

Abstract
The secretion superfamily ATPases are conserved motors in key microbial membrane transport and filament assembly machineries, including bacterial type II and IV secretion, type IV pilus assembly, natural competence, and archaeal flagellae assembly. We report here crystal structures and small angle X-ray scattering (SAXS) solution analyses of the Archaeoglobus fulgidus secretion superfamily ATPase, afGspE. AfGspE structures in complex with ATP analogue AMP-PNP and Mg2+ reveal for the first time, alternating open and closed subunit conformations within a hexameric ring. The closed-form active site with bound Mg2+ evidently reveals the catalytically active conformation. Furthermore, nucleotide binding results and SAXS analyses of ADP, ATPγS, ADP-Vi, and AMP-PNP-bound states in solution showed that asymmetric assembly involves ADP binding, but clamped closed conformations depend on both ATP γ-phosphate and Mg2+ plus the conserved motifs, arginine fingers, and subdomains of the secretion ATPase superfamily. Moreover, protruding N-terminal domain shifts caused by the closed conformation suggest a unified piston-like, push–pull mechanism for ATP hydrolysis-dependent conformational changes, suitable to drive diverse microbial secretion and assembly processes by a universal mechanism.
Keywords: archaea, crystal structure, hexameric ATPase, SAXS, secretion
Introduction
Bacterial and archaeal microbes use multi-protein translocation assembles to control the traffic out of cells by proteins and toxins (Remaut and Waksman, 2004). Type II secretion (T2S) is a major translocation mode, in which the secreted protein has a signal sequence and is transported from the cytoplasm to periplasmic space by Sec machinery that is called a general secretory pathway (GSP). So T2S is also termed the main terminal branch of GSP. Its protein components are designated in alphabetical order, for example, GspA-O in Escherichia coli (Sandkvist, 2001). Among them, the cytoplasmic ATPase, generally termed GspE, is especially important as the only protein able to utilize the energy of ATP hydrolysis and as belonging to the large superfamily of ‘type II/IV secretion NTPases', or ‘secretion superfamily ATPases'. This superfamily involves the ATPases, which function in multiple macromolecular transport systems in bacteria and archaea, such as bacterial T2S and type IV secretion (T4S), type IV pilus (T4 pilus) assembly, DNA uptake, and archaeal flagellae assembly systems (Planet et al, 2001; Peabody et al, 2003; Craig et al, 2004). The secretion ATPase superfamily proteins share similar amino-acids sequences along with the characteristic conserved motifs of the Walker box A, containing the P-loop GX4GK(S/T) of an NTP binding motif, an atypical Walker box B, plus Asp and His Boxes (Possot and Pugsley, 1994). They are subdivided into T2S and T4S classes by sequence similarity. Although both T2S and T4S are powered by secretion ATPases, their basic mechanisms are likely fundamentally different. The T2S system forms the pilin-like or ‘pseudopilin' filament, and secretion ATPase assembles this filament. For T4S, the secretion ATPase may be the protein translocator, which interacts with the secreted protein folded in the cytoplasm.
The archaeal secretion NTPase FlaI acts in the archaeal flagellar assembly system that resembles the better characterized bacterial T4 pilus and T2S systems (Patenge et al, 2001; Thomas et al, 2002). The characteristic N-terminal signal and conserved hydrophobic sequences of archaeal flagellins are remarkably similar to bacterial T4 pilin and T2S ‘pseudopilin' (Bardy et al, 2003; Peabody et al, 2003). The best characterized of these, the T4 pilus, is assembled from pilin subunits into a filament on the pathogenic bacteria cell surface, where it acts in attachment to human cells, host cell signaling, biofilm formation, and DNA uptake (Craig et al, 2004). The T2S ‘pseudopilus' is composed of subunits similar to T4P and may act as a molecular piston designed to push the secreted proteins (Sauvonnet et al, 2000; Sandkvist, 2001). Full-length T4P crystal structures show that the conserved hydrophobic sequence forms an extended α-helix (Parge et al, 1995; Craig et al, 2003), which assembles as an inner scaffold for filament assembly (Craig et al, 2006). Thus, the assembly utilizing conserved hydrophobic sequences may be universal to the archaeal flagellar, T2S pseudopilins, and T4 pilus (Kohler et al, 2004; Trachtenberg et al, 2005). These filament biogenesis mechanisms also resemble each other. Their filament subunits are transported by the Sec system, followed by signal sequence cleavage by a specific membrane-bound signal peptidase (Bardy and Jarrell, 2003). The matured subunits are then anchored in the inner membrane and assembled into a filament by the secretion ATPases (Turner et al, 1993). Notably, amino-acid sequences of FlaI and other archaeal secretion ATPases resemble bacterial T2S and T4S type ATPases, suggesting that the archaeal secretion ATPases may be a progenitor of the secretion ATPase superfamily.
Archaeal genomes encode multiple FlaI-like secretion superfamily NTPase genes. The flaI gene of the fla gene cluster is immediately followed by the flaJ gene that encodes a multi-transmembrane protein (Patenge et al, 2001; Thomas et al, 2002). Interestingly, most other archaeal secretion superfamily ATPase genes are also associated with FlaJ-like membrane protein genes (Albers and Driessen, 2005; Falb et al, 2005). These protein pairs may be functionally coupled and act in unique archaeal secretion systems that resemble T2S (Albers and Driessen, 2005; Albers et al, 2006). The Sulfolobus solfataricus sugar binding protein secreted from cytoplasm to periplasm harbors the archaeal flagellin-like hydrophobic sequence and signal sequence (Albers and Driessen, 2002). Secretion of these sugar binding proteins requires signal sequence cleavage by the membrane-bound signal peptidase, which also cleaves the flagellin signal sequence (Albers et al, 2003). Putative secreted protein genes with flagellin-like signal sequences also occur in other archaea (Albers and Driessen, 2005; Falb et al, 2005). So the secreted protein may be transported from cytoplasm to the inner membrane by Sec, and then extracted from the membrane by FlaI-like ATPase, coupled with the FlaJ-like membrane proteins (Albers and Driessen, 2005; Albers et al, 2006). These homologous secretion superfamily ATPase and membrane protein pairs also occur in bacteria. Secretion ATPase family phylogenetic analysis shows that TadA ATPase needed for the bacterial Flp T4 pilus assembly (Planet et al, 2001) resembles the archaeal secretion ATPases. This tadA gene cluster includes the tadB and tadC genes, that resemble FlaJ (Peabody et al, 2003; Planet et al, 2003). Thus, these NTPase/membrane protein pairs appear evolutionally related across bacteria and archaea.
Although the secretion superfamily ATPase is the critical conserved component responsible for archaeal flagellae assembly, bacterial T2S and T4S, and T4 pilus assembly, as well as twitching motility and biofilm formation, a unified molecular understanding of the mechanism and function of this ATPase superfamily resulting from defined conformation and assembly states has been elusive. At least some secretion superfamily NTPase family proteins assemble into hexameric rings, as seen by electron microscopy of T4S ATPase VirB11 (Krause et al, 2000) and of BfpD ATPase, which assembles the bundle-forming T4 pilin (Crowther et al, 2005). The crystal structure of the HP0525 VirB11 ATPase from Helicobactor pylori also revealed hexameric rings (Yeo et al, 2000; Savvides et al, 2003), and that of EpsE from Vibrio cholerae has a related six-fold helical assembly (Robien et al, 2003). As hexameric association can precede ATP hydrolysis, a key functional role is implied for ATP binding to GspE (Shiue et al, 2006). However, it has been unclear if other secretion superfamily NTPase proteins also form functionally-important rings, and furthermore, how their individual domain movements and overall quarternary assembly are coupled to ATP binding, hydrolysis, and biological function. This lack of unified mechanistic understanding reflects the fact that no hexameric ring structures in distinct functional conformations have been available for any secretion ATPase that functions by a T2S-like mechanism.
To help elucidate the secretion superfamily ATPase structure, conformational state, and dynamic assembly and their functional implications, we solved the crystal structure of the Archaeoglobus fulgidus GspE (afGspE) secretion ATPase with and without nucleotide and furthermore examined structures of the ADP, ATPγS, ADP plus vanadate (ADP-Vi), and AMP-PNP-bound states in solution by small angle X-ray scattering (SAXS). The crystal and solution structures reveal distinct open and closed subunit conformations and a new hexameric assembly. The resulting alternating conformational states and hexameric assembly have specific and general implications for domain movement and molecular rotation mechanisms upon ATP hydrolysis. Taken together, the results support a unified molecular push–pull model, whereby ATP hydrolysis-coupled rotation and domain translocation may be transmitted to distinct partner proteins as a universal molecular mechanism for assembling microbial filamentous pilin, pseudopilin, and archaeal flagilin subunits, and for promoting secretion by the microbial T2S and T4S systems.
