

Abstract
Stem cell factor (SCF) binds to and activates the KIT receptor, a class III receptor tyrosine kinase (RTK), to stimulate diverse processes including melanogenesis, gametogenesis and hematopoeisis. Dysregulation of KIT activation is associated with many cancers. We report a 2.5 Å crystal structure of the functional core of SCF bound to the extracellular ligand-binding domains of KIT. The structure reveals a ‘wrapping' SCF-recognition mode by KIT, in which KIT adopts a bent conformation to facilitate each of its first three immunoglobulin (Ig)-like domains to interact with SCF. Three surface epitopes on SCF, an extended loop, the B and C helices, and the N-terminal segment, contact distinct KIT domains, with two of the epitopes undergoing large conformational changes upon receptor binding. The SCF/KIT complex reveals a unique RTK dimerization assembly, and a novel recognition mode between four-helix bundle cytokines and Ig-family receptors. It serves as a framework for understanding the activation mechanisms of class III RTKs.
Keywords: cancer, growth factor, protein–protein interaction, signal transduction, X-ray crystallography
Introduction
Stem cell factor (SCF) binds to the extracellular domains of the KIT receptor tyrosine kinase (RTK) and plays a key role in diverse biological processes. KIT, the product of c-kit gene, was first identified as the cellular homologue of the transforming gene of Hardy-Zuckerman 4-feline sarcoma virus (Besmer et al, 1986; Yarden et al, 1987). Subsequently, KIT was mapped to the mouse White Spotting locus, followed by the identification of its ligand SCF (KitL) and the demonstration that it is allelic with the murine steel locus (for a review, see Besmer, 1991). These pioneer studies have brought to light the pleiotropic functions of the SCF/KIT system in melanogenesis, gametogenesis and hematopoiesis (Lennartsson et al, 2005). Recent findings of SCF and KIT's role in brain angiogenesis suggest additional function for this widely implicated system (Sun et al, 2006). Dysregulation of SCF–KIT signaling and gain-of-function KIT mutations contribute to the genesis of many cancers, with acute myeloid leukemia, gastrointestinal stromal tumors and mastocytosis being the most prevalent types (Lennartsson et al, 2005).
KIT is a member of the class III subfamily of RTKs that includes KIT (SCFR), FMS (MCSFR or CSF-1R), FLT3, and PDGFR-α and -β. This class of RTKs are also called PDGFR family and are key receptors in the regulation of hematopoiesis and embryonic development (Reilly, 2003; Tallquist and Kazlauskas, 2004). They are characterized by an extracellular fragment consisting of five immunoglobulin (Ig)-like domains, a single transmembrane domain, two intracellular tyrosine kinase domains divided by a kinase insert domain and a C-terminal domain. Among class III RTKs, KIT and FLT3 mediate most of the early hematopoietic signaling (Fichelson, 1998); modulating their activities has the potential to facilitate the regeneration, isolation and expansion of stem cells in clinical applications. In addition to their physiological roles, most class III RTKs are involved in the genesis and development of cancers, and are widely pursued targets in the development of anti-cancer drugs and therapies (Krause and Van Etten, 2005).
The ligands for class III RTKs are two groups of topologically unrelated growth factors. The first group, including SCF, the ligand for KIT, macrophage-colony stimulating factor (MCSF), the ligand for FMS, and FLT3L, the ligand for FLT3, are four-helix bundle type cytokines (Sprang and Bazan, 1993). They are non-covalently or covalently linked dimers existing naturally as membrane-anchored and soluble isoforms as a result of alternative RNA splicing and proteolytic processing. Their N-terminal global domains have been identified as a functional core, which includes the dimerization interface and portions that bind and activate receptors (Langley et al, 1994). The other group of class III RTK ligands, PDGF-AA, -AB, -BB, -CC and -DD, are VEGF-like cysteine-knot type growth factors. Class III RTKs therefore represent a rare receptor family that use the same scaffold to receive ligands in fundamentally different folds, prompting a fascinating structural question to be answered.
The pivotal roles of class III RTKs in hematopoiesis and tumorigenesis have led to extensive functional and biochemical investigations, but it has not been structurally understood how these receptors recognize ligands and become activated. Despite a handful of ligand structures (Oefner et al, 1992; Pandit et al, 1992; Jiang et al, 2000; Savvides et al, 2000; Zhang et al, 2000) and mutagenesis mapping studies on ligands (Taylor et al, 1994; Matous et al, 1996; Graddis et al, 1998; Hsu et al, 1998), a structure of ligand–receptor complex has been elusive for this important receptor family. Here, we present the first crystal structure of a class III RTK ligand–receptor complex, the core domain of SCF bound to the extracellular ligand-binding domains of KIT. The structure shows a unique RTK/ligand assembly, in which SCF dimer is ‘wrapped' by KIT domains to allow engagement of two receptors, providing a framework for understanding ligand–receptor interactions and activation mechanisms of class III RTKs. It also reveals a new recognition mode between four-helix bundle cytokines and Ig-family receptors.
Results
Overall structure of SCF/KIT complex
Crystals of SCF/KIT complex were prepared using refolded mouse SCF from Escherichia coli and soluble, baculovirus-expressed mouse KIT extracellular domains 1–3. Initial trials using fully glycosylated, heterogeneous KIT yielded poorly diffracting crystals. To improve crystal quality, we reduced KIT glycosylation by mutagenesis (N146Q). This material behaved identically to the fully glycosylated proteins and yielded crystals that diffracted to 2.5 Å resolution. The SCF/KIT structure was solved by single isomorphous replacement with anomalous scattering from an iodide derivative of the binary complex (Table I).
Table 1.
Data collection, phasing and refinement statistics
| SCF/KIT complex | SCF | ||
|---|---|---|---|
| Native | NaI | ||
| Wavelength (Å) | 0.9798 | 1.5498 | 0.9184 |
| Resolution range (Å) (highest resolution shell) | 20–2.5 (2.60–2.50) | 50–3.21 (3.31–3.21) | 50–2.18 (2.26–2.18) |
| Space group | P21 | P21 | P65 |
| Unique reflections | 82 592 | 40 563 | 13 625 |
| Completeness (%) | 96.8 (93.3) | 99.8 (99.8) | 98.9 (100.0) |
| I/σ(I) | 17.1 (2.6) | 22.5 (5.6) | 43.0 (5.0) |
| Redundancy | 2.9 | 9.3 | 13.0 |
| Rmerge (%) | 4.0 (38.9) | 8.6 (44.6) | 10.0 (49.0) |
| SIRAS phasing | |||
| Resolution (Å) | 20–3.2 | ||
| Number of heavy atom sites | 27 | ||
| Rano (%) | 5.8 | ||
| Riso (%) | 15.4 | ||
| Figure of merit | 0.41 | ||
| Refinement | |||
| SCF/KIT complex | SCF | ||
| Resolution range (Å) | 20–2.5 (2.59–2.50) | 15–2.2 (2.28–2.10) | |
| Rcryst | 0.237 (0.416) | 0.241 (0.267) | |
| Rfree | 0.270 (0.427) | 0.266 (0.286) | |
| Average B factor (Å2) | 97.4 | 54.5 | |
| R.m.s.d. bond length (Å) | 0.007 | 0.009 | |
| R.m.s.d. bond angle, dihedral, improper (deg) | 1.4, 25.3, 1.1 | 1.5, 22.2, 1.0 | |
| R.m.s.d. bonded B factor (Å) | 3.7 | 2.3 | |
| Ramachandran (favored, allowed, generally allowed, disallowed) (%) | 84.6, 15.3, 0.1, 0 | 85.3, 13.8, 0.9, 0 | |
The overall structure of the 2:2 SCF/KIT complex roughly resembles an ‘H' letter with its top pushed down (Figure 1A and B). The lower parts of KIT stand vertical to the long axis of a flat-lying SCF dimer, and the N-terminal parts of KIT are bent back like horns. The orientation of the complex in Figure 1 places the SCF dimer parallel to the cell surface and the KIT C-termini proximal to the membrane. The SCF structure is similar to the free human SCF structures (Jiang et al, 2000; Zhang et al, 2000), with a core of four α-helices (A, B, C and D) and two β-strands (one between helices A and B and one between helices C and D). The head-to-head dimerization mode of SCF is unchanged from free human SCF structures. Two KIT are tethered together entirely through SCF dimerization. Each KIT interacts with only one SCF, and no KIT-to-KIT interaction is observed. Each of the KIT domains, designated D1 (residues 33–114), D2 (residues 115–208) and D3 (residues 209–310), interacts with SCF through spatially adjacent, but largely separated, binding epitopes. To facilitate analysis, these epitopes are designated as sites 1, 2 and 3, respectively (Figure 1A). Overall, the binding of each KIT to an SCF monomer resembles a taco, with KIT wrapping around SCF in a half-closed fashion. This binding fashion differs substantially from the previous models of SCF/KIT complex (Jiang et al, 2000; Zhang et al, 2000) (Supplementary Figure S1). Jiang et al (2000) placed KIT-D2 at approximately correct positions, and successfully predicted the charge complementarity between SCF and KIT (discussed below), but the orientation of each modeled KIT domain (D2 and D3) is different from actual. Additionally, the modeled D2–D2 distance, mimicking the VEGF/FLT1 complex, is longer than the actual distance, and the modeled D2–D3 hinge angle, mimicking VCAM, is drastically different from that seen in this structure.
