

Abstract
Expression of AMAP1 correlates well with the invasive phenotypes and malignancy of human primary breast carcinomas. AMAP1 recruits its binding proteins, such as cortactin and paxillin, to sites of Arf6 activation to form invadopodia. A mouse ortholog of AMAP1, ASAP1, is known to bind to CIN85, a binding partner of an E3 ligase, Cbl. Here, we found that CIN85 colocalizes with AMAP1 at invadopodia, and binding of AMAP1 with CIN85 is important for the invasive activities of breast cancer cells, including MDA-MB-231. siRNA-mediated silencing of CIN85, as well as Cbl, also inhibited the invasion. We moreover found that AMAP1 is monoubiquitinated, rather than polyubiquitinated, by virtue of Cbl and provide evidence that the ability of AMAP1 to be monoubiquitinated is important for its involvement in invasion. Our results indicate that CIN85, as well as Cbl, which is a well-known suppressor of growth factor receptor signaling, can be positively involved in tumor invasion, and suggest that a complex epigenetic process is involved in AMAP1 function in breast cancer cell invasion.
Keywords: AMAP1, breast cancer, invasion, ubiquitination
Introduction
Invasive phenotypes of carcinomas are a major factor for the poor prognosis of patients. Extensive studies of gene expression profiles and genetic alterations of primary carcinomas of the human breast have indicated that there may be no clear universal expression signatures or mutations that accounts for the invasive phenotypes of breast carcinomas (Ma et al, 2003; Lacroix et al, 2004; Schuetz et al, 2006). The lack of clear ‘invasive' signatures or mutations may partly be because microenvironments often contribute to evoke the invasive phenotypes of breast carcinomas (Rizki and Bissell, 2004). However, a direct correlation between in vivo invasive phenotypes and in vitro invasion activities has been well documented with a number of different breast cancer cell lines, including MDA-MB-231 (Thompson et al, 1992; Coopman et al, 1998; Bowden et al, 2001). Therefore, such well-characterized breast cancer cells may provide excellent experimental model systems to identify intracellular events that cause these tumor cells to be invasive. The lack of clear ‘invasive' signatures and genetic mutations also implicate that epigenetic events contribute to processes generating invasive phenotypes of some breast cancer cells.
We have shown that Arf6 is localized to invadopodia and is crucial for the invasive activities of different breast cancer cells, including MDA-MB-231 (Hashimoto et al, 2004). Arf6 has also been shown to be necessary for the invasion of melanoma cells (Tague et al, 2004). We found that AMAP1 (also called DDEF1 and PAG2 in human, and ASAP1 in mouse) is another component of invadopodia, and acts as an effector for GTP-Arf6 in invasion (Hashimoto et al, 2005; Onodera et al, 2005). siRNA-mediated suppression of Arf6 and AMAP1 effectively blocks the invasive activities of different breast cancer cells. We have further shown that protein levels of Arf6 and AMAP1 are highly augmented in invasive breast cancer cells but not in weakly and non-invasive breast cancer cells, independent of their transcriptional upregulation (Hashimoto et al, 2004; Onodera et al, 2005). These results explain why genes for Arf6 and AMAP1 have not been nominated as being upregulated in invasive breast cancer cells by their gene expression profiling analysis. On the other hand, it has been reported that AMAP1 gene expression is transcriptionally upregulated in other types of invasive and malignant tumors, such as uveal melanomas (Ehlers et al, 2005), suggesting that fairly diversified mechanisms, even regarding those related to AMAP1, are involved in the development of invasive phenotypes of different cancers.
AMAP1 contains a proline-rich region. The fourth proline-rich sequence in this region is responsible for binding of AMAP1 to the src homology 3 (SH3) domain of cortactin (Onodera et al, 2005), which is a well-known component of invadopodia (Bowden et al, 1999). This binding of AMAP1 and cortactin is crucial for invasive activities in vitro and also metastasis in vivo of breast cancer cells (Onodera et al, 2005; Hashimoto et al, 2006). CIN85 (also called SETA (Bogler et al, 2000), Ruk (Gout et al, 2000) and SH3KBP1 (Narita et al, 2001)) is a ubiquitously expressed multi-adaptor protein and contains three SH3 domains (Take et al, 2000). It has been shown that the SH3 domains of CIN85 bind to the proline-rich region of ASAP1 through a proline-rich sequence that is distinct from the fourth proline-rich sequence (Kowanetz et al, 2004).
SH3 domains of CIN85 are also known to bind to Cbl, through the atypical SH3-ligand proline-rich sequence of Cbl, PxxxPR (Kowanetz et al, 2003). Cbl family proteins consist of three members in human, c-Cbl, Cbl-b and Cbl-3, each containing a RING finger domain, and associate with E2 ubiquitin-conjugating enzymes to direct protein ubiquitination (Thien and Langdon, 2001; Schmidt and Dikic, 2005). Each Cbl member also contains a single src homology 2 (SH2)-like module, namely the TKB (tyrosine kinase-binding) domain, by which Cbl binds to phosphorylated tyrosines of receptor tyrosine kinases (RTKs) in synergy with Grb2 (Huang and Sorkin, 2005). Binding of the CIN85 SH3 domains to Cbl has also been shown to be greatly enhanced by epidermal growth factor (EGF)-induced tyrosine phosphorylation of Cbl (Petrelli et al, 2002; Soubeyran et al, 2002). As a consequence, RTKs, such as EGF receptor (EGFR), bind to Cbl when they are activated, and hence acquire the ability to associate with CIN85. Cbl in this complex then mediates ubiquitination of RTKs and CIN85 (Haglund et al, 2002; Di Fiore et al, 2003; d'Azzo et al, 2005). In the same complex, CIN85, via its proline-rich region, may simultaneously interact with SH3 domains of endocytic proteins, such as endophilins (Petrelli et al, 2002; Soubeyran et al, 2002). It has been proposed that these Cbl-mediated ubiquitinations, as well as recruitment of endophilin to the activated RTKs, play crucial roles in recruiting activated RTKs to endocytic pathways to degrade these RTKs and downregulate their signaling (d'Azzo et al, 2005). It is also well known that internalized EGFR can be recycled back to the plasma membrane, although the mechanism remains largely elusive. On the other hand, Cbl and CIN85 are also known to regulate the actin cytoskeleton (Schmidt and Dikic, 2005).
In this paper, we found that CIN85 is a component of the AMAP1-mediated invasion machinery of several breast cancer cells, including MDA-MB-231. AMAP1 interacts with Cbl, through AMAP1's binding to CIN85, and we moreover show that Cbl also positively contributes to the invasive activities. Our results hence indicate that Cbl and CIN85 have the potential to contribute to tumor malignancy, although Cbl has previously been shown to act as a downregulator of RTK signaling and hence as a tumor suppressor. We furthermore found that AMAP1 can be monoubiquitinated by Cbl, and provide evidence that the E3 ligase function of Cbl on AMAP1 is necessary for AMAP1-mediated breast cancer cell invasion. Our results illustrate an example in which complicated epigenetic events participate in breast cancer cell invasion.
Results
CIN85 interacts with AMAP1 and is an integral component of invadopodia
We have shown previously that AMAP1 plays a crucial role in the invasive activities, such as matrix degradation activity and Matrigel chemoinvasion activity, of different breast cancer cells, including MDA-MB-231 cells (Onodera et al, 2005). The mouse ortholog of AMAP1, namely ASAP1, has been shown to bind to all three SH3 domains of CIN85, via a proline-rich sequence of ASAP1, PVPLPR (Kowanetz et al, 2004). AMAP1 contains the same proline-rich sequence, and we confirmed that AMAP1 also binds to all three SH3 domains of CIN85 in vitro, whereas its R1063A mutant, in which Arg1063 of PVPLPR is changed to alanine, does not (Supplementary Figure 1). Moreover, binding of endogenous AMAP1 to endogenous CIN85 was clearly detected in MDA-MB-231 cells, by their co-precipitation assays (Figure 1A). AMAP1 localizes to invadopodia of MDA-MB-231 cells (Onodera et al, 2005). Consistent with the above results, endogenous CIN85 was detected at invadopodia and colocalized well with endogenous AMAP1 (Figure 1B).