Results and discussion
GspE protein from A. fulgidus
To identify and purify a prototypic archaeal secretion ATPase, we searched the A. fulgidus genome and found four potential secretion ATPase genes, afgspE1–4. All translated AfGspE1–4 protein sequences are homologous to FlaI from Methanococcus voltae and Halobacterium salinarum, with sequence identities of ∼35–41% and similarities of ∼51–58% within coding sequences of similar length (see Figure 2), and belong to the archaeal secretion ATPase family (Supplementary Figure 1). As afGspE4 belongs to the fla gene cluster, it is classified as FlaI. In afgspE2, three genes are immediately followed by multi-transmembrane proteins, with marginal similarity to FlaJ/TadB/TadC. Recent analyses determined that S. solfataricus, Natronomonas pharaonis, and H. salinuram genomes also harbor these secretion NTPase and membrane protein couples (Albers and Driessen, 2005; Falb et al, 2005). Although putative secreted protein genes near the secretion NTPase gene were reported in S. solfataricus, we could not identify any candidate genes near afgspE1–3 genes (Albers and Driessen, 2005). The recombinant expression of the S. solfataricus secretion NTPases in E. coli was unsuccessful owing to inclusion body formation (Albers and Driessen, 2005). However, we successfully expressed and purified the afGspE2 ATPase, which provides a prototypic archaeal secretion superfamily ATPase for structure–function characterizations. As afGspE1–4 are highly homologous to each other, we designate afGspE2 as afGspE in the following sections.
Figure 2.
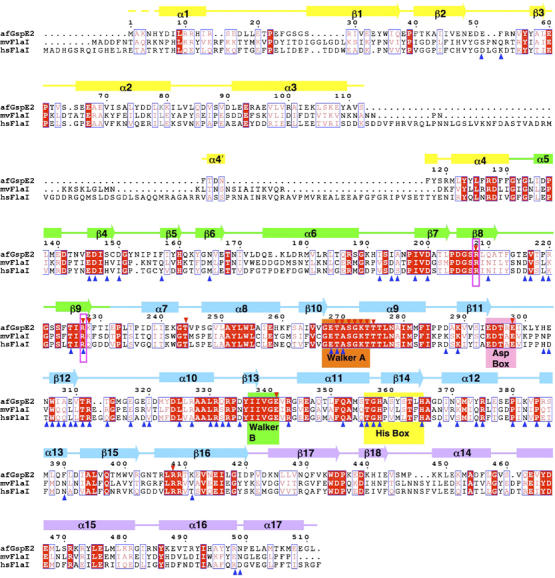
Sequence conservation, secondary structure and residue function. AfGspE sequence aligned with H. salinuram FlaI, and M. voltae FlaI reveals identical (white letters in red box) and homologous residues (red letters in blue box) with secondary structure helices (bars) and strands (arrows) colored as in Figure 1B. Conserved Walker motifs A (orange) and B (green) along with Asp (pink) and His (yellow) box motifs are labeled in boxes. Dual arginine fingers (Arg208 and 227) are highlighted by magenta boxes. Also shown as largely conserved are the residues binding AMP-PNP (red reverse triangles) and in subunit-subunit interactions (blue triangles).
Subunit structure and conformations
To reveal the atomic details for subunit structure and assembly, we determined the X-ray crystal structure of the afGspE ATPase. Crystals of selenomethionine (Se-Met)-substituted afGspE were grown in the presence of 10 mM AMP-PNP, a non-hydrolyzable ATP analog, and 10 mM Mg2+. Interpretable electron density maps generated by multi-wavelength anomalous dispersive phasing and subsequent solvent flipping (Figure 1A) allowed us to build the final models to the electron density for two molecules in the asymmetric unit. Both models are full-length, missing only four residues at the N terminus and a few disordered residues in the flexible regions (Figure 1). The resulting 2.95 Å resolution refined structure has R and Rfree factors of 22.6 and 23.3%, respectively (Supplementary Table I).
Figure 1.
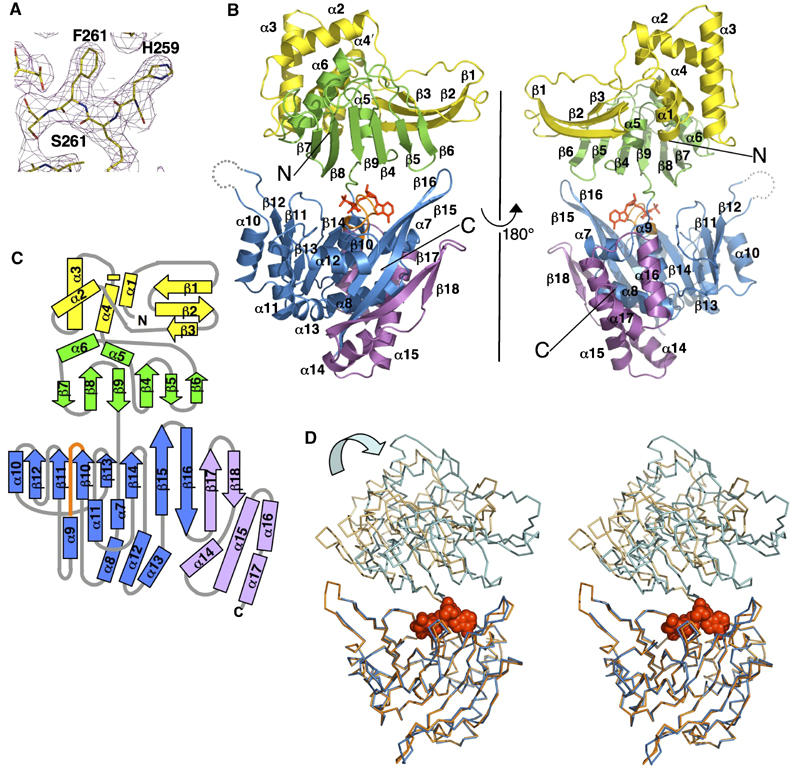
AfGspE subunit structure. (A) Experimental electron density (contoured at 1σ) overlaid with the final refined model. (B) AfGspE fold as ribbons (molecule B) with subdomains N1 (yellow), N2 (green), C1 (blue), and C2 (magenta) plus bound AMP-PNP (red sticks), and P-loop (orange, center). (C) Topology schematic, (D) Cα stereo diagram of two superimposed afGspE subunit structures with bound AMP-PNP (red, CPK spheres). The open form (molecule A, orange) needs NTD rotations to yield the closed form (molecule B, blue).
Each afGspE subunit has two overall domains: the N-terminal domain (NTD) and the C-terminal domain (CTD) (Figure 1B and C). The NTD is divided into two subdomains. The N1 subdomain consists of a 4-α-helix bundle (α1–4) and 3-stranded β-sheet (β1–3). Compared with other regions, the N1 electron density was less ordered, suggesting flexibility; in particular, molecule A N1 residues 105–113 were missing in the electron density maps and side chains for the α1–β2 loop and the α2–α3 loop in both molecules A and B also had poor electron density. These weak-electron density regions imply flexibility within N1, which may be stabilized through disorder–order transitions induced by binding to partner proteins. The evidently more ordered and rigid N2 subdomain is comprised of six antiparallel β-strands (β4–9), forming a flat face against the CTD, and two helices (α5–6) located on the opposite side (Figure 1B).
CTD also has two subdomains: C1 and C2 (Figure 1B and C). C1 is functionally critical with the Walker A and B motifs plus Asp box and His box motifs (Figures 1 and 2). C1 consists of a central β-strand core (β10–16) flanked on both sides by a total of seven α-helices (α7–13), with a topology similar to Walker type ATPases (Figure 1C), such as the RecA-like ATPases, including Rad51, F1-ATPase, the Holliday junction branch migration motor RuvB AAA+ ATPase, and the ABC ATPases, according to a DALI database search (Story and Steitz, 1992; Holm and Sander, 1993; Abrahams et al, 1994; Hopfner et al, 2000; Putnam et al, 2001; Shin et al, 2003). The molecule B β12–α10 loop was missing, although that in molecule A was well observed. One feature of afGspE C1 is that the two β-strands, β15–16, are extended from the central β-strand core toward the NTD (Figure 1B and C). C2 is composed of two β-strands (β17–18) and the four α-helices (α14–17). Significantly, the AMP-PNP nucleotide bound by Walker A motif in C1, lies in the interface between the NTD N2 and CTD C1 (Figure 1B–D), which contain the most conserved sequence blocks (Figure 2).