Figure 1.
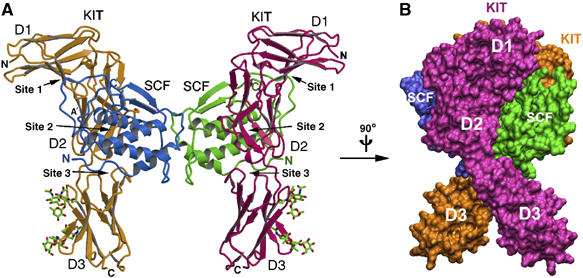
Structure of the SCF/KIT complex. (A) Ribbon model of the binary complex, SCF molecules are in cyan and green, and KIT molecules in orange and pink. (B) Side view of the complex in surface model, rotated 90° vertically relative to (A).
Structure of KIT
The extracellular SCF-binding region of KIT, comprising three Ig-like domains, resembles the perpendicular stroke of the letter ‘f' (Figure 2). It adopts an elongated, and surprisingly, bent conformation. Each D1–D2 or D2–D3 hinge angle is identical between different copies of KIT in the asymmetric unit, suggesting that this bent conformation is rigid rather than flexible. The D1–D2 hinge angle is abrupt (the β-strands directions of D1 and D2 are ∼80° relative to each other) and D1 folds back toward D2. The tight D1–D2 packing observed in KIT has also been observed among some cytokine receptors, Fc receptors and NK receptors, but has not observed in other RTKs. The D2–D3 hinge angle, in comparison, is much more linear (∼150°). D1 is a non-canonical intermediate between I-set and S-set Ig domains. Its first strand is divided into A and A′ strands and is shared by both layers of β-sheets (GFC face and BED face), characteristic of I-set configuration (Harpaz and Chothia, 1994), but its fourth strand switches from the BED face to the GFC face (D to C′), characteristic of S-type configuration (Bork et al, 1994) (Figure 2A). Unusually, D1's short CC′ loop and the C′ strand protrude from the domain core; they seem to be stabilized more by D1–D2 and D1–SCF interactions than by intra-domain interactions. D2 is a distorted I-set Ig domain. Its GFC face is twisted into two 90°-related β-sheets: the lower sheet (G, F and A′ strands) faces SCF to form D2–SCF interactions, and the upper sheet (C, G′ and F′ strands) faces outwards (Figure 2A). Domain D3 is a canonical I-set Ig domain. There are three predicted N-linked glycosylation sites in KIT, all on the opposite side of SCF-binding sites. Among these sites, Asn146 in D1 is mutated to Gln, whereas Asn296 and Asn303 in D3 appear fully glycosylated and three glycan residues were modeled for each site.
Figure 2.
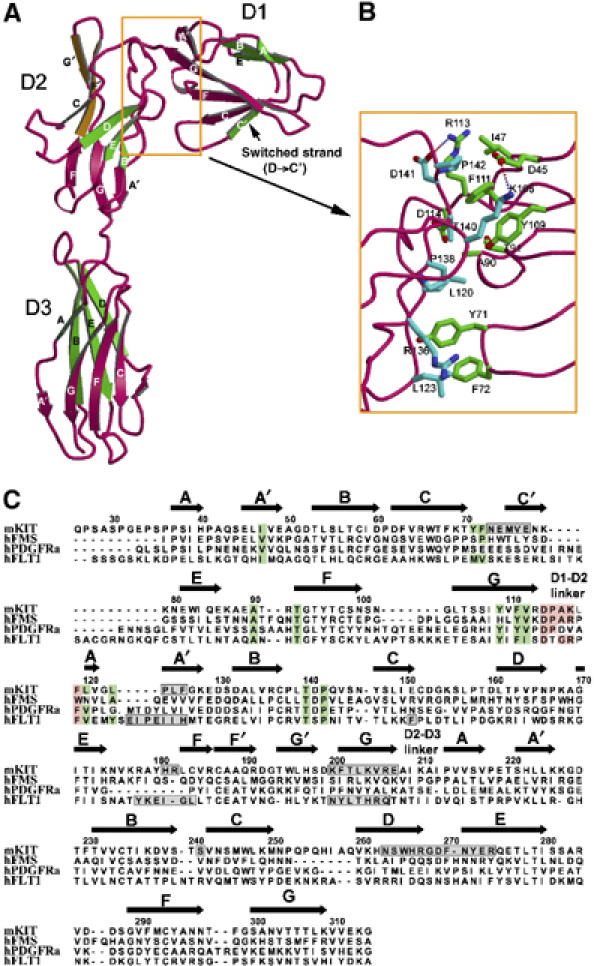
Structure of KIT. (A) Structural details of KIT domains. In each domain, the top layer of β-strands is colored in green and the bottom layer in red, with the exceptions that the switched strand (C′ strand) in D1 is green in color, and the upper part of the D2 bottom strands, vertical to the lower part, is colored in orange. (B) The D1–D2 interface. The side chains are colored in green for D1 residues and in cyan for D2 residues. (C) Structure-based sequence alignment of N-terminal three domains between KIT and related receptors. Residues involved in hydrophobic interaction at D1–D2 interface are shaded in green. Conserved residues at D1–D2 linker are shaded in pink. Residues involved in ligand binding as proven in structures of complexes are boxed in gray.
The segmental rigidity of KIT is ensured by the extensive inter-domain interactions. The D2–D3 junction is reinforced by both hydrophobic stacking and hydrogen bonds, with a total area of 630 Å2 buried in the interface. The D1–D2 interaction is even more extensive, burying a large area of the surface area (1380 Å2), which may suggest that D1 and D2 are integral and inseparable. The D1–D2 junction is exquisitely designed, encompassing primarily hydrophobic inter-domain interactions (Figure 2B). The D1–D2 interface can be divided into two areas. The first, larger area is a hydrophobic cluster, including Ile47, Ala90, Thr93 (Cγ2), Tyr109, Phe111, Asp114 (Cβ) from D1 and Lys117 (aliphatic part), Leu120, Thr140 (Cγ2), Pro142 from D2, further consolidated by two salt bridges (Arg113–Asp141 and Glu45–Lys168). The area is entirely conserved across mammalian KIT molecules (Supplementary Figure S2). The second, smaller area is formed between hydrophobic residues (Tyr71 and Phe72) extended from the D1 CC′ loop and the hydrophobic patch, Leu120, Leu123, Arg136 (aliphatic part), Pro138, from D2. These residues are also either conserved or similarly hydrophobic among mammalian species (Supplementary Figure S2). The sequence conservation of both patches suggests that the configuration between D1 and D2, and thereby the bent-back orientation of D1, is structurally conserved. The bent-back orientation of D1, together with the anchoring of the D1 CC′ loop to D2 through hydrophobic interaction, serves to position the D1 C′ strand, which is involved in SCF binding (discussed below).