Figure 1.

CIN85 is involved in AMAP1-mediated invasive activities of MDA-MB-231 cells. (A) Endogenous binding of AMAP1 and CIN85. Cell lysates (300 μg) were subjected to immunoprecipitation (IP) and subsequent immunoblotting using antibodies, as indicated. Total, 5 μg of total cell lysates. (B) Localization of CIN85 to invadopodia. Confocal projections of cells stained with CIN85 and AMAP1 are shown. Cells were cultured on fluorescently labeled, crosslinked gelatin matrix for 16 h; degraded gelatin zones are shown in black in the upper middle panel and in red in the upper merged image. Cells were stained with an anti-CIN85 antibody coupled with a Cy2-anti-mouse IgG antibody (upper panels) or with an anti-AMAP1 antibody coupled with a Cy2-anti-rabbit IgG antibody, and an anti-CIN85 antibody coupled with a Cy3-anti-mouse IgG antibody (lower panels). In the upper panels, z-sections of the images are also shown. Scale bar, 10 μm. Representative images are shown from more than 10 cells analyzed. (C, D) Involvement of CIN85 and its binding to AMAP1 in invasion. Cells were treated with CIN85 siRNA and cultured on Biocoat Matrigel chambers to measure chemoinvasion activity (C), or infected with a recombinant lentivirus expressing AMAP1 shRNA, then transfected with rescue constructs of wild-type AMAP1 (resWT) and the R1063A mutant (resR1063A), both tagged with HA, and cultured on fluorescently labeled, crosslinked gelatin to measure matrix degradation activity (D). siRNA and shRNA with irrelevant sequences (irr) were included as controls. Rescue constructs contained the Venus IRES sequence, and cells positive for Venus were analyzed. Data are presented as percentages calculated by normalizing the values obtained for the control cells as 100% (lower panels). Results represent the mean±s.e. of three independent experiments. Upper panels show protein expression in these cells by immunoblotting. (E, F) Effects of P17-TAT. Five μg of GST-CIN85, purified on glutathione beads, was incubated with 200 μg of lysates of HEK293T cells, expressing AMAP1-HA in the presence of 50 μM of P17-TAT, P4-TAT or scrambled TAT peptide, for 2 h at 4°C before subjected to washing and immunoblotting analysis (E). GST-cortactin was used instead of GST-CIN85 as a control. Amounts of GST fusion proteins are shown by Ponceau S staining. In (F), cells were cultured on Biocoat Matrigel chambers in the presence of 500 μM of the indicated peptides. Data are presented as in (C) and (D). Mock, cells untreated with peptides.
We then examined whether CIN85, and its binding to AMAP1, plays important roles in the invasive activities of breast cancer cells. For this, we first examined the effects of siRNA-mediated knockdown of CIN85 expression. We found that CIN85 expression cannot be suppressed very effectively in MDA-MB-231 cells (only about 50–60% suppression; Figure 1C). However, Matrigel chemoinvasion activity was inhibited also by ∼60% upon CIN85 siRNA treatment (Figure 1C). We also confirmed that matrix degradation activity was similarly inhibited by CIN85 siRNA (Supplementary Figure 2). We then examined whether the R1063A mutant can substitute for AMAP1 function in invasion. MDA-MB-231 cells were treated with an shRNA virus specific for AMAP1 and transfected with cDNAs encoding the wild type and the R1063A mutant of AMAP1 each tagged with hemagglutinin (HA), in which the shRNA target sites were mutated (resWT and resR1063A). The efficiency of AMAP1 expression by cDNA transfection was not very high in MDA-MB-231 cells under this condition (data not shown). We measured matrix degradation activity rather than Matrigel invasion activity in experiments using AMAP1 transfection, because the former assay was more suitable to detect cDNA transfection-positive cells and quantify their invasive activities. We found that although resWT restores matrix degradation activity, resR1063A does not (Figure 1D). We moreover generated a cell-permeable PVPLPR peptide, by fusing this sequence with the HIV TAT sequence (Vives et al, 1997; Hashimoto et al, 2006) (we call this peptide P17-TAT, as PVPLPR is the 17th proline-rich sequence in the proline-rich region of AMAP1). P17-TAT blocked binding of glutathione-S-transferase (GST)-tagged CIN85 with HA-tagged AMAP1 in vitro (Figure 1E). In this assay, we used the scrambled peptides, as well as P4-TAT peptide that was previously designed to inhibit AMAP1 binding with cortactin (Hashimoto et al, 2006), as negative controls (Figure 1E). Like P4-TAT (Hashimoto et al, 2006), P17-TAT also inhibited Matrigel chemoinvasion of MDA-MB-231 cells, whereas the control scrambled peptides did not (Figure 1F). These results, together with the above results, indicate that CIN85 is a component of invadopodia, and that CIN85 and its binding to AMAP1 are important for the invasive activities of MDA-MB-231 cells.
Cbl associates with AMAP1 and contributes to invasive activities
CIN85 is a multiadaptor protein strongly bound to Cbl (Petrelli et al, 2002; Soubeyran et al, 2002; Szymkiewicz et al, 2002). We next tested whether binding of AMAP1 to CIN85 leads to the association of AMAP1 with Cbl. HEK293T cells were transfected with plasmids encoding GST-tagged AMAP1, Xpress-tagged CIN85 and Cbl-b. Pull-down of GST-AMAP1 by glutathione-beads clearly co-precipitated Cbl-b in the presence of Xpress-CIN85 (Figure 2A), indicating that a complex of AMAP1/CIN85/Cbl is formed. A small amount of Cbl-b was also co-precipitated with GST-AMAP1 in the absence of Xpress-CIN85, possibly owing to the endogenous expression of CIN85 in HEK293T cells.
Figure 2.

Involvement of Cbl in invasion. (A) AMAP1 interacts with Cbl via CIN85. HEK293T cells were transfected with GST-AMAP1, Xpress-CIN85 and Cbl-b, as indicated. GST-AMAP2 was included as a control. GST proteins were pulled down from 200 μg of cell lysates and subjected to immunoblotting analysis using antibodies, as indicated. Total, 5 μg of total cell lysates. (B) EGF-dependent interaction of AMAP1 and Cbl in MDA-MB-231 cells. Cells were stimulated with or without 100 ng/ml EGF for 10 min. Cell lysates (600 μg) were then subjected to immunoprecipitation with an anti-AMAP1 antibody, followed by immunoblotting using antibodies, as indicated. Total, 10 μg of total cell lysates. (C, D) Involvement of Cbl and its binding to CIN85 in Matrigel chemoinvasion. MDA-MB-231 cells were treated with siRNAs for c-Cbl and/or Cbl-b (C), or treated with Cbl-b siRNA and transfected with rescue constructs of wild-type Cbl-b (Cbl/resWT) and its R911A mutant (resR911A) (D). Cells were then cultured on Biocoat Matrigel chambers to measure Matrigel chemoinvasion activity. siRNA with an irrelevant sequence (irr) was included as a control. Data collection and presentation are the same as in Figure 1C (lower panel). Results represent the mean±s.e. of three independent experiments. The upper panels show protein expression in these cells by immunoblotting.