Significantly, the two afGspE subunits within the asymmetric unit have surprisingly distinct conformations, involving a large rigid body movement of the NTDs about the bound nucleotide (Figure 1D). Molecule A reveals the open form (Figure 1D), in which NTD (light orange) twists and splays out from CTD (orange), while molecule B reveals the closed form (blue), in which NTD (light blue) closes against the CTD (blue). The range of the rotation between the open–closed forms is ∼16 Å, generating an overall ∼10-Å N1 shift. Conformational changes between the NTD and CTD also occur in HP0525 (Savvides et al, 2003). The Apo-HP0525 structure, which has six molecules with different conformations in the asymmetric unit, showed that the NTD is capable of 2° to 15° rotations. The open form of afGspE resembles the most open form of the apo-HP0525 (molecule F), whereas the afGspE's closed form resembles the apo-HP0525 closed form (molecule A). Also, the structure of a truncated GspE ATPase, V. cholerae EpsE, (Robien et al, 2003) has a single molecule in the asymmetric unit that adopts an ‘half-open' form, compared with the afGspE subunit structures. These results suggest the afGspE open to closed conformational changes and range of movement is a conserved functional feature for the secretion ATPase superfamily.
Secretion ATPase hexamer structure
Two afGspE subunits in the asymmetric unit form a hexameric ring structure via a crystallographic three-fold symmetry axes. The external ring diameter is ∼135 Å, with a height of ∼75 Å (Figure 3A). The inner diameter is ∼35 Å between the NTDs, which narrows to ∼25 Å at bottom side between the CTDs (Figure 3A). Similar appearing ring structures of afGspE in the presence of AMP-PNP were also observed by electron microscopy, with somewhat smaller diameters of about 100 Å, consistent with some dehydration (Figure 3B). Electron density for the upper side of the ring is sparse, as it is composed of the N1 helical region (Figure 3A). The core of the ring is composed of the N2 and C1 subdomains, which line the ∼50 Å height of the cylindrical core hole (Figure 3A). The bottom of the cylindrical hole is composed of C2 β17–18. The outside of the ring is surrounded by N1 β-strands and C2 α-helices. At the top of the afGspE hexameric ring, N1 changes promote distinct open and closed subunit conformations because the N1 α-helices show large nucleotide-dependent conformational changes relative to the ring's core (Figure 3A).
Figure 3.
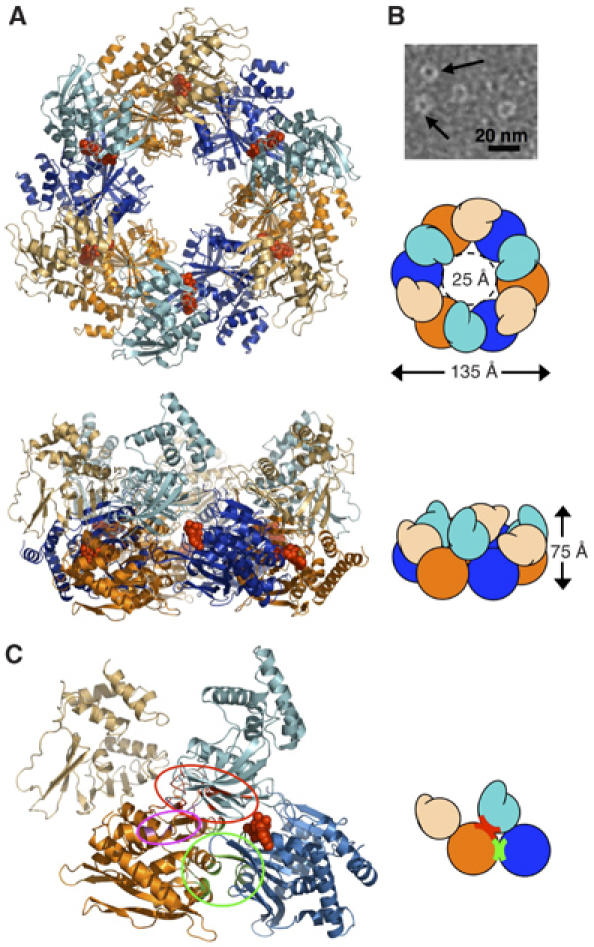
Hexameric ring structure, assembly, AMP-PNP binding, alternating subunit conformations, and subunit interactions. (A) AfGspE hexamer assembly and fold shown as ribbons and as schematic shapes viewed from top and side. Bound AMP-PNP (red, CPK spheres) in closed form molecules (blue, light blue NTD) and alternate open form molecules (orange, light orange NTD). (B) Electron micrograph of negatively stained AfGspE proteins with AMP-PNP. Ring structures are indicated by black arrows. (C) Subunit-subunit interactions with domain colors as in A, and encircled residues for N2:C1 (red), for C1:C1 (green), and N1:C2 (magenta) interactions.
Subunit–subunit interactions
The afGspE hexamer has three distinct subunit interfaces (Figure 3C). The dominant inter-subunit interaction is between the N2 of one molecule and the C1 of the adjacent molecule (Figure 3C, red elipse). Six N2 β-strands (β4–9) and their associated loops form a large interface with the C1 β12-loop-α10 and the N-terminal end of β11. Analogous interactions are formed within the N:C domain interface within the HP0525 ring and the N2:C1 domain interface of the EpsE helical assembly (Yeo et al, 2000; Robien et al, 2003). The ∼2000-Å2 buried surface of the afGspE major N2:C1 interface resembles the ∼1800-Å2 EpsE N2:C1 interface (Robien et al, 2003). Also FlaI family conserved residue pairs, such as Thr225:Lys289, Asp200:Arg331, and Glu216:Arg312, contribute key salt bridges and hydrogen bonds for the N2:C1 interface (Figure 2).
A second major but variable interface occurs between adjacent C1 domains and is conserved within the HP0525 and EpsE structures (Figure 3C, green circle). The C1:C1 interface buried surface area of ∼900 Å2 is half the size of the N2:C1 interface, but the conserved histidine residues (His 358 and 365) in His box are involved in C1:C1 interactions. Also, although the interacting residues pairs are consistent between both N2(molecule A):C1(molecule B) and N2(molecule B): C1(molecule A) pairs, the C1:C1 residue interactions are less conserved between the two different sets of intermolecular interactions. This, together with the smaller interface, suggests that the C1:C1 interactions alter when converting between the open and closed subunit forms. A third small interface forms between extended loop from the N1 domain β2 and the end of α16 from the C2 domain (Figure 3C, magenta elipse). This is the first observation of this interaction, as the N1 domain is truncated in the EpsE crystal structure and HP0525 lacks an N1 domain (Yeo et al, 2000; Robien et al, 2003).
The nucleotide binding site
Bound AMP-PNP has clear electron density in both molecules' active site; however, Mg2+ density was only observed in the closed form (molecule B) (Figure 4A and B). In molecule A, AMP-PNP is bound within the CTD by Walker A and P-loop residues (Figure 4A). The AMP-PNP triphosphates are hydrogen bonded by P-loop backbone nitrogens (Ala270 to Thr274) and Thr275, and Lys273 Nζ interacts with the β-phosphate. Side chains of Thr274 and 275 are also involved in interactions with the phosphates. Also, the conserved Glu298 and 341 interact with the γ-phosphate, although they are suitable for Mg2+ binding (see below). Notably, Glu298 is the conserved acidic residue in Asp box and Glu341 is the Walker motif B residue. The AMP-PNP adenine ring is stabilized by base stacking-like interactions with Arg408, which also interacts with ribose O4. The adenine N6 amide group interacts with Thr244 Oγ1, suggesting a preference for binding ATP rather than GTP. There are no interactions between NTD and AMP-PNP in molecule A.
Figure 4.