The D1–D2 configuration and the bent-back orientation of D1 may be a conserved feature among many Ig-containing RTKs, including other class III RTKs and closely related class V RTKs. Structure-based sequence alignment (Figure 2C) shows that the residues contributing to the hydrophobic interactions in the D1–D2 interface are highly conserved among helical ligand-binding class III RTKs (e.g., KIT and FMS), β-sheet ligand-binding class III RTKs (e.g., PDGFRα) and class V RTKs (e.g., FLT1). In particular, FLT1 shows the highest similarity to KIT in D1–D2 interface composition. In addition, the D1–D2 linker sequences are the same length and similar among these RTKs (Figure 2C). The conservation of both D1–D2 interface and D1–D2 linker suggests that many class III and class V RTKs share similar D1–D2 configuration. This configuration then dictates a bent-back D1, which can bear the potential to participate in ligand binding.
Interactions between SCF and KIT
Overall, about 2200 Å2 of solvent-accessible surface is buried between each pair of SCF and KIT, which can be subdivided into 610 Å2 for site 1, the D1/SCF epitope, 760 Å2 for site 2, the D2/SCF epitope, and 830 Å2 for site 3, the D3/SCF epitope. Consistent with early thermodynamic data showing that SCF/KIT binding is largely enthalpy-driven (Philo et al, 1996; Lemmon et al, 1997), the SCF/KIT-binding interface buries large areas of hydrophilic surface, with abundant salt bridges and hydrogen bonds. Only 12% (268 Å2) of the buried surface is hydrophobic. As predicted by Jiang et al (2000), charge complementarity appears to be an important feature of SCF/KIT recognition: KIT is positively charged at site 2 and slightly positively charged at site 3, whereas SCF is rich in negative charge at both these sites to meet KIT (Figure 3E). An exception is site 1, where the charge usage of receptor and ligand is reversed, that is, SCF uses positive charge and KIT uses negative charge (Figure 3E).
Figure 3.
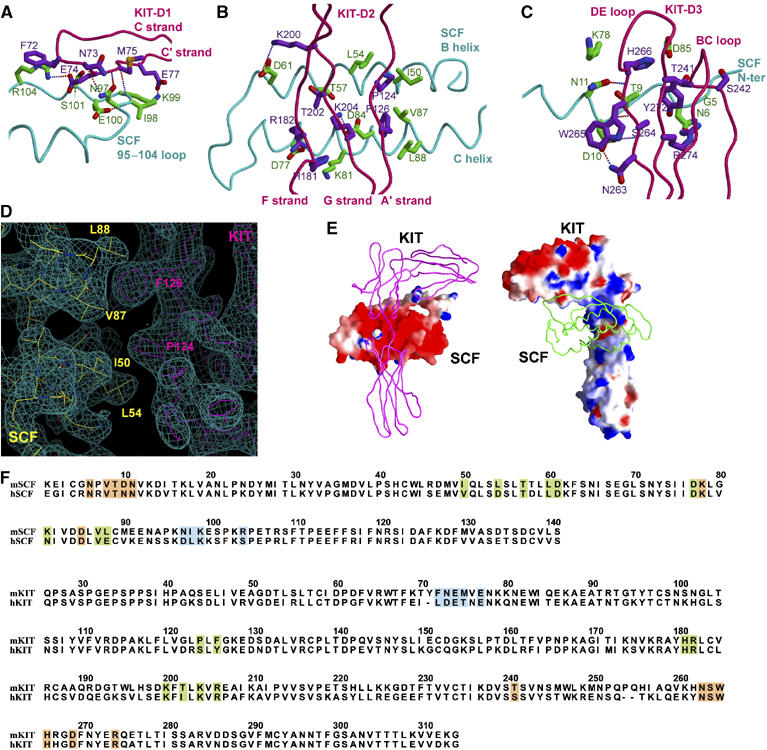
Interaction between SCF and KIT. (A) Site 1, the KIT-D1/SCF interface. SCF's main chain is colored in cyan, side chain in blue; KIT's main chain in pink, side chain in purple. (B) Site 2, the KIT-D2/SCF interface. (C) Site 3, the KIT-D3/SCF interface. (D) Composite 2Fo−Fc omit map contoured at 1.0σ showing a portion of the site 2 interface. (E) Charge complementarity between SCF and KIT. Left part is KIT as tube and SCF as GRASP surface colored with electrostatic potential, red being negative and blue being positive; right part vice versa. (F) Sequence alignment between mouse and human SCF/KIT. Site 1, 2 and 3 residues are shaded in blue, green and orange, respectively.
The small site 1 interface consists of the C′ strand (residues 73–77) from KIT-D1 and an extended loop (residues 95–104) from SCF (Figure 3A). It encompasses two salt bridges, three hydrogen bonds and a number of van der Waals interactions (Table II). The site 2 interface is formed by the lower part of the KIT-D2 bottom β-sheet, including the A′, F and G strands (residues 124–126, 181–182 and 200–206), crossing vertically above the B and C helices of SCF (residues 50–61 and 77–88) (Figure 3B). This interface can be divided into two side-by-side, chemically distinct patches, one hydrophobic and the other hydrophilic. The first, hydrophobic patch is formed between SCF and the A′ strand of KIT-D2, in which KIT inserts Pro124 and Phe126 into the cavity formed by SCF Ile50, Leu54, Val87 and Leu88 (Table II, Figure 3D). The second, hydrophilic patch is formed between SCF and the G and F strands of KIT-D2. This patch consists entirely of hydrophilic residues and is rich in charges (Table II). The charge complementarity at this patch is also clearly reflected in Figure 3E. The site 3 interface is formed between SCF N-terminal segment and the BC loop (residues 263–274) plus the DE loop (residues 239–243) of KIT-D3 (Figure 3C). Compared to sites 1 and 2, this interface has the largest buried area (830 Å2) and the largest number of SCF/KIT contacts (Table II).
Table 2.
Contacts between SCF and KIT
| KIT-D1 | SCF | Distance (Å) | ||
|---|---|---|---|---|
| Hydrogen bonds and salt bridges | ||||
| Asn3 | O | Asn97 | Nδ2 | 2.6 |
| Glu74 | Oɛ1 | Arg104 | Nη2 | 3.1 |
| Met75 | N | Asn97 | Oδ1 | 3.3 |
| Met75 | O | Lys99 | N | 2.9 |
| Glu77 | Oɛ1 | Lys99 | Nζ | 3.3 |
| van der Waals contacts | ||||
| Thr70 | Arg104 | |||
| Phe72 | Arg104 | |||
| Asn73 | Asn97 | |||
| Glu74 | Asn97 | |||
| Met75 | Ile98 | |||
| KIT-D2 | ||||
| Hydrogen bonds and salt bridges | ||||
| Arg182 | Nη2 | Asp77 | Oδ1 | 2.5 |
| Lys200 | Nζ | Asp61 | Oδ1 | 3.5 |
| Lys204 | Nζ | Asp84 | Oδ2 | 3.0 |
| van der Waals contacts | ||||
| Pro124 | Ile50 | |||
| Phe126 | Leu88, Val87 | |||
| His181 | Lys81 | |||
| Thr202 | Thr57 | |||
| Arg206 | Leu88 | |||
| KIT-D3 | ||||
| Hydrogen bonds and salt bridges | ||||
| Asn263 | Nδ2 | Asp10 | Oδ1 | 3.3 |
| Ser264 | N | Asp10 | Oδ2 | 3.3 |
| Ser264 | O | Asp10 | N | 2.7 |
| His266 | N | Asn11 | Oδ1 | 2.9 |
| Arg274 | Nη1 | Asn6 | Oδ1 | 3.3 |
| van der Waals contacts | ||||
| Thr241 | Asn6, Asp85 | |||
| Ser264 | Val8, Asn6, Thr9, Asp10 | |||
| Trp265 | Asp10 | |||
| His266 | Asn11, Thr9 | |||
| Asp269 | Lys81 | |||
The SCF/KIT structure can now be used to rationalize previous biochemical and functional data mapping SCF/KIT interaction. Mutagenesis and antibody mapping data on SCF implicate the first few N-terminal residues before Cys4 (Langley et al, 1994), the regions flanked by residues 61–65 and 91–95 (Mendiaz et al, 1996) or the region of residues 79–97 (Matous et al, 1996), being important for KIT binding. These regions, to some extent, differ from the SCF/KIT interfaces in the complex. Overall, these studies emphasize the end, or dimerization interface-distal, region of SCF being critical in KIT binding. In comparison, the SCF/KIT structure shows that the receptor-binding surface is located in the middle of SCF helices, and slightly closer to the dimerization interface than to the end. In another study, a quadruple mutant of SCF (R121N, D124N, K127D, D128K) is deficient in KIT binding (Matous et al, 1996). These residues, part of the D helix, are on the opposite side to the KIT-binding surface. This deficiency is likely due to indirect structural effect: Arg121 forms salt bridge with Asp37 and its long aliphatic region protects Met36 and Trp44 from the core; mutation of this residue may be structurally disruptive.
