for the production of virus were grown in DMEM (Gibco) supplemented with 10% heat inactivated FCS under the same conditions.
Western blot
Cells were harvested in loading buffer (60 mM Tris–HCl (pH 8.0), 6% SDS, 10 mM DTT, 6% β-mercaptoethanol, 40% glycerol and 0.1% bromophenol blue) and proteins were separated on 12–15% SDS–polyacrylamide gels and then blotted onto PVDF membranes (Millipore). Membranes were blocked for 1 h in 3% bovine serum albumin in Tris-buffered saline (TBS, pH 7.5), 0.1% Tween and then incubated overnight with respective antibodies in 3% bovine serum albumin in TBS, 0.1% Tween. The blots were washed three times for 10 min in TBS/0.1% Tween, incubated for another hour with peroxidase-coupled secondary antibody (Amersham), and bound antibody was detected by enhanced chemiluminescence (Western lightning) on Hyperfilm ECL (Amersham) or in a luminescent image analyzer LAS-3000 (Fujifilm). Antibodies/antisera used in this study: anti-β-actin (Sigma); anti-Bax NT (Upstate); anti-Bax (BD Pharmingen); anti-Bak NT (Upstate); anti-cleaved caspase-3 (Cell Signalling); anti-Mcl-1 (BD Pharmingen), anti-Tom-20 (Sigma), anti-ATP synthase a (BD Pharmingen), anti-Cox II (Molecular Probes) and anti-Caspase-9 (Cell Signalling). Equal loading was routinely confirmed by appropriate loading controls using anti-β-actin antibody (Sigma).
Fractionation of cells
To obtain mitochondria enriched fractions, HeLa cells were washed twice with ice-cold PBS, harvested and washed in M-Buffer (0.4 M sucrose, 50 mM Tris, 1 mM EGTA, 5 mM β-ME, 0.2% bovine serum albumin (BSA), 10 mM KH2PO4, pH 7.6). The cells were resuspended in M-Buffer and incubated for 20 min on ice. The cells were lysed using a Dounce homogenizer and the nuclei and debris were removed by centrifugation at 4000g for 1 min. The supernatant was carefully removed and the mitochondria containing heavy membranes were then obtained by centrifugation at 15 000g for 10 min. The cytoplasmic proteins contained in the supernatant were analyzed for cytochrome c by immunoblotting.
Subcellular fractionation experiments with the proteo-extraction kit (Merk Biosciences) following the manufacturers instructions. Fraction 1 is used as a cytosol and fraction 2 is used a mitochondria enriched fraction. The purity of cytosolic fractions was controlled by immunoblots using antibodies against mitochondrial marker proteins (COX II and ATPase Synthase α).
FACS analysis
Activation of Bak and Bax was measured by FACS analysis. Control and infected HeLa cells were harvested, washed with PBS (Gibco) and fixed in 2% PFA for 30 min at 4°C. After an additional wash in PBS, cells were incubated in Buffer S (1% FCS, 1% Saponin in PBS) with 0.5 μg anti-Bax 6A7 (BD Pharmingen) or anti-Bak AB1 (Oncogene) for 30 min at 4°C. Cells were washed with Buffer W (1% FCS in PBS) and stained in Buffer S with Cy2 conjugated anti-mouse or anti-rabbit secondary antibody (Jackson Immuno Research) for 30 min at 4°C. Cells were washed twice with Buffer W before to FACS analysis.
For the analysis of MMP, cells were harvested by trypsinization and washed with PBS before staining with 100 nM TMRE (Molecular Probes) in growth media at 37°C, 5% CO2 for 30 min. After staining, cells were washed twice with PBS and immediately analyzed by FACS analysis.