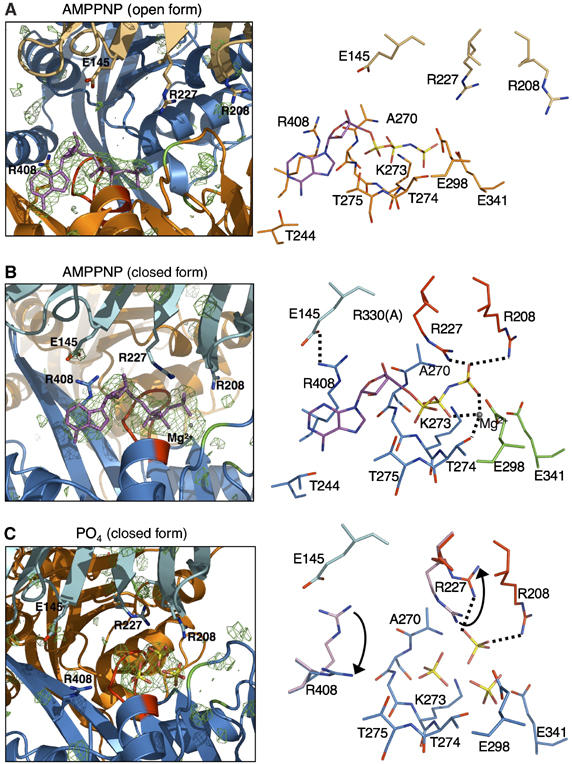
The nucleotide binding site. Left: Fo–Fc difference Fourier map (contoured at 3σ) was calculated with the protein residues and superimposed onto the refined structure with AMP-PNP (purple), P-loop residues (red), Glu298 and 341 (green) in Walker B motif. The active site residues are shown in sticks. Right: nucleotide binding site showing stick models (blue nitrogens and red oxygens) and Mg2+ (gray sphere). (A) The nucleotide binding site in molecule A (open form). (B) The nucleotide binding site in molecule B (closed form). Dual arginine fingers (Arg208 and 227, red) interact with AMP-PNP γ-phosphate (dotted lines) and Glu298/341 face Mg2+ ion. (C) Phosphate binding site in closed form (molecule B). Arg227 and 408 conformational changes from the AMP-PNP bound form (light magenta) to phosphate bound form are indicated by arrows.
In contrast, molecule B NTD makes additional contacts with AMP-PNP (Figure 4B). Dual arginine fingers formed by Arg208 and 227 interact with the AMP-PNP γ-phosphate. Also, the Thr230 Oγ1 and Lys228 main-chain amide contacts AMP-PNP 2′-O (not shown in Figure 4B). Dual arginine fingers are conserved in the FlaI family and most other secretion ATPase superfamily proteins (Figure 2). The bound nucleotide interactions with the dual arginine fingers also occur in the HP0525 structure, in which Arg103 and 113 interact with γ-phosphate in the ATPγS complex (Savvides et al, 2003). In the EpsE structure, the arginine fingers (Arg 210 and 227) do not contact the AMP-PNP phosphates as it is in the half-open form. Importantly, these dual arginine fingers may be distinguished as arginine clamps from the single arginine finger in other hexameric ATPases: all these secretion ATPase's dual arginine fingers:phosphate interactions occur within the same subunit, and not from the adjacent molecule, as do the distinct single arginine fingers in other hexameric ATPases, such as the F1-ATPase (Abrahams et al, 1994), the T7 gene 4 helicase (Singleton et al, 2000), and RuvB AAA+ ATPase (Putnam et al, 2001). No nucleotide contacts from adjacent molecules occur in either afGspE or HP0525.
The NTD residues interacting with AMP-PNP also form major contacts with CTD in the closed form. There are only a few NTD:CTD interactions except for these nucleotide-mediated interactions, suggesting that the nucleotide absence destabilizes the closed conformation. In the adenine ring moiety, Arg408 makes extensive adenine ring contacts and also can bind Glu145 from NTD ∼3.3 Å away (Figure 4B). In addition, Arg406 interacts with the NTD residues, Glu145, Asp146, and Tyr167 (not shown). Glu145 and Asp146 are conserved in the FlaI family (Figure 2).
Molecule B has clear electron density for Mg2+, liganded by the Walker A motif Thr 274 Oγ1 and two AMP-PNP phosphates. The side chains of Glu 298 (Asp box) and Glu 341 (Walker motif B) face the Mg2+ at distances of 3.6 and 3.8 Å, respectively, which are long for direct metal ion coordination. These Glu298/341 residues are equivalent to Glu209/248 in HP0525, and Glu296/334 in EpsE and analogous to RecA Glu96/Asp144 (Story and Steitz, 1992; Robien et al, 2003; Savvides et al, 2003). Taken together, these data suggest that afGspE Glu341 is a water activator for the in-line nucleophilic attack of the nucleotide γ-phosphate, and Glu298 stabilizes the hydrolytic intermediate by binding the water molecule liganded to Mg2+ ion.
Phosphate-bound afGspE hexamer structure
Surprisingly, apo afGspE crystallized in the same conditions as the AMP-PNP bound form, except without nucleotide and Mg2+ ion, still forming a hexameric ring with alternating subunit conformations. Both 2Fo–Fc and Fo–Fc difference Fourier maps showed an unexpected strong positive peak within the P-loop residues, which we interpret as phosphate ion scavenged from the crystallization conditions (Figure 4C). In the open form (molecule A), a single phosphate ion fits the density peak, whereas in the closed form (molecule B), the larger electron density peak fits three phosphate ions (see Supplementary data). As described below, the phosphate in molecule B interacts with the arginine fingers, which should stabilize the closed conformation and form the same hexameric structure as the AMP-PNP-bound form.
The overall structure of afGspE:phosphate matches afGspE:AMP-PNP, but Arg227 and Arg408 showed significant conformational changes. In the AMP-PNP-bound structure, Arg227 protrudes toward the γ-phosphate in the closed form (Figure 4B), whereas in the open form, it protrudes in the opposite direction (Figure 4A) to make contact with the main-chain Arg331 O (not shown in Figure 4A) in the adjacent molecule. In the afGspE:phosphate structure (closed form), the Arg227 side chain moves into a middle position between those of the open and closed forms in the AMP-PNP-bound structure, as it contacts both the bound phosphate and Arg331 from the adjacent molecule (Figure 4C). This suggests that Arg227 has a sufficiently flexible conformation to be a sensor for different phosphate states of the bound nucleotide. Phosphate absence may promote the open form Arg227 conformation, which then destabilizes the closed form NTD:CTD interactions. In the phosphate-bound structure, Arg408 moves to the adenine ring position in the AMP-PNP-bound structure, which breaks the Glu145 interaction seen in the AMP-PNP-bound form. Thus, nucleotide absence destabilizes closed conformations by breaking NTD:CTD interactions between phosphates and arginine fingers, and between Glu145 and Arg408.
ATP and Mg2+ binding and the catalytically active conformation
The remarkable differences in the nucleotide binding site structures between the open and closed forms led us to hypothesize that they have different ATP binding affinities. Indeed, the HP0525:ATPγS hexamer structure obtained by soaking ATPγS into HP0525:sulfate crystals revealed two three-fold symmetric related alternative occupancies of ATPγS, although all subunits have the same closed form (Savvides et al, 2003). To estimate afGspE affinity for AMP-PNP, we performed a crystallographic titration experiment for afGspE structures co-crystallized with 0.2, 0.5, 1 mM AMP-PNP and 10 mM Mg2+ (Supplementary Figure 2). Bound AMP-PNP was similar in the open and closed forms at about 0.5 mM AMP-PNP, but at 0.2 mM AMP-PNP (∼1:1 protein:nucleotide ratio), we measured a partial occupancy for nucleotide binding in the open form, whereas the closed form showed full occupancy. Thus, open and closed afGspE have surprisingly similar nucleotide occupancy at ∼mM levels of ATP.
Notably, afGspE structures with different AMP-PNP concentrations consistently showed that Mg2+ was bound only in the closed form. Even for Mg2+ concentration 10 times or greater than that of AMP-PNP in these structures, the bound nucleotide electron density in the open form shows only slight density at the closed form's γ-phosphate site. As the fitting of AMP-PNP plus Mg2+ model showed strong negative density at this site but positive density at the Mg2+ site in Fo–Fc difference Fourier map, we concluded that the open form cannot bind Mg2+. Indeed, the EpsE:AMP-PNP structure, which has a half-open conformation, also does not bind to Mg2+ even in the presence of 10 mM Mg2+ (Robien et al, 2003). Importantly, the selective Mg2+ binding to the closed form strongly suggests that only the closed form is catalytically active. This is analogous to the F1-ATPase, in which the most closed form, βDP, is considered the catalytically active subunit. Octahedral coordination of Mg2+ with protein residues and water molecules was optimal in βDP in the original F1-ATPase structure, suggesting it is the catalytic conformation (Abrahams et al, 1994; Senior et al, 2000). F1-ATPase structure inhibited by ADP and beryllium fluoride also revealed that only the βDP subunit has a water molecule suitable for nucleophilic attack of the beryllium atomic position, which mimics the ATP γ-phosphate (Kagawa et al, 2004).