Human SCF/KIT complex should have the same overall configuration between SCF and KIT as in mouse SCF/KIT complex in this study, given the high sequence similarity between species for both SCF and KIT (Figure 3F and Supplementary Figure S2). Although most SCF/KIT interactions are likely conserved, there is a notable difference between mouse and human complexes. In site 2, the hydrophobic interactions at the patch between mouse SCF (Ile50, Leu54, Val87 and Leu88) and mouse KIT (Pro124 and Phe126) are significantly altered in the human complex (Asp54 and Glu88 in human SCF, Ser 123 and Tyr125 in human KIT) (Figure 3F). This difference likely explains the species specificity in SCF/KIT binding. Human SCF does not detectably recognize murine KIT, but murine SCF can activate human KIT, albeit at a reduced affinity (Lev et al, 1992). This may be because Pro124 and Phe126 of mouse KIT are highly hydrophobic and require the corresponding SCF residues to be also hydrophobic. By comparison, Ser123 and Tyr125 in human KIT are less hydrophobic and can be more promiscuous at this site.
Glycosylation of SCF has been shown to affect its activity (Lu et al, 1992). Three of the four potential N-linked glycosylation sites, Asn65, Asn93 and Asn120, are occupied in the CHO-cell-derived human SCF. Asn120 glycosylation does not affect receptor binding. In the SCF/KIT complex, it is located on helix D of SCF, the back of the SCF/KIT interface. By comparison, the Asn65 and Asn93 sites, which reduce SCF's biological activity by 10-fold, are much closer to the SCF/KIT interface. The Asn93 site, being immediately before the flexible loop that binds KIT-D1, may interfere with re-ordering of this loop upon receptor binding (discussed below). The side chain of SCF Asn65 is adjacent to site 3 and points to KIT-D3, suggesting that the glycan on this site could hinder the incoming KIT-D3.
KIT-induced SCF conformational change
The mouse SCF structure in the complex is generally similar to the free human SCF structures reported previously (Jiang et al, 2000; Zhang et al, 2000), but differences exist in several surface segments. To facilitate comparison, we also crystallized free mouse SCF and solved its structure at 2.2 Å resolution (Table I). The free mouse SCF has a significant amount of surface regions disordered or poorly ordered, including the N-terminal polypeptide chain (up to residue 11), the 92–104 loop and the 129–136 loop (Figure 4A). The flexibility of these regions was also observed in free human SCF (Jiang et al, 2000; Zhang et al, 2000). In the SCF/KIT complex, however, this segmental flexibility is changed. One significant change is associated with the N-terminal segment. In the SCF/KIT complex, all four individual mouse SCF subunits have good electron densities at the N-terminal regions. But in free human SCF or mouse SCF, this segment is disordered in every subunit; even the disulfide bond (Cys4–Cys89) appears incapable of stabilizing its conformation. The disparity between unbound and bound states suggests that the structure of N-terminal segment in bound SCF is imposed by the receptor. Of particular importance are the hydrogen bonds between KIT main-chain atoms and SCF residues 10–11, as well as the hydrogen bond between KIT Arg274 and SCF Asn6, in orientating the exterior, hydrophilic side of the SCF N-terminal segment. Upon determination of the exterior side, SCF's Pro7, Val8 and Val12 side chains, the interior side, form hydrophobic interactions with SCF's core (Leu79, Ile82, Leu86, Phe119, Ile123 and Phe126), further stabilizing the local structure (Figure 4C).
Figure 4.
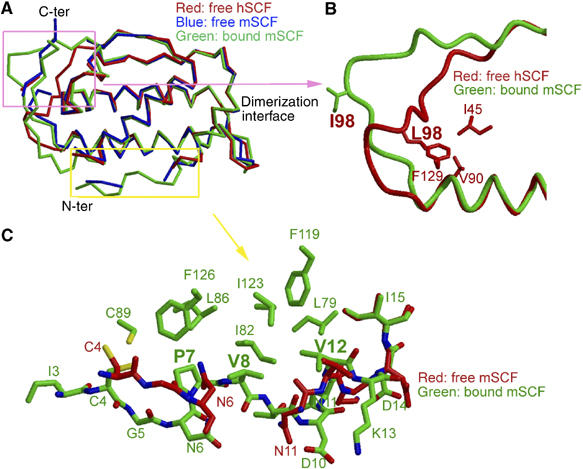
Conformational change of SCF. (A) Superimposition of the Cα atoms of free human SCF, free mouse SCF and bound SCF as in the SCF/KIT complex, highlighting the differences at the 92–104 loop and the N-terminal segment. (B) Comparison of the 92–104 loops of free human SCF and bound mouse SCF. (C) Comparison of the N-terminal segments of free and bound mouse SCF.
Another significant change is associated with the 92–104 loop of SCF. This loop is clearly visible and well defined in the SCF/KIT complex. It is, however, invisible in all but one of the four human SCF subunits (Jiang et al, 2000). In a different crystal form of human SCF, this loop is modeled, albeit with high temperature factors (Zhang et al, 2000). Comparison of the human SCF subunits containing visible 92–104 loops, solved independently in different crystal forms, shows that their 92–104 loops are roughly superimposable. This suggests that the 92–104 loop is only partially flexible and has a favorable conformation. As this loop in our free mouse SCF is disordered and does not support comparison, we compared one of the defined 92–104 loops in human SCF (Jiang et al, 2000) with that in our SCF/KIT complex (Figure 4B). The 92–104 loop appears to undergo a large conformational change upon receptor binding. In free SCF, the hydrophobic Leu98 side chain, being in the middle of this long loop, is anchored deeply to the SCF hydrophobic core (Figure 4B). This restraint limits the mobility of the 92–104 loop and brings it close to the end of the SCF B helix, adding numerous contacts between this loop and the SCF core structure. In bound SCF, however, the loop is deviated away from the core structure to form interactions with KIT-D1. The Cα atoms of residues 98–99 in bound SCF are 11–12 Å away from these atoms in free SCF. None of the residues 95–104 has contact with the rest of SCF; the loop is entirely stabilized by receptor–ligand interactions. Collectively, the conformational changes at both the N-terminal segment and the 92–104 loop of SCF are induced by KIT binding. The necessity of these changes for receptor binding adds another dimension to the specificity requirement in KIT activation.