Apoptosis assays
Control and infected cells were collected and washed twice with PBS and fixed with 70% ethanol at −20°C for 30 min. Cells were pretreated with zVAD-fmk (Bachem) as indicated. After removing ethanol, the cell pellets were stained with Nicoletti buffer (0.1% Triton X-100, 50 μg/ml propidium iodide in PBS, pH 7,4) at room temperature in the dark for 20 min and measured by FACS analysis. Caspase-3 and -7 activities were measured by CaspACE assay. Control and infected cells were collected and stained with 10 μM CaspACE (Promega) in growth media at 37°C, 5% CO2 for 20 min. After staining, cells were washed twice with PBS and immediately subjected to FACS analysis. Unless otherwise mentioned cells were induced to apoptosis, with cisplatin at a final concentration of 60 μM for 15 h, Brefeldin A at a final concentration of 1.5 μg/ml for 35 h, staurosporine at 1.5 μM for 6 h and 4 μM of doxorubicin for 40 h. For TNFα-induced apoptosis cells were treated with 25 ng/ml TNFα together with 10 μg/ml cycloheximid for 5 h. The cells were fixed with 3% PFA as mentioned before and stained with Hoechst 33342 as mentioned below.
Immunoprecipitation
Cells were washed in PBS and lysed in Buffer P (150 mM NaCl, 10 mM HEPES, 1% CHAPS, pH 7.4) containing Complete Protease Inhibitor (Roche). Cleared lysates were incubated over night with 2 μg of conformation specific anti-Bax 6A7 or anti-Bak Ab1 antibody in an overhead rotator at 4°C. Protein G-sepharose (20 μl) (Amersham) was added and incubation was continued for 2 h. After extensive washing with Buffer P, the precipitates were boiled in sample buffer at 95°C and the proteins were separated by SDS–PAGE. Western blots were performed using anti-Bax and anti-Bak antibodies (Upstate). For co-immunoprecipitation of Mcl-1 and Bak, control and infected cells were lysed in Buffer P and the lysates were cleared by centrifugation. Mcl-1 and Bak were immunoprecipitated with anti-Mcl-1 (BD Pharmingen) or anti-Bak (BD Pharmingen) antibodies, respectively, and detected by immunoblot.
RNA interference
HeLa cells were grown in 12-well plates for 24 h and then transfected with 1 μg of siRNA using RNAiFect transfection kit (Qiagen) according to the manufacturer's instructions. Efficiency of gene silencing was generally validated by real time PCR as previously described (Machuy et al, 2005) and by Western blot analysis 72 h post-transfection. Protein levels were routinely checked by Western blot analysis in experiments involving RNAi. The sequence targeted by siBax, siBak, siCaspase-9, siApaf-1 and siLuciferase are AAGGTGCCGGAACTGATCAGA, AAGCGAAGTCTTTGCCTTCTC, CAGTGACATCTTTGTGTCCTA, AAGAGCAGCTATGCTGATTAA and AACUUACGCUGAGUACUUCGA respectively.
shRNA oligonucleotides were annealed and cloned into the pLL3.7 or pLVTH-M vectors (Rubinson et al, 2003; Wiznerowicz and Trono, 2003), which carry a GFP cassette for monitoring virus production and infection. The following sequences were targeted to silence Bax and BAK by shRNA expression: GCUCUGAGCAGAUCAUGAA and CCCAUUCACUACAGGUGAA, respectively. Virus carrying Bak shRNAs or Bax shRNAs were produced by transfecting 293T cells with pLL3.7 shBak or pLVTH-M shBax together with viral packaging vectors (psPAX2, pMD2G) by calcium phosphate transfection. Virus were harvested from the supernatant 48 h post-transfection and concentrated by ultracentrifugation at 25 000 r.p.m. HeLa cells were infected with Bak shRNA or Bax shRNA containing virus. Three days postinfection, GFP positive cells were enriched by FACS sorting. Single clones of cells expressing GFP were lysed in sample buffer and the efficiency of gene silencing was checked by Western blot analysis as mentioned above. The cells transfected with the empty vector were used as control.