Binding of CIN85 with Cbl is mediated by SH3 domains of CIN85, and hence can occur constitutively when these proteins are overexpressed. However, binding of CIN85 with Cbl, both expressed endogenously, has been shown to be largely dependent on tyrosine phosphorylation of Cbl, which is induced by several stimuli including that of EGF (Petrelli et al, 2002; Soubeyran et al, 2002; Szymkiewicz et al, 2002). MDA-MB-231 cells express both c-Cbl and Cbl-b (see Figure 2B and C). We found that the association of endogenous AMAP1 with endogenous Cbl (c-Cbl and Cbl-b) was also dependent on EGF stimulation in MDA-MB-231 cells (Figure 2B). However, only very small amounts of AMAP1 appeared to be associated with Cbl even after cells were stimulated by EGF, whereas association AMAP1 with CIN85 was constitutive and robust. Consistent with this, endogenous Cbl (both c-Cbl and Cbl-b) was not found to be localized to invadopodia of MDA-MB-231 cells (data not shown). The lack of Cbl in invadopodia was also confirmed by use of myc-tagged c-Cbl and Cbl-b (data not shown). On the other hand, we nevertheless found that siRNA-mediated suppression of c-Cbl and Cbl-b inhibits Matrigel chemoinvasion activities of MDA-MB-231 cells (Figure 2C). Matrix degradation activity was similarly inhibited by Cbl knockdown (Supplementary Figure 2). The R911A mutant of Cbl-b, in which Arg911 is changed into alanine, does not bind to CIN85 (Kowanetz et al, 2003). To examine whether binding of Cbl to CIN85 is involved in invasion, we treated MDA-MB-231 cells with Cbl-b siRNA and transfected them with rescue constructs of wild type and the R911A mutant of Cbl-b (Cbl/resWT and resR911A). We found that although Cbl/resWT restores Matrigel chemoinvasion activity, resR911A does not (Figure 2D). These results indicate the involvement of Cbl in tumor invasion. Our results also suggest that only a small fraction of Cbl interacts with the CIN85/AMAP1 complex, and yet their interaction might be important for the function of Cbl in invasion.
AMAP1 is monoubiquitinated by virtue of CIN85 and Cbl
Cbl is an E3 component of ubiquitination. To assess the relevance of interaction of Cbl with AMAP1 in tumor invasion, we next investigated whether AMAP1 is ubiquitinated. HEK293T cells were transfected with FLAG-tagged AMAP1 and HA-tagged ubiquitin, with or without Xpress-CIN85 and/or Cbl-b. We found that dense ubiquitination of AMAP1-FLAG occurs when Xpress-CIN85 and Cbl-b are co-overexpressed (a major single band at ∼140 kDa in size), which was detected with precipitated AMAP1-FLAG proteins reactive with an anti-HA antibody (Figure 3A). Reblotting the filter with an anti-FLAG antibody, and judging from the amounts of the shifted band suggested that 10% of AMAP1-FLAG at most seems to be ubiquitinated under this condition (data not shown). On the other hand, amounts of this ubiquitinated band were greatly reduced when both Xpress-CIN85 and Cbl-b were not co-overexpressed (Figure 3A). Knockdown of endogenous Cbl substantially diminished ubiquitination of AMAP1-FLAG (Figure 3B; a higher concentration of an anti-HA antibody was used in this panel than in Figure 3A, to show AMAP1 ubiquitination more clearly, which was conducted in the absence of Xpress-CIN85 and Cbl-b overexpression). Essentially the same results were obtained with GST-AMAP1 (data not shown). These results suggest that AMAP1 can be ubiquitinated by virtue of CIN85 and Cbl.
Figure 3.

Monoubiquitination of AMAP1 by Cbl. (A) Ubiquitination of AMAP1. HEK293T cells were transfected with AMAP1-FLAG, Xpress-CIN85, Cbl-b and HA-ubiquitin, as indicated. FLAG-tagged proteins were then immunoprecipitated from 200 μg of cell lysates using an anti-FLAG antibody and subjected to blotting with an anti-HA antibody (at 1:20 000 dilution). (B) Requirement of Cbl for AMAP1 ubiquitination. HEK293T cells, treated with siRNA duplexes for c-Cbl and Cbl-b, or with an irrelevant sequence (irr) were transfected with AMAP1-FLAG and HA-ubiquitin. FLAG-tagged proteins were then immunoprecipitated from 200 μg of cell lysates and subjected to blotting with an anti-HA antibody (at 1:1000 dilution). (C) Evidence for the monoubiquitination of AMAP1. HEK293T cells were transfected with GST-AMAP1, Xpress-CIN85, Cbl-b and HA-ubiquitin. GST-fusion proteins were then pulled down from 200 μg of cell lysates and subjected to immunoblotting, as indicated. (D) Effects of proteasome inhibitors on AMAP1 and its ubiquitination. HEK293T cells, transfected with GST-AMAP1 and HA-ubiquitin, were treated with MG132 (20 μM), Epoxomicin (2 μM), Lactacystin (10 μg/ml), ZLLH (20 μM) or E64 (100 μg/ml) for 5 h. GST-AMAP1 was then pulled down from 200 μg of cell lysates and subjected to immunoblotting with an anti-HA antibody. (E) Effects of proteasome inhibitors on endogenous AMAP1 in MDA-MB-231 cells. Cells were treated with proteasome inhibitors or related compounds, as above. Total cell lysates were then subjected to immunoblotting, as indicated. (F) Ubiquitination of AMAP1 in MDA-MB-231 cells. Cells were transfected with AMAP1-FLAG and HA-ubiquitin. AMAP1-FLAG was then immunoprecipitated from 500 μg of cell lysates using an anti-FLAG antibody and subjected to immunoblotting with an anti-HA antibody. In (A–F), data are representative of more than three experiments. Total, 10 μg of total cell lysates. In (A–C), AMAP2 was included as a control.
The pattern of AMAP1 ubiquitination was quite different from that of usual protein polyubiquitination, which appears as high molecular weight smear bands on SDS–PAGE. To further investigate the nature of AMAP1 ubiquitination, we employed AMAP2, a highly structurally conserved isoform of AMAP1 that is yet not localized in invadopodia nor involved in invasion (Onodera et al, 2005), as a control. AMAP2 also contains the PVPLPR sequence, and binds to CIN85 and Cbl (Figure 2A). In sharp contrast to AMAP1-FLAG, we found that dense, slow-migrating smear bands of ubiquitinated AMAP2-FLAG appear when Xpress-CIN85 and Cbl-b are co-overexpressed in HEK293T cells (Figure 3A). On the other hand, a single band of ubiquitinated AMAP2-FLAG at ∼130 kDa in size was readily detectable in the absence of exogenous Xpress-CIN85 and Cbl-b, and this band was still clearly detected in the presence of exogenous Xpress-CIN85 and Cbl-b (Figure 3A). Knockdown of endogenous Cbl reduced overall ubiquitination of AMAP2-FLAG (Figure 3B). Essentially the same results were obtained with GST-AMAP2 (data not shown). FK1 is an antibody that recognizes only polymer forms of ubiquitin, but not its monomer form (Haglund et al, 2003b). We found that the single major band of ubiquitinated GST-AMAP1 does not react with the FK1 antibody (Figure 3C). On the other hand, most smear bands of ubiquitinated GST-AMAP2 were clearly reactive to this antibody, whereas the lowest band of ubiquitinated GST-AMAP2 (∼155 kDa in size as AMAP2 was tagged with GST) was not, suggesting that this band may represent a monoubiquitinated form of AMAP1 (Figure 3C). Therefore, ubiquitination of AMAP1 occurs primarily as the monomer form, whereas ubiquitination of AMAP2 occurs mostly as the polymer form. Judging from the molecular sizes, these monomer forms of ubiquitination seem to occur at a single site both in AMAP1 and AMAP2, rather than at multiple sites. We also noticed that very small amounts of polyubiquitination also seem to occur with AMAP1, by longer exposure of the anti-HA blot shown in Figure 3A and B (data not shown; also see later description).