Why does only the closed form bind Mg2+ ion? These afGspE structures suggest that the dual arginine fingers play a key role in sequestering Mg2+ ion. In the open form, γ-phosphate occupies the Mg2+ binding site. The interaction between the arginine fingers and γ-phosphate in the closed form promotes a γ-phosphate conformational change, allowing Mg2+ to bind in the active site. In addition, the arginine finger interactions with ATP phosphates may be essential for ATP hydrolysis. In the F1-ATPase, the arginine finger αArg373 may help position the γ-phosphate for hydrolysis (Kagawa et al, 2004). Indeed, mutation of each of the dual arginine fingers in HP0525 drastically reduced the ATP hydrolysis activity (Savvides et al, 2003). These observations suggest that afGspE co-crystallized with AMP-PNP, and Mg2+ represents the catalytically active form resulting from the dual arginines acting as clamps to close down the subunit NTD:CTD interactions around the bound ATP mimic.
Structural and functional insights from secretion ATPase comparisons
To test if afGspE shares conserved regions with other functionally related ATPases, we did a afGspE homology search with the DALI server that revealed very similar structures for the secretion ATPase subunits. HP0525 gave the highest Z-score (Z=23.1), followed by EpsE (Z=13.1) (Holm and Sander, 1993). HP0525 is smaller (330 residues) than afGspE (Figure 5A). Its N-terminal end has a long helix instead of the N1 domain, and lacks C2 domains. The full-length EpsE sequence is roughly of the same length as afGspE, and is divided into five subdomains, N1, N2, C1, C2, and CM domains. The CM domain has the added Zn2+ ion binding tetra cysteine motif, which only occurs in the GspE/PilB family (Robien et al, 2003) (Figure 5A).
Figure 5.
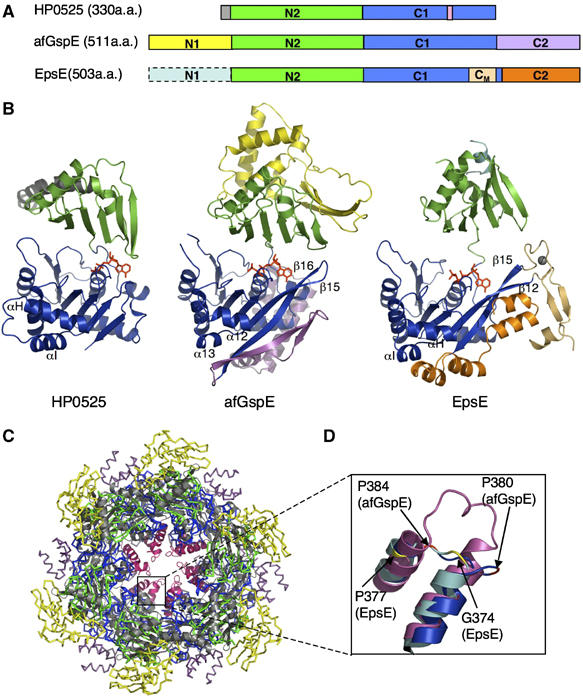
Secretion ATPase structural comparisons, subdomains, and a conserved functional core. (A) Schematic view of the three secretion ATPase structures. (B) Overall structures for HP0525 (left), afGspE (center), and EpsE (right) shown as ribbons with bound nucleotide (red sticks). Similar N2 (green) and C1 (blue) subdomains form the conserved core flanked by variable HP0525 N-terminal helix (gray), afGspE N1 (yellow) and C2 (magenta), and EpsE Zn2+ (gray sphere) region of the CM subdomain (light orange) and C2 (orange). (C) Structural comparison between afGspE (ribbons colored as in A) and HP0525 (gray) hexamers with pink HP0525 αH-I insertions. (D) Close up comparison of the closed site structure for HP0525 αH-I insertions (pink), afGspE α12–13 (blue), and EpsE (cyan). Colored residues kink the loop between helices in afGspE (red) and EpsE (yellow).
Thus, comparative analyses with our hexameric afGspE structures show that the N2 and C1 domains (Figure 5B, green and blue respectively) of the three secretion ATPases share a critical core of similarities, consistent with their blocks of sequence conservation (Figure 2). The N2 domain is commonly composed of six strands with two helices on one side. The afGspE N2 domain superimposes onto the HP0525 NTD with a root mean square deviation (r.m.s.d.) of 1.8 Å2 for 73 Cα atoms, and onto EpsE N2 with an r.m.s.d. of 1.5 Å2 for 54 Cα atoms. The first α-helix in HP0525 (Figure 5B, gray) was not fitted with N1 domain in afGspE (Figure 5B, yellow). The C1 domains of three secretion ATPases share the topology of seven β-strands and flanking seven helices. The afGspE C1 domain superimposes onto HP0525 CTD with an r.m.s.d.=1.3 Å2 for 152 Cα atoms, and onto that of EpsE with an r.m.s.d.=1.5 Å2 for 135 Cα atoms. Interestingly, EpsE β12/15 extend from the β-strand core, similar to afGspE β15–16. In afGspE, the extended region of β15–16 may be functionally important, as the extended region acts in the NTD:CTD interaction including Arg408 and 406. The Zn2+ ion (Figure 5B, gray ball) binding domain of the EpsE CM domain (Figure 5B, yellow) is absent in afGspE. The afGspE and EpsE C2 subdomains are different in both topology and relative positions in the overall structure (Figure 5B).
The EpsE N1 subdomain (N1-EpsE) forms a tight complex with the cytoplasmic domain of the membrane protein, EpsL (cyto-EpsL) (Sandkvist et al, 1995; Abendroth et al, 2005). The structural variable N1 subdomains of EpsE and afGspE share flexibility. N1-EpsE is evidently more stable in complex formation with cyto-EpsL, as the crystallizations of N1 truncated EpsE and of N1-EpsE bound to cyto-EpsL were successful, whereas that of full-length EpsE was unsuccessful (Abendroth et al, 2005). We therefore hypothesize that the disordered part of afGspE N1 undergoes a disorder to order transition in the context of partner proteins. If secretion NTPases N1 subdomains are generally responsible for binding to partner proteins, then the N1 domain structural variability may match the individual partner protein structures. Thus, these comparisons suggest that the secretion superfamily ATPases have a structurally conserved core of N2 and C1 subdomains, but exhibit functionally relevant diversity in the peripheral N1 and C2 domains. As the core N2–C1 domains are responsible for both nucleotide binding and subunit–subunit interactions, the basic functional mechanisms resulting from ATP hydrolysis-driven conformational change are probably conducted by this N2–C1 subdomain core conserved in all secretion ATPases, with the N1 and C2 subdomains diversities being critical for transmitting individual biological functions.
The afGspE hexamer is larger than the HP0525 ring (Yeo et al, 2000; Savvides et al, 2003), owing to the presence of the yellow N1 and magenta C2 domains (Figure 5C). The hexamer central holes also have different shapes. AfGspE has a central opening that tapers in width from top to bottom. HP0525 is open at the top and closed at the bottom by αH, αI, and the long loop between them (Figure 5C, pink). In afGspE, the equivalent α-helices (α12 and α13) are shorter and the loop joining these two helices is kinked by two conserved proline residues, Pro380 and 384 (Figures 2 and 5D). The same topology occurs in EpsE, in which the loop joining αH and αI is kinked by conserved Gly374 and Pro377 (Figure 5D). The EpsE six-fold helical assembly in crystals forms a central channel (Robien et al, 2003), of 37 Å diameter similar to the afGspE hexamer. This, together with the absence of the insertion loop for closed site, suggests that EpsE may form afGspE like hexamers in solution with the central cylindrical hole and without a closed end. The presence or the absence of the closed end may depend on whether the secretion ATPase functions as protein translocator or a filament assembly motor. In T2S and archaeal flagellar systems, the secretion ATPases are the filament assembly motor. On the other hand, HP0525 is proposed as a protein translocator, which may fold the secreted protein or machinery component in its cavity, and transport it to the inner membrane (Yeo et al, 2000). The absence of the closed end in the afGspE hexamer (and probable in EpsE hexamer) may facilitate a more dynamic conformational change, allowing them to serve as the powerful assembly motor in T2S-like machinery. Furthermore, the closed site in HP0525 hexamer (αH-loop-αI) may act as a ‘lid' to interact with or export the protein in one direction.
Dynamic hexameric assembly in solution
The open and closed afGspE subunit structures implicated these states as functionally important conformations for ATP binding and hydrolysis. The absence of the closed end of afGspE hexamer is also consistent with its dynamic conformational change. To define conformational changes upon ATP binding and post-hydrolysis in solution, we did SAXS analyses on afGspE hexamer in AMP-PNP-bound and ADP-bound states by adding 2 mM of each nucleotide plus 10 mM Mg2+. The scattering profiles of the two states altered remarkably, showing significant conformational change (Figure 6B, green and blue, for AMP-PNP- and ADP-bound states, respectively). The calculated gyration radii (Rg) and the particle intra-maximum distance (Dmax) are 48.5 and 151 Å for AMP-PNP- and 51.0 and 148 Å for ADP-bound states, respectively. Overall, these values resemble those from the crystal structure (47.3 and 146 Å) and indicate similar size hexameric rings in solution. The Rg decreases with AMP-PNP- compared with the ADP-bound form, as was also seen for the archaeal F1-like ATPase A1-ATPase suggesting related conformational change (Coskun et al, 2002). To further investigate the solution structure, we assessed SAXS data in comparison with the crystal structure plus calculated all-open and all-closed hexamer models (Figure 6A). The alignment of afGspE's closed form structure onto the HP0525 structure (Yeo et al, 2000) gave a geometrically reasonable all closed hexamer model without bad contacts. An all-open hexamer model, generated by replacement of closed form afGspE subunits with open form subunits, also gave a sterochemically reasonable model.
Figure 6.
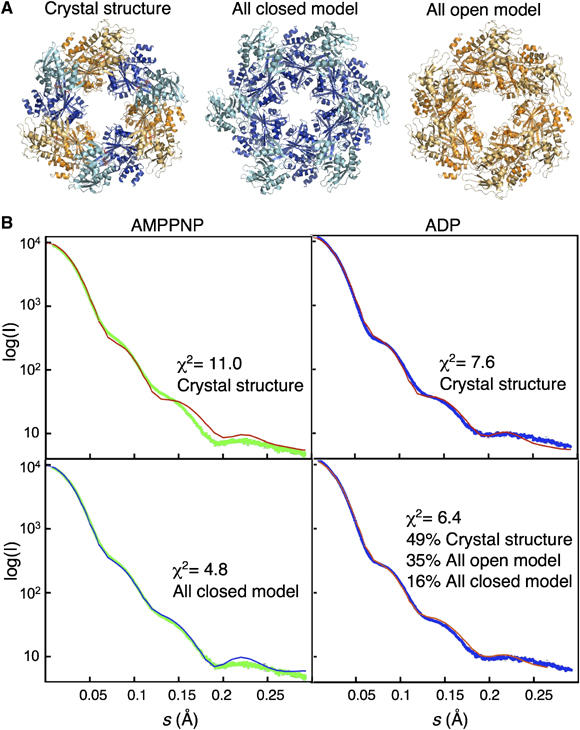
AfGspE hexamer structures in solution by SAXS. (A) Hexameric crystal structure (left), all-closed hexamer model (center), and all-open hexamer model (right). (B) X-ray scattering of the AMP-PNP bound (green) and ADP bound (blue) hexamers compared to profiles calculated from the crystal structure (red line) (top, left and right), the all-closed model (bottom left, blue line) and the mixture of crystal structure, all open hexamer, and all closed hexamer models (bottom right, orange line). The scattering data includes the error bars (gray), however, as the error is about ∼4% even in the highest angle region, it is almost invisible.
The AMP-PNP-afGspE scattering curve shows differences from that calculated from the crystal structure (Figure 6B, top left). Surprisingly, the AMP-PNP-bound state showed an excellent and complete overall fit to the calculated scattering curve of the all-closed hexamer model (Figure 6B, bottom left), indicating AMP-PNP-afGspE can form the symmetrical all-closed hexamer in solution. We further tested if a mixture of multiple conformations is possible, by fitting of the scattering curve with those calculated from three models: the crystal structure, all-closed hexamer, and all-open hexamer models. This analysis resulted a 100% of all-closed hexamer, ruling out the other two conformations. The closed conformation in the AMP-PNP state is consistent with the observation from the crystal structure that the interaction between the γ-phosphate of AMP-PNP and arginine fingers from NTD can ‘lock' the closed form. SAXS curves of afGspE with ATPγS or ADP-Vi, also show the AMP-PNP type scattering curves and excellent fit to the all-closed model with χ2 of ∼5 (Supplementary Figure 3A), suggesting the γ-phosphate can lock an all-closed hexamer.
ADP-afGspE exhibits a scattering profile resembling that calculated from the crystal structure with χ2 of ∼7.6 (Figure 6B top right). Thorough analysis suggests three models in solution consisting of 49% of the crystal structure, 35% of the all-open hexamer, and only 16% of the all closed hexamer, with the slightly improved χ2 of ∼6.4 (Figure 6B bottom right). Yet, even the calculated curve from the three states is not a perfect fit to the experimental ADP data suggesting that the ADP state is more flexible than our models, or that they do not exactly model the ADP state. However, these results show that ADP, which lacks the ATP γ-phosphate, is unable to lock the closed form, but does relax afGspE to a flexible state including a mixture of open and closed forms. Surprisingly, SAXS data in the presence of ATP more closely resembles the ADP rather than the AMP-PNP SAXS results (Supplementary Figure 3C). Thus, SAXS data for the ATP state supports an ADP-type asymmetric hexamer with open-closed (catalytic and non-catalytic) subunits.
Nucleotide binding stoichiometry to afGspE
The all closed hexamer formed in the AMP-PNP state, as shown from SAXS analysis, suggests that all subunits bind to AMP-PNP and Mg2+. Our crystal structure shows that distinct open and closed conformations do not significantly alter nucleotide binding, so all subunits in the ADP-afGspE hexamer may have equal ability to bind nucleotide. We therefore investigated the binding stoichiometry of afGspE to nucleotide using fluorescence nucleotide analogs, TNP-ATP and TNP-ADP. TNP-nucleotide, which generally acts as a non-hydrolyzable nucleotide analog with the tighter binding affinity than natural nucleotide, has weak fluorescence in aqueous solution that is greatly enhanced in the hydrophobic environment provided by protein binding. As no hydrolysis activity for TNP-ATP by afGspE was observed (data not shown), the binding of afGspE to TNP-ATP reflects the AMP-PNP bound state. By increasing TNP-ATP concentration against afGspE, the fluorescence enhancement (ΔF) upon binding of TNP-ATP to afGspE linearly increased (Figure 7A, closed circle), indicating that all of the added TNP-ATP can tightly bind to afGspE. Also, the ΔF saturated, after TNP-ATP concentration reached that of afGspE showing that TNP-PNP binds to afGspE with a 1:1 binding stoichiometry. As expected, the mutant afGspE K273A for the conserved Walker A motif lysine drastically reduced its to TNP-ATP binding ability (open circle in Figure 7A).
Figure 7.
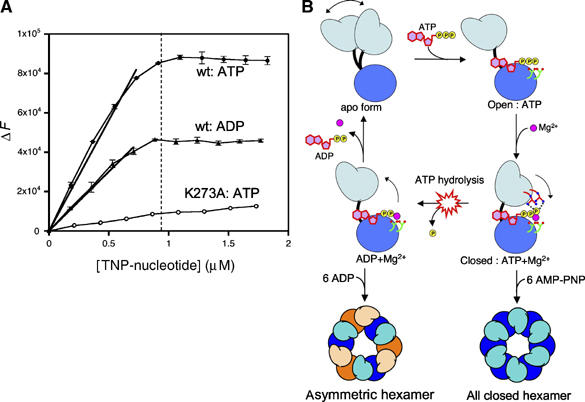
Nucleotide binding and the catalytic mechanism of afGspE. (A) The titration of fluorescent TNP-ATP (closed circles) and TNP-ADP (closed triangles) against the fixed afGspE concentration (0.94 μM, dotted line) is shown. The titration of TNP-ATP against the mutant afGspE K273A is shown by open circles. (B) Proposed catalytic cycle with experimentally defined open–closed conformational changes upon ATP binding and hydrolysis. In the flexible apo form (top left), the NTD (light blue) and CTD (blue) are in equilibrium between open and closed conformations. The more solvent-accessible open conformation (top right) favors ATP (red nucleotide and yellow phosphates) binding. The interaction between the dual arginine clamps (red tubes and blue nitrogen atoms) and the ATP g-phosphate locks the catalytically active closed conformation (middle right), and the resulting g-phosphate shifts allow Mg2+ (magenta sphere) to interact with the two conserved Glu residues (green tubes, red oxygen atoms). The g-phosphate release after ATP hydrolysis unlocks the closed form and accelerates the conformational change to the open form (middle left), suitable for ADP release. The primarily rigid-body movement of the top domain shown here prompts our proposal of a universal piston-like push-pull mechanism for the secretion superfamily ATPases, suitable to drive diverse microbial secretion and assembly processes. The symmetrical closed hexamer, as induced by non-hydrolysable ATP analogs such as AMP-PNP, and the asymmetrical hexamer in the ADP-bound state are shown at the bottom.