Discussion
A unique RTK dimerization assembly
Ligand-induced receptor dimerization is believed to be the triggering step for RTK activation (Schlessinger, 2000). Indeed, clustering of two receptors is a common feature of all known structures of RTK/ligand complexes. However, among different RTK subfamilies, the scheme used to achieve receptor clustering is widely diverse: each subfamily appears to have its own configuration of assembling receptor/ligand complex. In general, the dimerized RTK complexes can be divided into two groups: dimeric ligand-driven and monomeric ligand-driven. The SCF/KIT complex, together with VEGF/FLT1 (Wiesmann et al, 1997) and NGF/TrkA (Wiesmann et al, 1999), defines a group of RTK complexes that are dimerized primarily by the dimeric nature of their ligands, although receptor-to-receptor contacts can add to dimerization in some cases (Blechman et al, 1995; Barleon et al, 1997). This group may also include Ret, Tie and Met subfamilies, for which structures of fully associated, dimerized complexes remain to be determined. The other group of RTK complexes induced by monomeric ligands are dimerized by either ligand–receptor crosslinking, for example, Ephrin/Eph (Himanen et al, 2001), gas6/Axl (Sasaki et al, 2006) and FGF/FGFR (Mohammadi et al, 2005), or by ligand-induced and conformation-regulated receptor–receptor contact, for example, EGF/EGFR (Schlessinger, 2002). A third possible assembly, in which two receptors bind to different sites on a single ligand, has not been found for RTKs, although has been reported redundantly for type I cytokine receptors (Stroud and Wells, 2004). Within the group of dimeric ligand-driven RTK complexes (Figure 5A), SCF/KIT distinguishes itself from VEGF/FLT1 and NGF/TrkA by three major differences. First, SCF/KIT is the only one that uses a four-helix bundle ligand, whereas the other two complexes use cysteine-knot β-sheet ligands. The four-helix bundle fold, in general, is used in cytokine receptor recognition that leads to JAK–STAT pathways; its usage by SCF/MCSF/FLT3L in RTK recognition is an exception. Second, the presentation of receptor-binding surface on SCF is fundamentally different from other dimeric ligands: each SCF binds a separate KIT, but VEGF and NGF both use dimerization seams to bind receptors, with each receptor binding to two ligands. SCF/KIT therefore stands for the most simplified paradigm of receptor dimerization, in which dimerization and receptor recognition are separate issues. Third, although these receptors all use Ig domains to bind ligands, their usages of Ig domains are strikingly different. KIT uses three domains to bind SCF, and the ligand-contacting regions range from an edge β-strand from D1, CFG β-sheet from D2, and BC and DE loops from D3. TrkA uses a single Ig domain to bind NGF, with primarily the membrane-proximal loops contacting NGF. FLT1-D2 uses its CFG sheet, as KIT-D2 does, but adds a CD loop to bind VEGF (Figures 2C and 5A). Notably, the orientations of KIT-D2 and FLT1-D2 in their complexes are roughly related but clearly different: FLT1-D2 is ∼30° tilted, and KIT-D2 is vertical, to the cell membrane (Figure 5A). In addition, as judged by the distance between two ligand-binding surfaces on D2, FLT1-to-FLT1 spacing in VEGF/FLT1 is ∼8 Å longer than the KIT-to-KIT distance in SCF/KIT. Reportedly, FLT1-D3, and possibly FLT1-D1, also participates in VEGF binding (Fuh et al, 1998); whether this configuration is similar to that in SCF/KIT remains to be demonstrated. The significant differences of SCF/KIT, a class III RTK, from other classes of RTK complexes further demonstrates the diversity of RTK signaling assemblies, reflecting the early emergence and divergence of RTKs during metazoan evolution (Ben-Shlomo et al, 2003).
Figure 5.
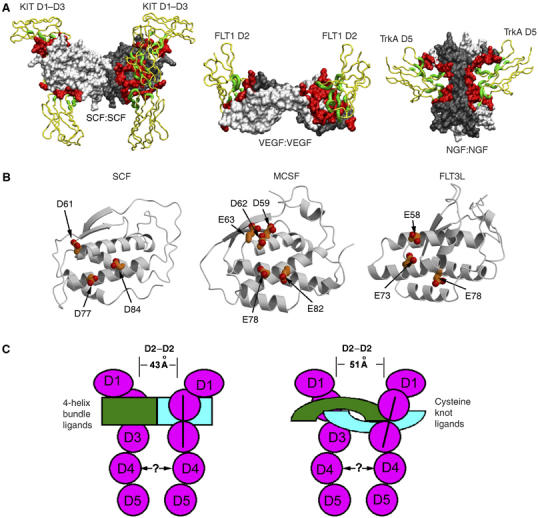
Activation of class III RTKs. (A) Comparison of SCF/KIT with VEGF/FLT1 and NGF/TrkA. Regions involved in ligand–receptor binding are highlighted in green for receptors and in red for ligands. (B) Similarity of four-helix bundle ligands in presenting acidic patches to their class III RTK receptors. (C) Cartoon representation of class III RTK signaling assemblies. Four-helix bundle ligand-binding receptors have closer D2–D2 distance and vertical D2–D3 domains, whereas cysteine-knot ligand-binding receptors have farther D2–D2 distance and tilted D2–D3 domains.
Activation of class III RTKs
The SCF/KIT complex explains how two KIT receptors can be engaged at a specific distance, mechanistically parallel with other classes of RTKs. As a structural template, it can now lend insights into other class III RTKs. Among class III RTKs, KIT, FMS and FLT3 have ligands with the similar dimeric structures. Superimposition of SCF, MCSF and FLT3L shows root mean square (r.m.s.) deviations of 2–3 Å for their Cα atoms (Pandit et al, 1992; Jiang et al, 2000; Savvides et al, 2000; Zhang et al, 2000). More importantly, the receptor-binding sites of these ligands have been mapped to roughly similar regions on the dimer scaffold (Taylor et al, 1994; Matous et al, 1996; Graddis et al, 1998; Hsu et al, 1998). In accord with the ligand similarities, the ligand-binding regions on the receptors have been similarly confined to the N-terminal three domains (Blechman et al, 1993; Lev et al, 1993; Wang et al, 1993). It is likely that the MCSF/FMS complex and the FLT3L/FLT3 complex resemble the SCF/KIT complex in the shape of receptor/ligand assembly. However, given the wide sequence disparity both among ligands (MCSF versus SCF is only 14% identical, MCSF versus FLT3L is 12%, and SCF versus FLT3 is 8%) (Supplementary Figure S3) and among receptors (<25% identity) (Figure 2C), most of the specific interactions at the binding interface may not be conserved. Nevertheless, they may similarly use charge attraction (receptor being positive versus ligand being negative) as the driving force for ligand–receptor D2 interactions. Overlay of MCSF to the SCF/KIT complex reveals that MCSF may present highly enriched negative charges, a cluster of acidic residues including Asp59, Asp62, Glu63, Glu78 and Glu82, to domain D2 of its receptor FMS (Figure 5B). These residues are located on helices B and C and correspond well to site 2 contributing patches on SCF. Similarly, on FLT3L, Glu58, Glu73 and Glu78 are at the center of putative FLT3-D2-binding surface (Figure 5B). Another similarity may be the reduction of flexibility for the ligand N-terminal segments upon receptor binding. In the free ligand structures, both MCSF and FLT3L N-terminal segments, when not involved in crystal packing, have significant flexibility. These segments may become more ordered in their respective complexes by interacting with receptor D3 domains.
PDGFRα and PDGFRβ, unlike KIT that binds a helical ligand, have cysteine-knot growth factor-type ligands, which are related with VEGF but not SCF. Assuming that PDGFR-D2 takes FLT1-D2's position as in VEGF/FLT1-D2 complex (Wiesmann et al, 1997), the PDGF/PDGFR complexes would have two PDGF-binding sites on receptor D2 domains ∼51 Å away. In comparison, the distance between two SCF-binding sites on KIT-D2 in the SCF/KIT complex is only 43 Å. Given the close, sometimes functionally interchangeable, relationship among class III RTKs, it is likely that their transmembrane segments in signaling complexes are tethered at a similar distance. Therefore, the distance disparity between four-helical bundle ligand-bound complexes and cysteine-knot ligand-bound complexes at D2 level needs to be compensated at membrane-proximal D3–D5 levels. Interestingly, D2 in VEGF/FLT1, and deducibly in PDGF/PDGFR, is tilted, with closer distance between membrane-proximal ends than between membrane-distal ends. In comparison, D2 in SCF/KIT is vertical. Consequently, the PDGFR D3 domains could be drawn closer, back to a distance as seen in SCF/KIT (illustrated in Figure 5C).