The above results suggest that most of the AMAP1 ubiquitination is non-canonical, and may not be directly leading to its rapid proteasomal degradation. To investigate this issue, we examined the effects of proteasomal inhibitors on AMAP1 ubiquitination. HEK293T cells expressing GST-AMAP1 and HA-ubiquitin were treated with MG132, epoxomicin and lactacystin, or with an inactive derivative of MG132, ZLLH, and the calpain inhibitor E64. We found that none of these compounds notably increase the amount of the protein bands corresponding to the monoubiquitinated form of GST-AMAP1, whereas smear bands of GST-AMAP1, which are otherwise very faint, become evident when cells are treated with these proteasomal inhibitors (Figure 3D). Amounts of the non-ubiquitinated GST-AMAP1, as detected by an anti-GST immunoblot, were also not increased notably by these proteasomal inhibitors (Figure 3D). We confirmed essentially the same results with AMAP1-FLAG (data not shown). Moreover, amounts of endogenous AMAP1 in MDA-MB-231 cells were also not increased by these proteasomal inhibitors (Figure 3E). We used β-catenin as an internal control (Figure 3D and E), which is under constitutive polyubiquitination and rapid proteasomal degradation (Aberle et al, 1997). These results suggest that monoubiquitination of AMAP1 does not lead to its rapid proteasomal degradation. However, we could not clearly detect monoubiquitination (and also polyubiquitination) of endogenous AMAP1 in MDA-MB-231 cells (data not shown). Lack of detectable levels of endogenous ubiquitination of AMAP1 seems to be consistent with our results that very small amounts of endogenous AMAP1 and Cbl are associated in MDA-MB-231 cells, as described earlier. However, a single band of AMAP1 ubiquitination was detected in MDA-MB-231 cells, when AMAP1-FLAG (and also GST-AMAP1; data not shown) and HA-ubiquitin were co-overexpressed (Figure 3F), providing evidence for the occurrence of AMAP1 monoubiquitination in MDA-MB-231 cells.
Evidence that the function of Cbl on AMAP1 is necessary for breast cancer cell invasion
We then sought to obtain evidence that the activity of Cbl on AMAP1 is involved in tumor invasion. For this, as AMAP1 is primarily monoubiquitinated, rather than polyubiquitinated by Cbl, we examined whether the ability of AMAP1 to be monoubiquitinated is necessary for tumor invasion. We first analyzed the possible structural basis as to how different patterns of ubiquitination are evoked between AMAP1 and AMAP2. AMAP1 and AMAP2 share significant similarity in their primary sequence, whereas their proline-rich regions show high diversity (see Figure 4A). Moreover, AMAP1 has a 20-amino-acid sequence at the N-terminal end that is absent in AMAP2. We made a series of swap mutants between AMAP1 and AMAP2 (Figure 4A) and tested their ubiquitination in HEK293T cells. We found that swapping of the PH domain, ArfGAP domain, ankyrin repeats, proline-rich regions and the SH3 domain does not notably alter the ubiquitination pattern of GST-AMAP1 (Figure 4B). However, swapping the N-terminal regions (amino acids 1–332; NT2 mutant) caused a large increase in the smear bands of ubiquitination in GST-AMAP1, in addition to the monoubiquitination band, although deletion of the N-terminal 20 amino acids (ΔN20 mutant) causes only a marginal increase in the smear bands in GST-AMAP1 (Figure 4B). Essentially the same results were obtained with equivalent mutants of AMAP1-FLAG (data not shown). These results suggest that the N-terminal regions of AMAP1 and AMAP2 have a role in determining the ubiquitination patterns of these AMAP family proteins, regarding whether to be primarily monoubiquitinated or polyubiquitinated.
Figure 4.
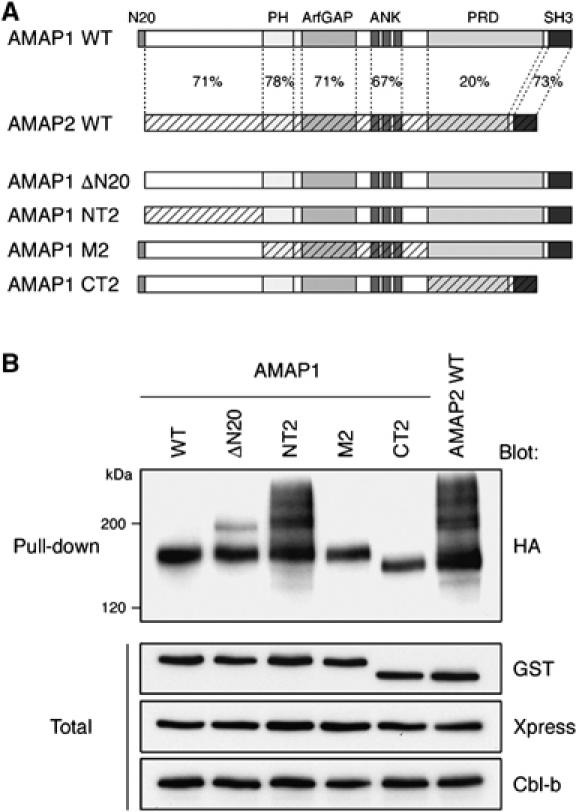
Possible involvement of the N-terminal region of AMAP1 in its preferential monoubiquitination. (A) Schematic representation of AMAP1 and AMAP2, and their swapping mutants. Amino-acid sequence homology is indicated by percentages. (B) HEK293T cells were transfected with GST-AMAP1/2 swap mutants, together with Xpress-CIN85, Cbl-b and HA-ubiquitin. Wild-type GST-AMAP2 was included as a control. GST-fusion proteins, pulled down from 200 μg of cell lysates using glutathione-beads, were subjected to immunoblotting with an anti-HA antibody. Total, 5 μg of total cell lysates. Data are representative of three experiments.
We then examined whether these N-terminal mutants of AMAP1 are involved in invasion. For this, we again used AMAP1 tagged with HA, as in Figure 1D. In a separate experiment, we confirmed that the ΔN20 and NT2 mutants of AMAP1-HA exhibit essentially the same patterns of ubiquitination as observed with each of the equivalent mutants of GST-AMAP1 (data not shown). Then MDA-MB-231 cells, treated with an shRNA virus specific for AMAP1, were transfected with cDNAs encoding rescue constructs of wild type, ΔN20 and NT2 mutants of AMAP1-HA (resWT, resΔN20 and resNT2). We found that although resΔN20 restores matrix degradation activity similar to resWT, the resNT2 mutant does not (Figure 5A). Moreover, overexpression of the NT2 mutant in MDA-MB-231 cells, which were not treated with AMAP1 shRNA, significantly blocked the degradation activity, whereas similar levels of overexpression of the wild type and ΔN20 mutant did not (Figure 5B). These results are consistent with the notion that the property of AMAP1 to be primarily monoubiquitinated, rather than polyubiquitinated, is necessary for this protein to be involved in the invasive activity of MDA-MB-231 cells. These results also support the notion that Cbl's function through modification of AMAP1 is necessary for tumor invasion.
Figure 5.