Immunofluorescence microscopy
Cells seeded on coverslips were infected for 15 h or treated with cisplatin for 20 h. Cells were fixed in 3.7% PFA sometimes followed by staining with 150 nM MitoTracker CMTMRos (M7510) (Molecular Probes). Fixed cells were permeabilized using 0.2% Triton X-100 and nonspecific binding was blocked by using 1% goat serum. Samples were stained using anticytochrome c (BD Pharmingen), anti-Tom-20 (Sigma) or anti-Bax-NT antibody (Upstate) followed by detection with fluorochrome-coupled secondary antibodies (Jackson Immuno Research). Samples were routinely costained with Hoechst 33342 to visualize DNA fragmentation and were analyzed under a Leica confocal microscope using TCS software or a Zeiss immunofluorescence microscope using ACT software.
Supplementary Material
Supplementary Figures
Supplementary Figure Legends
Acknowledgments
We thank Kathleen Gottschalk for excellent technical assistance, Dr Anna Walduck for critically reading the manuscript and Dr Eyal Gottlieb (Beatson Institute, Glagow. UK) for his helpful comments and suggestions. We are grateful to Dr Nikolaus Machuy and the EURIT team for validating the siRNAs. This work was supported by grant SPP1131 from the Deutsche Forschungsgemeinschaft (DFG) to TR.
References
- Bouillet P, Strasser A (2002) BH3-only proteins – evolutionarily conserved proapoptotic Bcl-2 family members essential for initiating programmed cell death. J Cell Sci 115: 1567–1574 [DOI] [PubMed] [Google Scholar]
- Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M, Green DR (2004) Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 303: 1010–1014 [DOI] [PubMed] [Google Scholar]
- Deng Y, Ren X, Yang L, Lin Y, Wu X (2003) A JNK-dependent pathway is required for TNFalpha-induced apoptosis. Cell 115: 61–70 [DOI] [PubMed] [Google Scholar]
- Fischer U, Janicke RU, Schulze-Osthoff K (2003) Many cuts to ruin: a comprehensive update of caspase substrates. Cell Death Differ 10: 76–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DR, Kroemer G (2004) The pathophysiology of mitochondrial cell death. Science 305: 626–629 [DOI] [PubMed] [Google Scholar]
- Korsmeyer SJ, Wei MC, Saito M, Weller S, Oh KJ, Schlesinger PH (2000) Pro-apoptotic cascade activates BID, which oligomerizes BAK or BAX into pores that result in the release of cytochrome c. Cell Death Differ 7: 1166–1173 [DOI] [PubMed] [Google Scholar]
- Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, Green DR, Newmeyer DD (2002) Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell 111: 331–342 [DOI] [PubMed] [Google Scholar]
- Lakhani SA, Masud A, Kuida K, Porter GA Jr, Booth CJ, Mehal WZ, Inayat I, Flavell RA (2006) Caspases 3 and 7: key mediators of mitochondrial events of apoptosis. Science 311: 847–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leu JI, Dumont P, Hafey M, Murphy ME, George DL (2004) Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex. Nat Cell Biol 6: 443–450 [DOI] [PubMed] [Google Scholar]
- Li H, Zhu H, Xu CJ, Yuan J (1998) Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 94: 491–501 [DOI] [PubMed] [Google Scholar]
- Luo X, Budihardjo I, Zou H, Slaughter C, Wang XD (1998) Bid, a bcl2 interacting protein, mediates cytochrome-c release from mitochondria in response to activation of cell-surface death receptors. Cell 94: 481–490 [DOI] [PubMed] [Google Scholar]
- Machuy N, Thiede B, Rajalingam K, Dimmler C, Meyer TF, Rudel T (2005) A global approach to explore receptor-induced apoptosis by proteome analysis and phenotypic screening with RNA interference. Mol Cell Proteomics 4: 44–55 [DOI] [PubMed] [Google Scholar]