Our SCF/KIT complex includes only the first three domains of KIT, and the role of D4 and D5 domains in receptor activation remains to be addressed. It has been suggested for both class III and class V RTKs that D4, by forming D4–D4 interaction, plays a role in receptor dimerization (Blechman et al, 1995; Barleon et al, 1997), but this suggestion has been questioned (Philo et al, 1996; Lemmon et al, 1997). In our SCF/KIT complex, the D3 domains point to the opposite directions. The distance between two C-termini of KIT-D3 (Cα atoms of residues 280) is 60 Å, about 1.5 times the length of a typical Ig domain. The opposite direction and wide distance between KIT-D3 C-termini do not favor D4–D4 interaction, but cannot rule out the possibility of a highly bent D3–D4 hinge that allows two D4 domains to reach each other. A definitive answer about D4–D4 interaction still awaits a structure encompassing entire KIT extracellular region.
A new recognition mode between four-helix bundle cytokines and Ig-like domain-containing receptors
Most helical cytokines are recognized by JAK–STAT-activating receptors, instead of RTKs, to regulate various cellular functions. The helical cytokines can be divided into long chain, short chain and interferon like (Sprang and Bazan, 1993). Both long-chain and short-chain helical cytokines have a four-helix bundle fold with up-up–down-down topology, and activate the family of class I hematopoietic cytokine receptors. Several of these cytokine/receptor complexes, including both long chain (e.g., hGH/hGHR, EPO/EPOR, GCSF/GCSFR, IL-6/gp130/IL-6Rα) and short chain (e.g., IL-4/IL-4Rα, IL-2/Rα/Rβ/γc), have been solved (reviewed in Stroud and Wells, 2004; Wang et al, 2005). Notably, all these complexes follow the cytokine-recognition paradigm established by hGH/hGHR (de Vos et al, 1992). In this paradigm, a conserved cytokine-binding homology region (CHR), consisting of two Ig-like fibronectin-III domains, recognizes distinct sites (sites 1 and 2) on the four-helix bundle cytokines (Bazan, 1990; Sprang and Bazan, 1993). These sites can be used to recruit the same or different receptors, one site can be unoccupied and an additional site can be used, resulting in variation of receptor activation schemes (Stroud and Wells, 2004). Nevertheless, the recognition mode at these sites, especially site 1, is rigorously kept. Being also a four-helix bundle cytokine/Ig-family receptor complex, SCF/KIT is proximal to the site 1 interactions, but not to site 2, of other cytokine complexes (Figure 6). But even at site 1, the SCF/KIT recognition has little similarity to these classical cytokine receptor complexes. First, the ligands use largely different regions to bind their receptors. SCF uses the N-terminal segment, helices B and C, and a flapping loop to bind KIT (Figure 6A). Other four-helical bundle cytokines, whether long chain (e.g., hGH complex) or short chain (e.g., IL-2 complex), use helices A and C to bind receptor at site 1 (Figure 6B and C). Second, the receptors use different regions to bind their ligands. The KIT-D1 contribution in ligand recognition is unseen in other cytokine complexes; KIT-D2, corresponding to the D1 domains of hGHR and IL-2Rβ, uses strands from a β-sheet to form face-to-face interactions with SCF, whereas hGHR-D1 uses AB and EF loops, and IL-2Rβ-D1 domains use CD and EF loops to contact their ligands; KIT-D3, oriented similarly to hGHR and IL-2Rβ D2 domains, is much higher up relative to ligand and uses mainly DE loop to contact SCF, whereas hGHR and IL-2Rβ D2 domains use BC and FG loops to contact hGH and IL2 (Figure 6). Noticeably, in all short-chain and long-chain cytokine complexes, receptors at site 1 show an ∼90° bending between two Ig-like, fibronectin-III domains, to allow the outside of the elbow to contact ligands. This configuration, repeatedly observed for the CHR module, is not observed in KIT, whose D2 and D3 Ig domains are, in contrast, linearly related. Third, the chemistry of SCF/KIT binding is unique when compared to other cytokine complexes. None of the classical cytokine complexes presents spatially discontinuous and mostly charge-driven interface at site 1 as in SCF/KIT; their binding sites are continuous, often presenting a small group of dominating hydrophobic side chains at the center of site 1 as the ‘hot-spot' (Stroud and Wells, 2004). The major differences between SCF/KIT and these complexes indicate that SCF/KIT, as an RTK complex, has evolved a new recognition mode from classical hematopoietic cytokine/receptor recognition, despite their structural kinships on both ligand and receptor sides.
Figure 6.
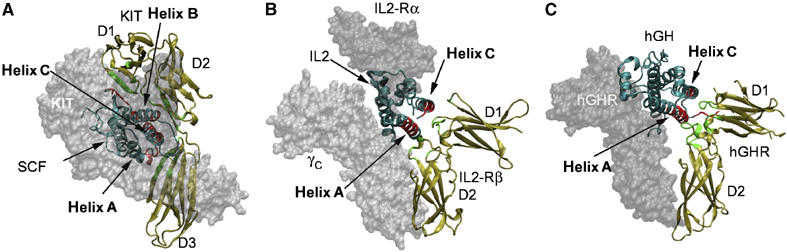
Comparison of (A) SCF/KIT with (B) short-chain and (C) long-chain four-helix bundle cytokine/receptor complexes. The copy of ligand and the copy of receptor for comparison are depicted in ribbons, the other parts in transparent surface. Regions involved in ligand–receptor binding are highlighted in red for ligands and in green for receptors.
Materials and methods
Cloning and expression
Recombinant mouse SCF was produced in E. coli. The coding sequence for residues 1–141 plus C-terminal 6-His-tag was subcloned into pGEX4T1, and the construct was transformed into Rosetta-gami2 cells (EMD Biosciences). Overnight culture was scaled to 4 l and induced with 1 mM IPTG at 37°C. Inclusion bodies were refolded in direct dilution following the protocol for human SCF (Zhang et al, 2000). Refolded GST-fusion protein was captured with GST-affinity resin, cleaved with thrombin and further purified with size-exclusion chromatography.
Recombinant mouse KIT extracellular domains were produced using the baculovirus system in insect cells. The coding sequence for mouse KIT residues 24–314 plus C-terminal 7-His-tag was subcloned into the pAcGp67A (Pharmingen). Recombinant baculovirus was produced and amplified using sf9 cells in serum-containing media. For large-scale expression of the protein, Tn5 cells (6 l) were grown to a density of 1.8 × 106 cells/ml in HyQ SFX media (HyClone) in shaking Fernbach flasks and were infected with recombinant virus at an MOI of 10. Cultures were allowed to progress for 66 h before the cells were pelleted by centrifugation. The supernatant was concentrated and exchanged into HBS (Hepes buffered saline, 10 mM Hepes pH 7.2, 150 mM NaCl). The protein was captured by Ni-NTA resin and eluted with 300 mM imidazole pH 7.5. The C-terminal His-tags of proteins were removed with bovine carboxypeptidase-A. The proteins were further purified with size-exclusion chromatography.
Deglycosylation mutants
Mutagenesis was carried out using overlap extension PCR. Seven KIT glycosylation mutants, N146Q, N296Q, N303Q, N146Q–N296Q, N146Q–N303Q, N296Q–N303Q, and N146Q–N296Q–N303Q, were prepared. The mutant N146Q was expressed at half the expression level of wild-type KIT, and showed a significant reduction of apparent size as observed in SDS–PAGE. Other mutants were expressed at substantially lower level and aggregated in gel filtration analysis.