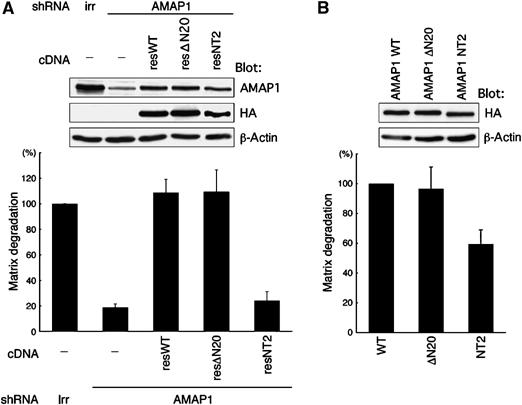
The NT2 mutant does not substitute for AMAP1 function in invasion. MDA-MB-231 cells were infected with AMAP1 shRNA lentivirus and transfected with rescue constructs of wild-type AMAP1 (resWT), the ΔN20 mutant (resΔN20) or NT2 mutant (resNT2), all tagged with HA (A), or transfected with AMAP1 WT HA-IRES-Venus AMAP1 ΔN20 HA-IRES-Venus, or AMAP1 NT2 HA-IRES-Venus (B). Cells were then cultured on fluorescently labeled, crosslinked gelatin. A lentivirus expressing shRNA with an irrelevant sequence (irr) was included as a control. Data collection and presentation are the same as in Figure 1D (upper panels). Results represent the mean±s.e. of three independent experiments. The upper panels show protein expression in these cells by immunoblotting.
Requirement for CIN85, Cbl, and binding of CIN85 with AMAP1 in invasion of different breast cancer cells
Tumor cells are highly diverse in their ways of invasion (Friedl and Wolf, 2003). Invasive characters are also diverse even among the different cell lines of breast tumors (Bowden et al, 1999; Zajchowski et al, 2001). We have shown previously that in addition to MDA-MB-231 cells, AMAP1 is also involved in the invasive activities of highly invasive human breast cancer cells, including MDA-MB-435s and Hs578T, whereas AMAP1 expression is only marginal in weakly and non-invasive breast cancer cells, such as MCF7 (Onodera et al, 2005). We finally examined whether CIN85, binding of CIN85 with AMAP1, and Cbl are also involved in the invasive activities of these breast cancer cells. We found that these invasive cells generally express higher amounts of CIN85 and Cbl-b proteins than those observed in weakly and non-invasive breast cancer cells, whereas all of them express almost comparable levels of c-Cbl (Figure 6A). However, it should be noted that weakly invasive MDA-MB-468 cells express significant amounts of CIN85 and Cbl-b, suggesting that the high level of expression of CIN85 and Cbl is not a major determinant of the invasiveness. Among invasive cells, siRNA-mediated knockdown of CIN85 and Cbl (both c-Cbl and Cbl-b) very effectively blocks Matrigel invasion activity of MDA-MB-435s cells (Figure 6B). P17-TAT was also very effective in inhibiting the invasion of MDA-MB-435s cells (Figure 6C). On the other hand, siRNA-mediated knockdown of CIN85, as well as Cbl, was not very effective in inhibiting Matrigel invasion of Hs578T cells (Figure 6B). P17-TAT was also not very effective in Hs578T cells, although it had a slight inhibitory effect (Figure 6C). These results suggest the highly diverse characters of invasion even among different breast cancer cell lines, as mentioned above.
Figure 6.
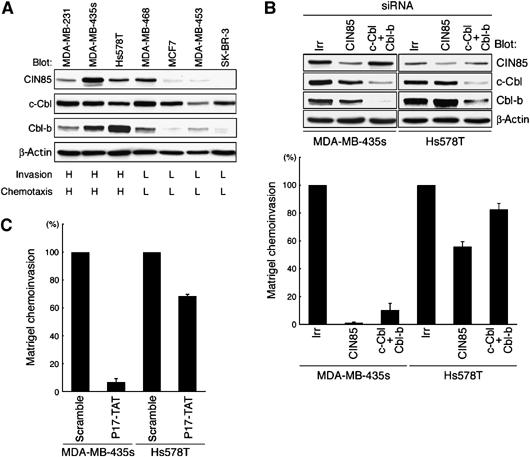
Requirement for CIN85, Cbl and binding of CIN85 with AMAP1 in the invasion of different breast cancer cells. (A) Protein expression of CIN85, c-Cbl and Cbl-b. Total cell lysates (10 μg) were subjected to immunoblotting analysis using antibodies, as indicated. Invasion and chemotactic activities from the literature are shown below, in which activities graded as % MDA-MB-231; L, 0–40%; M, 40–60%; and H, more than 60%. (B) Effects of knockdown of CIN85 and Cbl on Matrigel chemoinvasion. Cells were treated with siRNA duplexes for CIN85 or Cbl (both c-Cbl and Cbl-b), or with an irrelevant sequence (irr), and cultured on Biocoat Matrigel chambers. (C) Effects of P17-TAT. Cells were cultured on Biocoat Matrigel chambers in the presence of 250 μM of P17-TAT or scramble TAT peptide. In (B) and (C) data collection and presentation are the same as in Figure 1C and F, respectively. Results represent the mean±s.e. of three independent experiments. In (B) the upper panels show protein expression in these cells by immunoblotting.
Discussion
AMAP1 plays an important role in the invasive activities of different breast cancer cells, including MDA-MB-231 cells (Onodera et al, 2005). In this paper, we show that CIN85 is a component of invadopodia, and that CIN85, as well as its binding to AMAP1, is important for the invasive activities of several different breast cancer cells, including MDA-MB-231. We also found that through the binding of AMAP1 to CIN85, AMAP1 can be associated with Cbl. By siRNA-mediated knockdown, we show that Cbl also participates in the invasive activity of MBA-MB-231 cells. However, our results indicated that only a very small amount of AMAP1 can be associated with Cbl endogenously in MDA-MB-231 cells. Nevertheless, we show that AMAP1 is primarily monoubiquitinated by virtue of Cbl in a reconstituted system using HEK293T and MDA-MB-231 cells, and moreover suggest that such a property of AMAP1 to be monoubiquitinated is crucial for the invasive activities of MDA-MB-231 cells (also see below). Altogether, our results indicate that in addition to CIN85, Cbl also contributes to the invasive activity of MDA-MB-231 cells, and provide evidence that this function of Cbl occurs at least partly through its binding to the CIN85/AMAP1 complex and via the E3 ligase function on AMAP1.
Cbl-mediated ubiquitination has been well documented to downregulate RTK signalings, such as those of EGFR, CSF-1R and c-Met, all of which have been highly implicated to contribute to the malignancy of many tumors, including breast carcinomas (Sapi et al, 1996). Binding of Cbl with CIN85, expressed endogenously, has been shown to be largely dependent on tyrosine phosphorylation of Cbl, as mentioned earlier. Consistent with this, we found that association of Cbl with the CIN85/AMAP1 complex is primarily dependent on EGF stimulation in MDA-MB-231 cells. EGF is known to enhance the invasive activities of many breast cancer cells, including MDA-MB-231 (Aaronson, 1991; Price et al, 1999; Wells et al, 2002), which we have also confirmed with our culture of MDA-MB-231 cells (data not shown). Our results illustrate an aspect of the mechanisms as to how EGF stimulation is linked to the enhancement of invasive activities in breast cancer cells, and hence point out that CIN85 and Cbl (c-Cbl and Cbl-b) have a potential to positively contribute to EGF-induced tumor malignancy.