We found the same 1:1 binding stoichiometry for TNP-ADP:afGspE binding (closed triangle in Figure 7A). The fluorescence enhancement upon TNP-ADP:afGspE binding is about half of that upon TNP-ATP:afGspE binding. A similar fluorescence enhancement profile for the binding of BfpD ATPase to TNP-nucleotides was also observed, with same 1:1 binding stoichiometry (Crowther et al, 2005). Since TNP is attached to the ribose in the TNP-nucleotide, there should be no significant difference in the fluorescence properties between TNP-ATP and TNP-ADP. Thus, the observed half fluorescence enhancement of the TNP-ADP–afGspE complex may indicate that the environment of TNP-ADP binding site in its complex with afGspE is less hydrophobic than that with TNP-ATP, consistent with the ADP form having the more open, solvent exposed conformation and the ATP form having the closed, solvent excluded conformation.
A unified mechanism for the secretion ATPase superfamily
Based on the afGspE crystal structures complexed with AMP-PNP or phosphate, and on solution structures with AMP-PNP and ADP by SAXS, we propose the following unified mechanism for ATP-mediated functional conformational change (Figure 7B). Subunits are in dynamic equilibrium between open and closed conformations. The open form facilitates active site access by ATP and can bind to it. The binding to ATP γ-phosphate plus Mg2+ drives the open to closed transition by increasing NTD:CTD interactions, converting binding energy into conformation change. Mg2+ binding and thus ATP hydrolysis requires this closed form. After hydrolysis, ATP γ-phosphate release destabilizes the interaction with arginine finger Arg227 to trigger the closed to open form conformational change. The HP0525 crystal structure showed the all-closed form can occur with Arg130 (one of the dual arginine fingers) binding to ADP β-phosphate (Yeo et al, 2000). However, the afGspE SAXS analyses argue that ATP γ-phosphate plus Mg2+, but not ADP, locks or clamps the closed form, and that ADP allows, but does not lock the closed form, and is sufficiently unclamped to allow the transition to open form. Non-hydrolysable nucleotide analogues all give the symmetrical all closed hexamer by SAXS results, in which all subunits bind to nucleotide, as also supported from the fluorescence nucleotide binding experiment. Also the ADP bound state, which cannot hold the closed conformation, forms an asymmetric hexamer with the mixture of open-closed conformations, although all subunits can equally binds to nucleotide.
Three ATP hydrolysis mechanisms by hexameric ATPases have thus far been proposed: concerted, sequential, and probabilistic mechanisms (Martin et al, 2005). The combined crystallographic, EM, and SAXS results for afGspE suggest that ATP state forms the asymmetric hexamer similar to ADP-type with open-closed (catalytic and non-catalytic) subunits, supporting the sequential or probabilistic mechanism. Yet, the all closed symmetrical hexamer in AMP-PNP state is consistent with the concerted mechanism, it does not directly reflect the actural hydrolysis state. This may be happened due to the extensive afGspE hexamer flexibility, derived in part from its unique inter-subunit interactions. AfGspE and other secretion ATPases commonly have N2:C1 and C1:C1 interfaces, but no nucleotide dependent inter-subunit interactions. Yet, in other hexameric ATPases, represented by F1-ATPase or AAA+ ATPases, nucleotide bound in one subunit interacts with the arginine finger of the adjacent subunit. In contrast, the dual intra-subunit arginine clamps in secretion ATPases may provide an extensively flexible movement between NTD:CTD, that is less coupled to subunit interactions than has been found for other hexameric ATPases.
The afGspE structure reveals a large N1 movement from the open-closed conformational transitions promoted by binding ATP γ-phosphate and Mg2+ and involving motifs, features, and subdomains conserved across the entire secretion ATPase superfamily (Figures 1D, 2 and 3). In the closed form, N1 four helix bundles (α1–4) ‘stand up', while they ‘lie down' in the open form. AfGspE N1 may provide the interaction with partner proteins, as secretion ATPases evidently form multi-protein complexes with membrane partner proteins to transmit ATP hydrolysis for function in filament assembly and secretion. The existence of the conserved FlaJ/TadB/TadC related membrane proteins coupled with the archaeal secretion NTPases suggest they are partner proteins. Thus, the large piston-like N1 shift upon open-closed conformational changes may be transmitted by the bound partner protein movement (Figure 7B), as proposed for EPEC bundle forming pilus (Crowther et al, 2005) and Neisseria T4 pilus assembly (Craig et al, 2006). For the bundle forming pilus assembly, BfpD ATPase may shift its binding site on membrane partner protein BfpE upon ATP hydrolysis to translate BfpE and thereby extract the pilin subunit hydrophobic sequence from the inner membrane into the growing pilus filament (Crowther et al, 2005). The cryo-EM structure of the T4 pilus indicates assembly required a 10.5 Å shift for each subunit (Craig et al, 2006), consistent with the 10 Å N1 shifts defined here. Although new biochemical and genetic experiments suggested by these structures will be important to evaluate function for the secretion NTPases, these afGspE enzyme assemblies appear to be prototypic systems for elucidating functional mechanisms of secretion ATPase superfamily motors.
Materials and methods
Cloning, expression, and purification of afGspE
Cloning, expression, and purification are described in Supplementary data.
Crystallization, crystallographic data collection, structure determination and refinement
Crystallization, data collection and refinement are described in Supplementary data.
Electron microscopy
The glow-discharged carbon-coated grids were floated onto 10 μl of 0.1–0.2 mg/ml afGspE with 10 mM AMP-PNP and 25 mM MgCl2 for 2 min. Grids were stained in 3% uranyl acetate and viewed on a Philips CM100 microscope at 100 kV with 52 000 × magnification (see Supplementary data).
Small angle X-ray scattering (SAXS)
SAXS data for afGspE hexamer were collected at the SIBYLS beamline at ALS using MAR 165 CCD area detector (165 mm diameter) (see Supplementary data).
Fluorescence nucleotide binding
Fluorescence nucleotide binding methods are described in Supplementary data.
Coordinates
The atomic coordinates of the afGspE with and without bound nucleotide are deposited in the Protein Data Bank with the accession numbers 2OAP and 2OAQ.
Figure preparation
See Supplementary data.
Supplementary Material
Supplementary Legends and Table I
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Acknowledgments
We thank the staffs of SSRL beamline 9-1, ALS beamline 5.0.2 and the ALS SIBYLS beamline 12.3.1 for diffraction facilities. We thank Chris Putnam, Lisa Craig, David Shin, and Scott Williams for discussions and Greg Hura for aiding SAXS data collection. This work was supported by NIH grant AI22160 (JAT) and by US. Department of Energy support of the SIBYLS beamline and SAXS characterizations of archaeal assemblies under Contract Number DE-AC02-05CH11231. AY was supported in part by a Skaggs Institute of Chemical Biology fellowship.
References
- Abendroth J, Murphy P, Sandkvist M, Bagdasarian M, Hol WG (2005) The X-ray structure of the type II secretion system complex formed by the N-terminal domain of EpsE and the cytoplasmic domain of EpsL of Vibrio cholerae. J Mol Biol 348: 845–855 [DOI] [PubMed] [Google Scholar]
- Abrahams JP, Leslie AG, Lutter R, Walker JE (1994) Structure at 2.8 Å resolution of F1-ATPase from bovine heart mitochondria. Nature 370: 621–628 [DOI] [PubMed] [Google Scholar]
- Albers SV, Driessen AJ (2005) Analysis of ATPases of putative secretion operons in the thermoacidophilic archaeon Sulfolobus solfataricus. Microbiology 151: 763–773 [DOI] [PubMed] [Google Scholar]
- Albers SV, Driessen AM (2002) Signal peptides of secreted proteins of the archaeon Sulfolobus solfataricus: a genomic survey. Arch Microbiol 177: 209–216 [DOI] [PubMed] [Google Scholar]
- Albers SV, Szabo Z, Driessen AJ (2003) Archaeal homolog of bacterial type IV prepilin signal peptidases with broad substrate specificity. J Bacteriol 185: 3918–3925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albers SV, Szabo Z, Driessen AJ (2006) Protein secretion in the Archaea: multiple paths towards a unique cell surface. Nat Rev Microbiol 4: 537–547 [DOI] [PubMed] [Google Scholar]
- Bardy SL, Jarrell KF (2003) Cleavage of preflagellins by an aspartic acid signal peptidase is essential for flagellation in the archaeon Methanococcus voltae. Mol Microbiol 50: 1339–1347 [DOI] [PubMed] [Google Scholar]
- Bardy SL, Ng SY, Jarrell KF (2003) Prokaryotic motility structures. Microbiology 149: 295–304 [DOI] [PubMed] [Google Scholar]
- Coskun U, Gruber G, Koch MH, Godovac-Zimmermann J, Lemker T, Muller V (2002) Cross-talk in the A1-ATPase from Methanosarcina mazei Go1 due to nucleotide binding. J Biol Chem 277: 17327–17333 [DOI] [PubMed] [Google Scholar]
- Craig L, Pique ME, Tainer JA (2004) Type IV pilus structure and bacterial pathogenicity. Nat Rev Microbiol 2: 363–378 [DOI] [PubMed] [Google Scholar]
- Craig L, Taylor RK, Pique ME, Adair BD, Arvai AS, Singh M, Lloyd SJ, Shin DS, Getzoff ED, Yeager M, Forest KT, Tainer JA (2003) Type IV pilin structure and assembly: X-ray and EM analyses of Vibrio cholerae toxin-coregulated pilus and Pseudomonas aeruginosa PAK pilin. Mol Cell 11: 1139–1150 [DOI] [PubMed] [Google Scholar]
- Craig L, Volkmann N, Arvai AS, Pique ME, Yeager M, Egelman EH, Tainer JA (2006) Type IV pilus structure by cryo-electron microscopy and crystallography: implications for pilus assembly and functions. Mol Cell 23: 651–662 [DOI] [PubMed] [Google Scholar]
- Crowther LJ, Yamagata A, Craig L, Tainer JA, Donnenberg MS (2005) The ATPase activity of BfpD is greatly enhanced by zinc and allosteric interactions with other Bfp proteins. J Biol Chem 280: 24839–24848 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Falb M, Pfeiffer F, Palm P, Rodewald K, Hickmann V, Tittor J, Oesterhelt D (2005) Living with two extremes: conclusions from the genome sequence of Natronomonas pharaonis. Genome Res 15: 1336–1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L, Sander C (1993) Protein structure comparison by alignment of distance matrices. J Mol Biol 233: 123–138 [DOI] [PubMed] [Google Scholar]
- Hopfner KP, Karcher A, Shin DS, Craig L, Arthur LM, Carney JP, Tainer JA (2000) Structural biology of Rad50 ATPase: ATP-driven conformational control in DNA double-strand break repair and the ABC-ATPase superfamily. Cell 101: 789–800 [DOI] [PubMed] [Google Scholar]
- Kagawa R, Montgomery MG, Braig K, Leslie AG, Walker JE (2004) The structure of bovine F1-ATPase inhibited by ADP and beryllium fluoride. EMBO J 23: 2734–2744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler R, Schafer K, Muller S, Vignon G, Diederichs K, Philippsen A, Ringler P, Pugsley AP, Engel A, Welte W (2004) Structure and assembly of the pseudopilin PulG. Mol Microbiol 54: 647–664 [DOI] [PubMed] [Google Scholar]
- Krause S, Barcena M, Pansegrau W, Lurz R, Carazo JM, Lanka E (2000) Sequence-related protein export NTPases encoded by the conjugative transfer region of RP4 and by the cag pathogenicity island of Helicobacter pylori share similar hexameric ring structures. Proc Natl Acad Sci USA 97: 3067–3072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin A, Baker TA, Sauer RT (2005) Rebuilt AAA+ motors reveal operating principles for ATP-fuelled machines. Nature 437: 1115–1120 [DOI] [PubMed] [Google Scholar]
- Parge HE, Forest KT, Hickey MJ, Christensen DA, Getzoff ED, Tainer JA (1995) Structure of the fibre-forming protein pilin at 2.6 A resolution. Nature 378: 32–38. [DOI] [PubMed] [Google Scholar]
- Patenge N, Berendes A, Engelhardt H, Schuster SC, Oesterhelt D (2001) The fla gene cluster is involved in the biogenesis of flagella in Halobacterium salinarum. Mol Microbiol 41: 653–663 [DOI] [PubMed] [Google Scholar]
- Peabody CR, Chung YJ, Yen MR, Vidal-Ingigliardi D, Pugsley AP, Saier MH Jr (2003) Type II protein secretion and its relationship to bacterial type IV pili and archaeal flagella. Microbiology 149: 3051–3072 [DOI] [PubMed] [Google Scholar]
- Planet PJ, Kachlany SC, DeSalle R, Figurski DH (2001) Phylogeny of genes for secretion NTPases: identification of the widespread tadA subfamily and development of a diagnostic key for gene classification. Proc Natl Acad Sci USA 98: 2503–2508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planet PJ, Kachlany SC, Fine DH, DeSalle R, Figurski DH (2003) The widespread colonization Island of actinobacillus actinomycetemcomitans. Nat Genet 34: 193–198 [DOI] [PubMed] [Google Scholar]
- Possot O, Pugsley AP (1994) Molecular characterization of PulE, a protein required for pullulanase secretion. Mol Microbiol 12: 287–299 [DOI] [PubMed] [Google Scholar]
- Putnam CD, Clancy SB, Tsuruta H, Gonzalez S, Wetmur JG, Tainer JA (2001) Structure and mechanism of the RuvB Holliday junction branch migration motor. J Mol Biol 311: 297–310 [DOI] [PubMed] [Google Scholar]
- Remaut H, Waksman G (2004) Structural biology of bacterial pathogenesis. Curr Opin Struct Biol 14: 161–170 [DOI] [PubMed] [Google Scholar]
- Robien MA, Krumm BE, Sandkvist M, Hol WG (2003) Crystal structure of the extracellular protein secretion NTPase EpsE of Vibrio cholerae. J Mol Biol 333: 657–674 [DOI] [PubMed] [Google Scholar]
- Sandkvist M (2001) Biology of type II secretion. Mol Microbiol 40: 271–283 [DOI] [PubMed] [Google Scholar]
- Sandkvist M, Bagdasarian M, Howard SP, DiRita VJ (1995) Interaction between the autokinase EpsE and EpsL in the cytoplasmic membrane is required for extracellular secretion in Vibrio cholerae. Embo J 14: 1664–1673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauvonnet N, Vignon G, Pugsley AP, Gounon P (2000) Pilus formation and protein secretion by the same machinery in Escherichia coli. EMBO J 19: 2221–2228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savvides SN, Yeo HJ, Beck MR, Blaesing F, Lurz R, Lanka E, Buhrdorf R, Fischer W, Haas R, Waksman G (2003) VirB11 ATPases are dynamic hexameric assemblies: new insights into bacterial type IV secretion. EMBO J 22: 1969–1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senior AE, Nadanaciva S, Weber J (2000) Rate acceleration of ATP hydrolysis by F(1)F(o)-ATP synthase. J Exp Biol 203: 35–40 [DOI] [PubMed] [Google Scholar]
- Shin DS, Pellegrini L, Daniels DS, Yelent B, Craig L, Bates D, Yu DS, Shivji MK, Hitomi C, Arvai AS, Volkmann N, Tsuruta H, Blundell TL, Venkitaraman AR, Tainer JA (2003) Full-length archaeal Rad51 structure and mutants: mechanisms for RAD51 assembly and control by BRCA2. EMBO J 22: 4566–4576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiue SJ, Kao KM, Leu WM, Chen LY, Chan NL, Hu NT (2006) XpsE oligomerization triggered by ATP binding, not hydrolysis, leads to its association with XpsL. EMBO J 25: 1426–1435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton MR, Sawaya MR, Ellenberger T, Wigley DB (2000) Crystal structure of T7 gene 4 ring helicase indicates a mechanism for sequential hydrolysis of nucleotides. Cell 101: 589–600 [DOI] [PubMed] [Google Scholar]
- Story RM, Steitz TA (1992) Structure of the recA protein-ADP complex. Nature 355: 374–376 [DOI] [PubMed] [Google Scholar]
- Thomas NA, Mueller S, Klein A, Jarrell KF (2002) Mutants in flaI and flaJ of the archaeon Methanococcus voltae are deficient in flagellum assembly. Mol Microbiol 46: 879–887 [DOI] [PubMed] [Google Scholar]
- Trachtenberg S, Galkin VE, Egelman EH (2005) Refining the structure of the Halobacterium salinarum flagellar filament using the iterative helical real space reconstruction method: insights into polymorphism. J Mol Biol 346: 665–676 [DOI] [PubMed] [Google Scholar]
- Turner LR, Lara JC, Nunn DN, Lory S (1993) Mutations in the consensus ATP-binding sites of XcpR and PilB eliminate extracellular protein secretion and pilus biogenesis in Pseudomonas aeruginosa. J Bacteriol 175: 4962–4969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo HJ, Savvides SN, Herr AB, Lanka E, Waksman G (2000) Crystal structure of the hexameric traffic ATPase of the Helicobacter pylori type IV secretion system. Mol Cell 6: 1461–1472 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Legends and Table I
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3