Crystallization, data collection and processing
To prepare SCF/KIT complex for crystallization, excess SCF was mixed with wild-type KIT or KIT N146Q mutant and the mixtures were re-purified with size exclusion chromatography. Stoichiometric SCF/KIT complex was concentrated to 10 mg/ml in HBS. The crystals were obtained through vapor diffusion in sitting drops containing equal volumes of protein and the well solution containing 15% PEG1000, 0.2 M NaCl, buffered with 0.1 M MES, pH 6.0. The free mouse SCF was crystallized using similar vapor-diffusion procedures, and the well solution contained 1.8 M ammonium sulfate, buffered with 0.1 M Hepes, pH 8.0. Data sets were collected at beamlines DND-CAT, IMCA-CAT and NE-CAT at the Advanced Photon Source, Argonne, IL. Crystals were cryoprotected before being cooled to 100 K in the presence of 25% glycerol in mother liquor. The SCF/KIT crystals have a spacegroup of P21, with cell dimensions a=76.85 Å, b=200.15 Å, c=82.02 Å, β=91.42°. The free SCF crystals have a spacegroup of P65, with cell dimensions a=b=43.27 Å, c=248.43 Å. The data were indexed, integrated and scaled with HKL2000 (Otwinowski and Minor, 1997). To prepare a heavy atom derivative for the complex, crystals were quick-soaked with a cryo-solution containing 0.2 M NaI before being flash-cooled for X-ray diffraction. Statistics of data collection are summarized in Table I.
Phase determination, model building and refinement
The structure of free mouse SCF was determined using molecular replacement in CNS (Brunger et al, 1998), with human SCF (PDB ID 1SCF) as a search model (Jiang et al, 2000). The SCF/KIT complex was phased with an NaI derivative using programs CNS (Brunger et al, 1998) and SOLVE (Terwilliger and Berendzen, 1999). The phases were improved with density modification and four-fold non-crystallographic symmetry averaging in CNS. The models were traced and built with the program O (Jones et al, 1991). The structures were refined using CNS maximum-likelihood simulated annealing with torsion angle dynamics protocol, and repeated iterations between manual rebuilding and minimization. Water molecules were automatically included with CNS and manually edited with electron density maps. A summary of the refinement statistics and the stereochemistry analysis is given in Table I.
Graphics
The figures were produced with the programs MOLSCRIPT (Kraulis, 1991), VMD (Humphrey et al, 1996) and GRASP (Nicholls et al, 1991).
Accession codes
Coordinates and structural factors for SCF/KIT complex and free mouse SCF have been deposited in the Protein Data Bank, with accession codes 2O26 and 2O27, respectively.
Supplementary Material
Supplementary Figures
Acknowledgments
We thank A Gross and W Anderson for sharing beamtime, and staff members at the APS DND-CAT, IMCA-CAT and NE-CAT beamlines for assistance. XH is supported by the NIH grant 1R01GM078055-01. The Structural Biology Facility is supported by the RH Lurie Comprehensive Cancer Center of Northwestern University. DND-CAT is supported by DuPont, Dow, the US National Science Foundation and the State of Illinois. Use of the IMCA-CAT was supported by the companies of the Industrial Macromolecular Crystallography Association through a contract with the Center for Advanced Radiation Sources at the University of Chicago. Use of NE-CAT was supported by award RR-15301 from NCRR at NIH.
References
- Barleon B, Totzke F, Herzog C, Blanke S, Kremmer E, Siemeister G, Marme D, Martiny-Baron G (1997) Mapping of the sites for ligand binding and receptor dimerization at the extracellular domain of the vascular endothelial growth factor receptor FLT-1. J Biol Chem 272: 10382–10388 [DOI] [PubMed] [Google Scholar]
- Bazan JF (1990) Structural design and molecular evolution of a cytokine receptor superfamily. Proc Natl Acad Sci USA 87: 6934–6938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Shlomo I, Yu Hsu S, Rauch R, Kowalski HW, Hsueh AJ (2003) Signaling receptome: a genomic and evolutionary perspective of plasma membrane receptors involved in signal transduction. Sci STKE 2003: RE9. [DOI] [PubMed] [Google Scholar]
- Besmer P (1991) The kit ligand encoded at the murine Steel locus: a pleiotropic growth and differentiation factor. Curr Opin Cell Biol 3: 939–946 [DOI] [PubMed] [Google Scholar]
- Besmer P, Murphy JE, George PC, Qiu FH, Bergold PJ, Lederman L, Snyder HW Jr, Brodeur D, Zuckerman EE, Hardy WD (1986) A new acute transforming feline retrovirus and relationship of its oncogene v-kit with the protein kinase gene family. Nature 320: 415–421 [DOI] [PubMed] [Google Scholar]
- Blechman JM, Lev S, Barg J, Eisenstein M, Vaks B, Vogel Z, Givol D, Yarden Y (1995) The fourth immunoglobulin domain of the stem cell factor receptor couples ligand binding to signal transduction. Cell 80: 103–113 [DOI] [PubMed] [Google Scholar]
- Blechman JM, Lev S, Brizzi MF, Leitner O, Pegoraro L, Givol D, Yarden Y (1993) Soluble c-kit proteins and antireceptor monoclonal antibodies confine the binding site of the stem cell factor. J Biol Chem 268: 4399–4406 [PubMed] [Google Scholar]
- Bork P, Holm L, Sander C (1994) The immunoglobulin fold. Structural classification, sequence patterns and common core. J Mol Biol 242: 309–320 [DOI] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D 54: 905–921 [DOI] [PubMed] [Google Scholar]
- de Vos AM, Ultsch M, Kossiakoff AA (1992) Human growth hormone and extracellular domain of its receptor: crystal structure of the complex. Science 255: 306–312 [DOI] [PubMed] [Google Scholar]
- Fichelson S (1998) The FLT3/FLK2 ligand: structure, functions and prospects. Eur Cytokine Netw 9: 7–22 [PubMed] [Google Scholar]
- Fuh G, Li B, Crowley C, Cunningham B, Wells JA (1998) Requirements for binding and signaling of the kinase domain receptor for vascular endothelial growth factor. J Biol Chem 273: 11197–11204 [DOI] [PubMed] [Google Scholar]
- Graddis TJ, Brasel K, Friend D, Srinivasan S, Wee S, Lyman SD, March CJ, McGrew JT (1998) Structure-function analysis of FLT3 ligand-FLT3 receptor interactions using a rapid functional screen. J Biol Chem 273: 17626–17633 [DOI] [PubMed] [Google Scholar]
- Harpaz Y, Chothia C (1994) Many of the immunoglobulin superfamily domains in cell adhesion molecules and surface receptors belong to a new structural set which is close to that containing variable domains. J Mol Biol 238: 528–539 [DOI] [PubMed] [Google Scholar]
- Himanen JP, Rajashankar KR, Lackmann M, Cowan CA, Henkemeyer M, Nikolov DB (2001) Crystal structure of an Eph receptor-ephrin complex. Nature 414: 933–938 [DOI] [PubMed] [Google Scholar]
- Hsu YR, Chang WC, Mendiaz EA, Hara S, Chow DT, Mann MB, Langley KE, Lu HS (1998) Selective deamidation of recombinant human stem cell factor during in vitro aging: isolation and characterization of the aspartyl and isoaspartyl homodimers and heterodimers. Biochemistry 37: 2251–2262 [DOI] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, Schulten K (1996) VMD: visual molecular dynamics. J Mol Graph 14: 33–38, 27–38 [DOI] [PubMed] [Google Scholar]