We used AMAP2, a close isoform of AMAP1 not involved in invasion, as a control when necessary, and showed that AMAP2 also binds to CIN85 and Cbl, but undergoes mostly polyubiquitination. We made the NT2 mutant of AMAP1, by swapping the N-terminal regions between AMAP1 and AMAP2, and showed that this mutant becomes primarily polyubiquitinated and does not substitute AMAP1's function in invasion. Moreover, we show that this mutant acts as a dominant-negative form and blocks invasion when overexpressed. This may be because the NT2 mutant retains its activity to interact with binding partners of wild-type AMAP1, including paxillin and cortactin, which are essential for invasion; overexpressed NT2 may sequester these proteins from binding to endogenous AMAP1. However, although our results suggest that the property of AMAP1 to be monoubiquitinated, but not polyubiquitinated, is necessary for AMAP1 to be involved in invasive activities, we do not know yet whether monoubiquitination of AMAP1 is directly involved in the processes of tumor invasion.
Important questions that remain to be solved are the precise biological function and fate of monoubiquitinated AMAP1. In addition, it is of interest as to how different modes of ubiquitination can be evoked on AMAP1 and AMAP2 by Cbl. Several reports have documented that Cbl can mediate either mono- or polyubiquitination depending on the nature of substrates, in which polyubiquitination of substrates, such as Sprouty, Src and Abl, leads to proteasomal degradation (Harris et al, 1999; Echarri and Pendergast, 2001; Hall et al, 2003). In the case of Mdm2, this E3 ligase has been shown to evoke either mono- or polyubiquitination on the same substrate, p53 tumor suppressor protein, presumably depending on the concentration of cellular Mdm2 (Li et al, 2003). In this case, polyubiquitination of p53, but not monoubiquitination, also leads to rapid degradation. Our results suggest that monoubiquitination of AMAP1 may not lead to its rapid proteasomal degradation. In contrast, polyubiquitination of AMAP2 seems to be linked to its proteasomal degradation, as we observed an apparent increase in the smear bands of AMAP2 ubiquitination upon treatment with proteasomal inhibitors (our unpublished results). Moreover, we noticed that very small amounts of AMAP1 undergo polyubiquitination, and the amount of this polyubiquitination is augmented by proteasomal inhibitors. Therefore, AMAP1, when polyubiquitinated, may be destined to proteasomal degradation, as in the case of AMAP2. Consistent with this notion, we have observed that the polyubiquitination bands of the NT2 mutant are also increased by proteasomal inhibitors (our unpublished results). Invadopodia are sites of phagocytosis where integrins bind to degraded matrix fragments to endocytose them (Coopman et al, 1996; Bowden et al, 1999). Many protein monoubiquitinations are recognized by ubiquitin receptors to be recruited to endocytic pathways, which are mostly destined to lysosomal degradation (Haglund et al, 2003a; Chen and De Camilli, 2005; Sigismund et al, 2005). It hence awaits to be clarified whether monoubiquitination of AMAP1 is required for AMAP1 to enter such endocytic pathways, which may be necessary for the lysosomal degradation of endocytosed matrix fragments in invasion. On the other hand, it has been shown that the monoubiquitination of proteins containing ubiquitin-binding devices converts these proteins to a default state, by masking their ubiquitin-binding devices intramolecularly by their binding to the monoubiquitin and thus the protein becomes unable to interact in trans with outer ubiquitins (Hoeller et al, 2006). Although AMAP1 does not contain such typical ubiquitin-binding devices (we also found that the N-terminal region of AMAP1 does not exhibit affinity to ubiquitin (our unpublished results)), it may also be worthy to examine whether monoubiquitination of AMAP1 is a default state, preventing AMAP1 from being polyubiquitinated and degraded proteasomally.
Overexpression of ASAP1 has been shown to enhance the recycling-back of EGFR overexpressed in CHO cells, in a manner dependent on its CIN85-binding motif (Kowanetz et al, 2004). In this study, whether ubiquitination of ASAP1 is involved in this recycling was not clarified, although it was shown that very small amounts of EGFR could be co-precipitated with ASAP1/CIN85 in CHO cells. In MDA-MB-231 cells, we could not detect such a co-precipitation of EGFR with the AMAP1/CIN85 complex (our unpublished results). It has been shown that Cbl is involved in the initial step of EGFR internalization without ubiquitination, whereas subsequent Cbl-mediated ubiquitination of EGFR is closely linked to the lysosomal sorting of ubiquitinated EGFR (Waterman et al, 2002; Jiang and Sorkin, 2003; Sigismund et al, 2005). Although further analyses are required, it is possible that high levels of overexpression of ASAP1 in CHO cells may have sequestered its binding partner CIN85 from interacting with ubiquitin-binding/lysosomal sorting proteins, or from interacting with the EGFR/Cbl complex, both of which may have resulted in blocking the lysosomal targeting of internalized EGFR. Such blockage may in turn lead to the enhanced recycling-back of EGFR. In any case, it will be interesting to investigate whether increased expression of AMAP1 in invasive breast cancer cells contributes to the enhanced recycling of EGFR, which potentially contributes to the development of malignancy.
An increasing number of reports have shown that monoubiquitination, and also other non-canonical polyubiquitinations have more diverse cellular functions in addition to vesicular trafficking (Polo et al, 2002; Duncan et al, 2006). Roles of monoubiquitination may also depend on the binding partners (Haglund et al, 2002) and also on cell types. Given that the property of AMAP1 to be monoubiquitinated is crucially required for AMAP-mediated invasive activities of several breast cancer cells, clarifying the mechanism as to how AMAP1 is primarily monoubiquitinated by Cbl, together with the function and fate of monoubiquitinated AMAP1 in such tumor cells will contribute to the further understanding of molecular processes as to how invasive phenotypes arise epigenetically during the development of malignancy in breast carcinomas.
Materials and methods
Cells
Human breast cancer cell line, MDA-MB-231, was obtained from the American Type Culture Collection and cultured in a 1:1 mixture of DMEM and RPMI 1640 supplemented with 10% FCS (HyClone), 5% NuSerum (Becton Dickinson) and 2 mM L-glutamine under 7.5% CO2 at 37°C, as described (Bowden et al, 1999). HEK293T cells were cultured in DMEM with 10% FCS (HyClone) under 5% CO2 at 37°C.
Peptides
P17-TAT-peptide, LTPTLPETPVPLPRKINTGKNK-GRKKRRQRRR, was synthesized and purified by Sigma-Aldrich (Sigma Genosis, Japan). P4-TAT and control scramble peptides were described previously (Hashimoto et al, 2006).
Ubiquitination assay
For the ubiquitination assay, cells were lysed in RIPA buffer (1% Nonidet P-40, 150 mM NaCl, 20 mM Tris–HCl, pH 7.4, 5 mM EDTA, 1% Na-deoxycholate, 0.1% SDS, 1 mM Na3VO4, 10 μM Na2MoO4, 5 μg/ml aprotinin, 2 μg/ml leupeptin, 3 μg/ml pepstatin A and 1 mM phenylmethylsulfonyl fluoride), containing 200 μM iodoacetamide and 10 μg/ml N-ethylmaleimide. Concentrations of other inhibitors were MG132 (20 μM), epoxomicin (2 μM), lactacystin (10 μg/ml), ZLLH (20 μM), E64 (100 μg/ml) and chloroquine (1 μM). Cells were treated with these inhibitors for 5 h before analyses, as described previously (Nakayama et al, 2004).
GST pull-down, immunoprecipitation and immunoblotting
Protein precipitation and binding assays were performed as described previously (Onodera et al, 2005; Hashimoto et al, 2006), using cell lysates prepared with 1% Nonidet P-40 buffer (1% Nonidet P-40, 150 mM NaCl, 20 mM Tris–HCl, pH 7.4, 5 mM EDTA, 1 mM Na3VO4, 10 μM Na2MoO4, 5 μg/ml aprotinin, 2 μg/ml leupeptin, 3 μg/ml pepstatin A and 1 mM phenylmethylsulfonyl fluoride). Co-precipitation of endogenous AMAP1 with CIN85 was performed using a polyclonal AMAP1 antibody bound to Protein A-sepharose beads, or using a CIN85 antibody bound to Protein G-Sepharose beads (Amersham Biosciences). Immunoprecipitation for FLAG-tagged protein was performed using an anti-FLAG antibody bound to Protein G-Sepharose beads, and for GST proteins using glutathione-sepharose beads (Amersham Biosciences). Proteins retained on the beads were subjected to immunoblotting analysis after separation by SDS–PAGE and detected by an enzyme-linked chemiluminescence method (Amersham Biosciences).
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Information
Acknowledgments
We are grateful to Manami Hiraishi and Tomoko Yoneda for their technical assistance, Mayumi Iwahara and Mika Miyoshi for their secretarial work and Helena Akiko Popiel for critical reading of the paper. This work was supported in part by Grants-In-Aid from the Ministry of Education, Science, Sports and Culture of Japan.
References
- Aaronson SA (1991) Growth factors and cancer. Science 254: 1146–1153 [DOI] [PubMed] [Google Scholar]
- Aberle H, Bauer A, Stappert J, Kispert A, Kemler R (1997) β-Catenin is a target for the ubiquitin–proteasome pathway. EMBO J 16: 3797–3804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogler O, Furnari FB, Kindler-Roehrborn A, Sykes VW, Yung R, Huang HJ, Cavenee WK (2000) SETA: a novel SH3 domain-containing adapter molecule associated with malignancy in astrocytes. Neuro-oncology 2: 6–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowden ET, Barth M, Thomas D, Glazer RI, Mueller SC (1999) An invasion-related complex of cortactin, paxillin and PKCμ associates with invadopodia at sites of extracellular matrix degradation. Oncogene 18: 4440–4449 [DOI] [PubMed] [Google Scholar]
- Bowden ET, Coopman PJ, Mueller SC (2001) Invadopodia: unique methods for measurement of extracellular matrix degradation in vitro. Methods Cell Biol 63: 613–627 [DOI] [PubMed] [Google Scholar]
- Chen H, De Camilli P (2005) The association of epsin with ubiquitinated cargo along the endocytic pathway is negatively regulated by its interaction with clathrin. Proc Natl Acad Sci USA 102: 2766–2771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coopman PJ, Do MT, Thompson EW, Mueller SC (1998) Phagocytosis of cross-linked gelatin matrix by human breast carcinoma cells correlates with their invasive capacity. Clin Cancer Res 4: 507–515 [PubMed] [Google Scholar]
- Coopman PJ, Thomas DM, Gehlsen KR, Mueller SC (1996) Integrin α3β1 participates in the phagocytosis of extracellular matrix molecules by human breast cancer cells. Mol Biol Cell 7: 1789–1804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- d'Azzo A, Bongiovanni A, Nastasi T (2005) E3 ubiquitin ligases as regulators of membrane protein trafficking and degradation. Traffic 6: 429–441 [DOI] [PubMed] [Google Scholar]
- Di Fiore PP, Polo S, Hofmann K (2003) When ubiquitin meets ubiquitin receptors: a signalling connection. Nat Rev Mol Cell Biol 4: 491–497 [DOI] [PubMed] [Google Scholar]
- Duncan LM, Piper S, Dodd RB, Saville MK, Sanderson CM, Luzio JP, Lehner PJ (2006) Lysine-63-linked ubiquitination is required for endolysosomal degradation of class I molecules. EMBO J 25: 1635–1645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echarri A, Pendergast AM (2001) Activated c-Abl is degraded by the ubiquitin-dependent proteasome pathway. Curr Biol 11: 1759–1765 [DOI] [PubMed] [Google Scholar]
- Ehlers JP, Worley L, Onken MD, Harbour JW (2005) DDEF1 is located in an amplified region of chromosome 8q and is overexpressed in uveal melanoma. Clin Cancer Res 11: 3609–3613 [DOI] [PubMed] [Google Scholar]
- Friedl P, Wolf K (2003) Tumour-cell invasion and migration: diversity and escape mechanisms. Nat Rev Cancer 3: 362–374 [DOI] [PubMed] [Google Scholar]
- Gout I, Middleton G, Adu J, Ninkina NN, Drobot LB, Filonenko V, Matsuka G, Davies AM, Waterfield M, Buchman VL (2000) Negative regulation of PI 3-kinase by Ruk, a novel adaptor protein. EMBO J 19: 4015–4025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haglund K, Di Fiore PP, Dikic I (2003a) Distinct monoubiquitin signals in receptor endocytosis. Trends Biochem Sci 28: 598–603 [DOI] [PubMed] [Google Scholar]
- Haglund K, Shimokawa N, Szymkiewicz I, Dikic I (2002) Cbl-directed monoubiquitination of CIN85 is involved in regulation of ligand-induced degradation of EGF receptors. Proc Natl Acad Sci USA 99: 12191–12196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haglund K, Sigismund S, Polo S, Szymkiewicz I, Di Fiore PP, Dikic I (2003b) Multiple monoubiquitination of RTKs is sufficient for their endocytosis and degradation. Nat Cell Biol 5: 461–466 [DOI] [PubMed] [Google Scholar]
- Hall AB, Jura N, DaSilva J, Jang YJ, Gong D, Bar-Sagi D (2003) hSpry2 is targeted to the ubiquitin-dependent proteasome pathway by c-Cbl. Curr Biol 13: 308–314 [DOI] [PubMed] [Google Scholar]
- Harris KF, Shoji I, Cooper EM, Kumar S, Oda H, Howley PM (1999) Ubiquitin-mediated degradation of active Src tyrosine kinase. Proc Natl Acad Sci USA 96: 13738–13743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto S, Hashimoto A, Yamada A, Onodera Y, Sabe H (2005) Assays and properties of the ArfGAPs, AMAP1 and AMAP2, in Arf6 function. Methods Enzymol 404: 216–231 [DOI] [PubMed] [Google Scholar]
- Hashimoto S, Hirose M, Hashimoto A, Morishige M, Yamada A, Hosaka H, Akagi K, Ogawa E, Oneyama C, Agatsuma T, Okada M, Kobayashi H, Wada H, Nakano H, Ikegami T, Nakagawa A, Sabe H (2006) Targeting AMAP1 and cortactin binding bearing an atypical src homology 3/proline interface for prevention of breast cancer invasion and metastasis. Proc Natl Acad Sci USA 103: 7036–7041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto S, Onodera Y, Hashimoto A, Tanaka M, Hamaguchi M, Yamada M, Sabe H (2004) Requirement for Arf6 in breast cancer invasive activities. Proc Natl Acad Sci USA 101: 6647–6652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeller D, Crosetto N, Blagoev B, Raiborg C, Tikkanen R, Wagner S, Kowanetz K, Breitling R, Mann M, Stenmark H, Dikic I (2006) Regulation of ubiquitin-binding proteins by monoubiquitination. Nat Cell Biol 8: 163–169 [DOI] [PubMed] [Google Scholar]
- Huang F, Sorkin A (2005) Growth factor receptor binding protein 2-mediated recruitment of the RING domain of Cbl to the epidermal growth factor receptor is essential and sufficient to support receptor endocytosis. Mol Biol Cell 16: 1268–1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Sorkin A (2003) Epidermal growth factor receptor internalization through clathrin-coated pits requires Cbl RING finger and proline-rich domains but not receptor polyubiquitylation. Traffic 4: 529–543 [DOI] [PubMed] [Google Scholar]
- Kowanetz K, Husnjak K, Holler D, Kowanetz M, Soubeyran P, Hirsch D, Schmidt MH, Pavelic K, De Camilli P, Randazzo PA, Dikic I (2004) CIN85 associates with multiple effectors controllong intracellular trafficking of epidermal growth factor receptors. Mol Biol Cell 15: 3155–3166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowanetz K, Szymkiewicz I, Haglund K, Kowanetz M, Husnjak K, Taylor JD, Soubeyran P, Engstrom U, Ladbury JE, Dikic I (2003) Identification of a novel proline-arginine motif involved in CIN85-dependent clustering of Cbl and down-regulation of epidermal growth factor receptors. J Biol Chem 278: 39735–39746 [DOI] [PubMed] [Google Scholar]
- Lacroix M, Toillon RA, Leclercq G (2004) Stable ‘portrait' of breast tumors during progression: data from biology, pathology and genetics. Endocr Relat Cancer 11: 497–522 [DOI] [PubMed] [Google Scholar]
- Li M, Brooks CL, Wu-Baer F, Chen D, Baer R, Gu W (2003) Mono- versus polyubiquitination differential control of p53 fate by Mdm2. Science 302: 1972–1975 [DOI] [PubMed] [Google Scholar]
- Ma XJ, Salunga R, Tuggle JT, Gaudet J, Enright E, McQuary P, Payette T, Pistone M, Stecker K, Zhang BM, Zhou YX, Varnholt H, Smith B, Gadd M, Chatfield E, Kessler J, Baer TM, Erlander MG, Sgroi DC (2003) Gene expression profiles of human breast cancer progression. Proc Natl Acad Sci USA 100: 5974–5979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama K, Frew IJ, Hagensen M, Skals M, Habelhah H, Bhoumik A, Kadoya T, Erdjument-Bromage H, Tempst P, Frappell PB, Bowtell DD, Ronai Z (2004) Siah2 regulates stability of prolyl-hydroxylases, controls HIF1α abundance, and modulates physiological responses to hypoxia. Cell 117: 941–952 [DOI] [PubMed] [Google Scholar]
- Narita T, Amano F, Yoshizaki K, Nishimoto N, Nishimura T, Tajima T, Namiki H, Taniyama T (2001) Assignment of SH3KBP1 to human chromosome band Xp22.1 → p21.3 by in situ hybridization. Cytogenet Cell Genet 93: 133–134 [DOI] [PubMed] [Google Scholar]
- Onodera Y, Hashimoto S, Hashimoto A, Morishige M, Mazaki Y, Yamada A, Ogawa E, Adachi M, Sakurai T, Manabe T, Wada H, Matsuura N, Sabe H (2005) Expression of AMAP1, an ArfGAP, provides novel targets to inhibit breast cancer invasive activities. EMBO J 24: 963–973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrelli A, Gilestro GF, Lanzardo S, Comoglio PM, Migone N, Giordano S (2002) The endophilin–CIN85–Cbl complex mediates ligand-dependent downregulation of c-Met. Nature 416: 187–190 [DOI] [PubMed] [Google Scholar]
- Polo S, Sigismund S, Faretta M, Guidi M, Capua MR, Bossi G, Chen H, De Camilli P, Di Fiore PP (2002) A single motif responsible for ubiquitin recognition and monoubiquitination in endocytic proteins. Nature 416: 451–455 [DOI] [PubMed] [Google Scholar]
- Price JT, Tiganis T, Agarwal A, Djakiew D, Thompson EW (1999) Epidermal growth factor promotes MDA-MB-231 breast cancer cell migration through a phosphatidylinositol 3′-kinase and phospholipase C-dependent mechanism. Cancer Res 59: 5475–5478 [PubMed] [Google Scholar]
- Rizki A, Bissell MJ (2004) Homeostasis in the breast: it takes a village. Cancer Cell 6: 1–2 [DOI] [PubMed] [Google Scholar]
- Sapi E, Flick MB, Rodov S, Gilmore-Hebert M, Kelley M, Rockwell S, Kacinski BM (1996) Independent regulation of invasion and anchorage-independent growth by different autophosphorylation sites of the macrophage colony-stimulating factor 1 receptor. Cancer Res 56: 5704–5712 [PubMed] [Google Scholar]
- Schmidt MH, Dikic I (2005) The Cbl interactome and its functions. Nat Rev Mol Cell Biol 6: 907–919 [DOI] [PubMed] [Google Scholar]
- Schuetz CS, Bonin M, Clare SE, Nieselt K, Sotlar K, Walter M, Fehm T, Solomayer E, Riess O, Wallwiener D, Kurek R, Neubauer HJ (2006) Progression-specific genes identified by expression profiling of matched ductal carcinomas in situ and invasive breast tumors, combining laser capture microdissection and oligonucleotide microarray analysis. Cancer Res 66: 5278–5286 [DOI] [PubMed] [Google Scholar]
- Sigismund S, Woelk T, Puri C, Maspero E, Tacchetti C, Transidico P, Di Fiore PP, Polo S (2005) Clathrin-independent endocytosis of ubiquitinated cargos. Proc Natl Acad Sci USA 102: 2760–2765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soubeyran P, Kowanetz K, Szymkiewicz I, Langdon WY, Dikic I (2002) Cbl–CIN85–endophilin complex mediates ligand-induced downregulation of EGF receptors. Nature 14: 183–187 [DOI] [PubMed] [Google Scholar]
- Szymkiewicz I, Kowanetz K, Soubeyran P, Dinarina A, Lipkowiz S, Dikic I (2002) CIN85 participates in Cbl-b-mediated down-regulation of receptor tyrosine kinases. J Biol Chem 277: 39666–39672 [DOI] [PubMed] [Google Scholar]
- Tague SE, Muralidharan V, D'Souza-Schorey C (2004) ADP-ribosylation factor 6 regulates tumor cell invasion through the activation of the MEK/ERK signaling pathway. Proc Natl Acad Sci USA 101: 9671–9676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Take H, Watanabe S, Takeda K, Yu ZX, Iwata N, Kajigaya S (2000) Cloning and characterization of a novel adaptor protein, CIN85, that interacts with c-Cbl. Biochem Biophys Res Commun 268: 321–328 [DOI] [PubMed] [Google Scholar]
- Thien CB, Langdon WY (2001) Cbl: many adaptations to regulate protein tyrosine kinases. Nat Rev Mol Cell Biol 2: 294–307 [DOI] [PubMed] [Google Scholar]
- Thompson EW, Paik S, Brunner N, Sommers CL, Zugmaier G, Clarke R, Shima TB, Torri J, Donahue S, Lippman ME, Martin GR, Dickson RB (1992) Association of increased basement membrane invasiveness with absence of estrogen receptor and expression of vimentin in human breast cancer cell lines. J Cell Physiol 150: 534–544 [DOI] [PubMed] [Google Scholar]
- Vives E, Brodin P, Lebleu B (1997) A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J Biol Chem 272: 16010–160107 [DOI] [PubMed] [Google Scholar]
- Waterman H, Katz M, Rubin C, Shtiegman K, Lavi S, Elson A, Jovin T, Yarden Y (2002) A mutant EGF-receptor defective in ubiquitylation and endocytosis unveils a role for Grb2 in negative signaling. EMBO J 21: 303–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells A, Kassis J, Solava J, Turner T, Lauffenburger DA (2002) Growth factor-induced cell motility in tumor invasion. Acta Oncol 41: 124–130 [DOI] [PubMed] [Google Scholar]
- Zajchowski DA, Bartholdi MF, Gong Y, Webster L, Liu HL, Munishkin A, Beauheim C, Harvey S, Ethier SP, Johnson PH (2001) Identification of gene expression profiles that predict the aggressive behavior of breast cancer cells. Cancer Res 61: 5168–5178 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Information