- Massari P, Ho Y, Wetzler LM (2000) Neisseria meningitidis porin PorB interacts with mitochondria and protects cells from apoptosis. Proc Natl Acad Sci USA 97: 9070–9075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, Moll UM (2003) p53 has a direct apoptogenic role at the mitochondria. Mol Cell 11: 577–590 [DOI] [PubMed] [Google Scholar]
- Müller A, Gunther D, Brinkmann V, Hurwitz R, Meyer TF, Rudel T (2000) Targeting of the pro-apoptotic VDAC-like porin (PorB) of Neisseria gonorrhoeae to mitochondria of infected cells. EMBO J 19: 5332–5343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller A, Rassow J, Grimm J, Machuy N, Meyer TF, Rudel T (2002) VDAC and the bacterial porin PorB of Neisseria gonorrhoeae share mitochondrial import pathways. EMBO J 21: 1916–1929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller A, Rudel T (2001) Modification of host cell apoptosis by viral and bacterial pathogens. Int J Med Microbiol 291: 197–207 [DOI] [PubMed] [Google Scholar]
- Naumann M, Rudel T, Meyer TF (1999) Host cell interactions and signalling with Neisseria gonorrhoeae. Curr Opin Microbiol 2: 62–70 [DOI] [PubMed] [Google Scholar]
- Ricci JE, Munoz-Pinedo C, Fitzgerald P, Bailly-Maitre B, Perkins GA, Yadava N, Scheffler IE, Ellisman MH, Green DR (2004) Disruption of mitochondrial function during apoptosis is mediated by caspase cleavage of the p75 subunit of complex I of the electron transport chain. Cell 117: 773–786 [DOI] [PubMed] [Google Scholar]
- Rubinson DA, Dillon CP, Kwiatkowski AV, Sievers C, Yang L, Kopinja J, Rooney DL, Ihrig MM, McManus MT, Gertler FB, Scott ML, Van Parijs L (2003) A lentivirus-based system to functionally silence genes in primary mammalian cells, stem cells and transgenic mice by RNA interference. Nat Genet 33: 401–406 [DOI] [PubMed] [Google Scholar]
- Rudel T, Schmid A, Benz R, Kolb HA, Lang F, Meyer TF (1996) Modulation of Neisseria porin (PorB) by cytosolic ATP/GTP of target cells: parallels between pathogen accommodation and mitochondrial endosymbiosis. Cell 85: 391–402 [DOI] [PubMed] [Google Scholar]
- Scaffidi C, Fulda S, Srinivasan A, Friesen C, Li F, Tomaselli KJ, Debatin KM, Krammer PH, Peter ME (1998) Two CD95 (APO-1/Fas) signaling pathways. EMBO J 17: 1675–1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaux DL, Korsmeyer SJ (1999) Cell death in development. Cell 96: 245–254 [DOI] [PubMed] [Google Scholar]
- Verma S, Zhao LJ, Chinnadurai G (2001) Phosphorylation of the pro-apoptotic protein BIK—mapping of phosphorylation sites and effect on apoptosis. J Biol Chem 276: 4671–4676 [DOI] [PubMed] [Google Scholar]
- Weel JF, van Putten JP (1991) Fate of the major outer membrane protein P.IA in early and late events of gonococcal infection of epithelial cells. Res Microbiol 142: 985–993 [DOI] [PubMed] [Google Scholar]
- Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ (2001) Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292: 727–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinrauch Y, Zychlinsky A (1999) The induction of apoptosis by bacterial pathogens. Annu Rev Microbiol 53: 155–187 [DOI] [PubMed] [Google Scholar]
- Willis SN, Chen L, Dewson G, Wei A, Naik E, Fletcher JI, Adams JM, Huang DC (2005) Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev 19: 1294–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiznerowicz M, Trono D (2003) Conditional suppression of cellular genes: lentivirus vector-mediated drug-inducible RNA interference. J Virol 77: 8957–8961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ (1996) Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L). Cell 87: 619–628 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures
Supplementary Figure Legends