- Jiang X, Gurel O, Mendiaz EA, Stearns GW, Clogston CL, Lu HS, Osslund TD, Syed RS, Langley KE, Hendrickson WA (2000) Structure of the active core of human stem cell factor and analysis of binding to its receptor kit. EMBO J 19: 3192–3203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M (1991) Improved methods for binding protein models in electron density maps and the location of errors in these models. Acta Crystallogr A 47: 110–119 [DOI] [PubMed] [Google Scholar]
- Kraulis PJ (1991) MOLSCRIPT. J Appl Crystallogr 24: 946–950 [Google Scholar]
- Krause DS, Van Etten RA (2005) Tyrosine kinases as targets for cancer therapy. N Engl J Med 353: 172–187 [DOI] [PubMed] [Google Scholar]
- Langley KE, Mendiaz EA, Liu N, Narhi LO, Zeni L, Parseghian CM, Clogston CL, Leslie I, Pope JA, Lu HS, Zsebo KM, Boone TC (1994) Properties of variant forms of human stem cell factor recombinantly expressed in Escherichia coli. Arch Biochem Biophys 311: 55–61 [DOI] [PubMed] [Google Scholar]
- Lemmon MA, Pinchasi D, Zhou M, Lax I, Schlessinger J (1997) Kit receptor dimerization is driven by bivalent binding of stem cell factor. J Biol Chem 272: 6311–6317 [DOI] [PubMed] [Google Scholar]
- Lennartsson J, Jelacic T, Linnekin D, Shivakrupa R (2005) Normal and oncogenic forms of the receptor tyrosine kinase kit. Stem Cells 23: 16–43 [DOI] [PubMed] [Google Scholar]
- Lev S, Blechman J, Nishikawa S, Givol D, Yarden Y (1993) Interspecies molecular chimeras of kit help define the binding site of the stem cell factor. Mol Cell Biol 13: 2224–2234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lev S, Yarden Y, Givol D (1992) Dimerization and activation of the kit receptor by monovalent and bivalent binding of the stem cell factor. J Biol Chem 267: 15970–15977 [PubMed] [Google Scholar]
- Lu HS, Clogston CL, Wypych J, Parker VP, Lee TD, Swiderek K, Baltera RF Jr, Patel AC, Chang DC, Brankow DW, Liu XD, Ogden SG, Karkare SB, Hu SS, Zsebo KM, Langley KE (1992) Post-translational processing of membrane-associated recombinant human stem cell factor expressed in Chinese hamster ovary cells. Arch Biochem Biophys 298: 150–158 [DOI] [PubMed] [Google Scholar]
- Matous JV, Langley K, Kaushansky K (1996) Structure–function relationships of stem cell factor: an analysis based on a series of human-murine stem cell factor chimera and the mapping of a neutralizing monoclonal antibody. Blood 88: 437–444 [PubMed] [Google Scholar]
- Mendiaz EA, Chang DG, Boone TC, Grant JR, Wypych J, Aguero B, Egrie JC, Langley KE (1996) Epitope mapping and immunoneutralization of recombinant human stem-cell factor. Eur J Biochem 239: 842–849 [DOI] [PubMed] [Google Scholar]
- Mohammadi M, Olsen SK, Ibrahimi OA (2005) Structural basis for fibroblast growth factor receptor activation. Cytokine Growth Factor Rev 16: 107–137 [DOI] [PubMed] [Google Scholar]
- Nicholls A, Sharp KA, Honig B (1991) Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins 11: 281–296 [DOI] [PubMed] [Google Scholar]
- Oefner C, D'Arcy A, Winkler FK, Eggimann B, Hosang M (1992) Crystal structure of human platelet-derived growth factor BB. EMBO J 11: 3921–3926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276: 307–326 [DOI] [PubMed] [Google Scholar]
- Pandit J, Bohm A, Jancarik J, Halenbeck R, Koths K, Kim SH (1992) Three-dimensional structure of dimeric human recombinant macrophage colony-stimulating factor. Science 258: 1358–1362 [DOI] [PubMed] [Google Scholar]
- Philo JS, Wen J, Wypych J, Schwartz MG, Mendiaz EA, Langley KE (1996) Human stem cell factor dimer forms a complex with two molecules of the extracellular domain of its receptor, Kit. J Biol Chem 271: 6895–6902 [DOI] [PubMed] [Google Scholar]
- Reilly JT (2003) Receptor tyrosine kinases in normal and malignant haematopoiesis. Blood Rev 17: 241–248 [DOI] [PubMed] [Google Scholar]
- Sasaki T, Knyazev PG, Clout NJ, Cheburkin Y, Gohring W, Ullrich A, Timpl R, Hohenester E (2006) Structural basis for Gas6-Axl signalling. EMBO J 25: 80–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savvides SN, Boone T, Andrew Karplus P (2000) Flt3 ligand structure and unexpected commonalities of helical bundles and cystine knots. Nat Struct Biol 7: 486–491 [DOI] [PubMed] [Google Scholar]
- Schlessinger J (2000) Cell signaling by receptor tyrosine kinases. Cell 103: 211–225 [DOI] [PubMed] [Google Scholar]
- Schlessinger J (2002) Ligand-induced, receptor-mediated dimerization and activation of EGF receptor. Cell 110: 669–672 [DOI] [PubMed] [Google Scholar]
- Sprang SR, Bazan JF (1993) Cytokine structural taxonomy and mechanisms of receptor engagement. Curr Opin Struct Biol 3: 815–827 [Google Scholar]
- Stroud RM, Wells JA (2004) Mechanistic diversity of cytokine receptor signaling across cell membranes. Sci STKE 2004: re7. [DOI] [PubMed] [Google Scholar]
- Sun L, Hui AM, Su Q, Vortmeyer A, Kotliarov Y, Pastorino S, Passaniti A, Menon J, Walling J, Bailey R, Rosenblum M, Mikkelsen T, Fine HA (2006) Neuronal and glioma-derived stem cell factor induces angiogenesis within the brain. Cancer Cell 9: 287–300 [DOI] [PubMed] [Google Scholar]
- Tallquist M, Kazlauskas A (2004) PDGF signaling in cells and mice. Cytokine Growth Factor Rev 15: 205–213 [DOI] [PubMed] [Google Scholar]
- Taylor EW, Fear AL, Bohm A, Kim SH, Koths K (1994) Structure-function studies on recombinant human macrophage colony-stimulating factor (M-CSF). J Biol Chem 269: 31171–31177 [PubMed] [Google Scholar]
- Terwilliger TC, Berendzen J (1999) Automated structure solution for MIR and MAD. Acta Crystallogr D 55: 849–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Rickert M, Garcia KC (2005) Structure of the quaternary complex of interleukin-2 with its alpha, beta, and gammac receptors. Science 310: 1159–1163 [DOI] [PubMed] [Google Scholar]
- Wang ZE, Myles GM, Brandt CS, Lioubin MN, Rohrschneider L (1993) Identification of the ligand-binding regions in the macrophage colony-stimulating factor receptor extracellular domain. Mol Cell Biol 13: 5348–5359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesmann C, Fuh G, Christinger HW, Eigenbrot C, Wells JA, de Vos AM (1997) Crystal structure at 1.7 Åresolution of VEGF in complex with domain 2 of the Flt-1 receptor. Cell 91: 695–704 [DOI] [PubMed] [Google Scholar]
- Wiesmann C, Ultsch MH, Bass SH, de Vos AM (1999) Crystal structure of nerve growth factor in complex with the ligand-binding domain of the TrkA receptor. Nature 401: 184–188 [DOI] [PubMed] [Google Scholar]
- Yarden Y, Kuang WJ, Yang-Feng T, Coussens L, Munemitsu S, Dull TJ, Chen E, Schlessinger J, Francke U, Ullrich A (1987) Human proto-oncogene c-kit: a new cell surface receptor tyrosine kinase for an unidentified ligand. EMBO J 6: 3341–3351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Zhang R, Joachimiak A, Schlessinger J, Kong XP (2000) Crystal structure of human stem cell factor: implication for stem cell factor receptor dimerization and activation. Proc Natl Acad Sci USA 97: 7732–7737 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures