

Abstract
Background
The influence of the membrane-bound AAA+ protease FtsH on membrane and cytoplasmic proteins of Corynebacterium glutamicum was investigated in this study. For the analysis of the membrane fraction, anion exchange chromatography was combined with SDS-PAGE, while the cytoplasmic protein fraction was studied by conventional two-dimensional gel electrophoresis.
Results
In contrast to the situation in other bacteria, deletion of C. glutamicum ftsH has no significant effect on growth in standard minimal medium or response to heat or osmotic stress. On the proteome level, deletion of the ftsH gene resulted in a strong increase of ten cytoplasmic and membrane proteins, namely biotin carboxylase/biotin carboxyl carrier protein (accBC), glyceraldehyde-3-phosphate dehydrogenase (gap), homocysteine methyltransferase (metE), malate synthase (aceB), isocitrate lyase (aceA), a conserved hypothetical protein (NCgl1985), succinate dehydrogenase A (sdhA), succinate dehydrogenase B (sdhB), succinate dehydrogenase CD (sdhCD), and glutamate binding protein (gluB), while 38 cytoplasmic and membrane-associated proteins showed a decreased abundance. The decreasing amount of succinate dehydrogenase A (sdhA) in the cytoplasmic fraction of the ftsH mutant compared to the wild type and its increasing abundance in the membrane fraction indicates that FtsH might be involved in the cleavage of a membrane anchor of this membrane-associated protein and by this changes its localization.
Conclusion
The data obtained hint to an involvement of C. glutamicum FtsH protease mainly in regulation of energy and carbon metabolism, while the protease is not involved in stress response, as found in other bacteria.
Background
Corynebacterium glutamicum, is a Gram-positive soil bacterium, which is used for the industrial production of different amino acids, mainly L-glutamate and L-lysine, and of nucleotides [1,2]. As a member of the Corynebacterinae, C. glutamicum is closely related to other mycolic acid-containing bacteria, e. g. to the amino acids producer Corynebacterium efficiens and to important pathogens such as Corynebacterium diphtheriae, Mycobacterium tuberculosis and Mycobacterium leprae [3]. Due to the enormous industrial importance of C. glutamicum, this bacterium is very well investigated. Its genome was sequenced and published independently by different industry-supported groups recently [4,5] and different global analyses techniques are available including transcriptome [6], metabolome [7], flux [8] and proteome analyses [9].
We are interested in nitrogen metabolism and nitrogen control in C. glutamicum (for review, see [10-12]) and recently identified proteolysis as a new regulatory mechanism in nitrogen regulation [13]. Different proteases, namely ClpXP and ClpCP [14] as well as FtsH are involved in the degradation of nitrogen signal transduction protein GlnK [13]. The identified enzymes are members of the AAA+ protease family. These proteases and protein disassembly machines are found in all kingdoms of life and often exhibit crucial regulatory functions (for recent reviews, see [15,16]).
In C. glutamicum, an effect of FtsH on the degradation of nitrogen signal control protein GlnK was reported [13]. The deletion of the ftsH gene is very well tolerated by C. glutamicum cells and obvious detrimental effects of an ftsH deletion could not be observed. Since we were interested in the function of this protease, we initiated a proteomic study and investigated the influence of an ftsH deletion on membrane and cytoplasmic protein profiles.
Results
Influence of FtsH on growth of C. glutamicum strains
Mutations of ftsH were described in different bacteria. The effect of these mutations are remarkably species-specific and range from drastic growth impairment in Escherichia coli [17] to effects on sporulation, development and stress response in Bacillus subtilis [18,19] and Caulobacter crescentus [20]. When growth of C. glutamicum wild type strains ATCC 13032 and strain ΔftsH was analyzed, only a minor effect of the ftsH deletion was observed (Fig. 1A). Doubling times of wild type and mutant strain were very similar in standard minimal medium (2 h 18 min versus 2 h 38 min). Next, the influence of increased temperature on growth was tested. Neither growth at increased temperature (37°C instead of 30°C) had a significant detrimental effect nor exposure of cells grown at 30°C to a sudden heat shock of 37 and 39°C, respectively (data not shown). Also when the ftsH mutant was exposed to osmotic stress applied either by growth in medium with increased osmolarity or as sudden osmotic shock due to sodium chloride addition, no significant growth defect compared to the wild type was detectable (S. Morbach and U. Meyer, personal communication). Obviously, FtsH plays a less crucial role in C. glutamicum compared to other bacteria. To identify FtsH targets in the cell, proteome studies were carried out.
Figure 1.
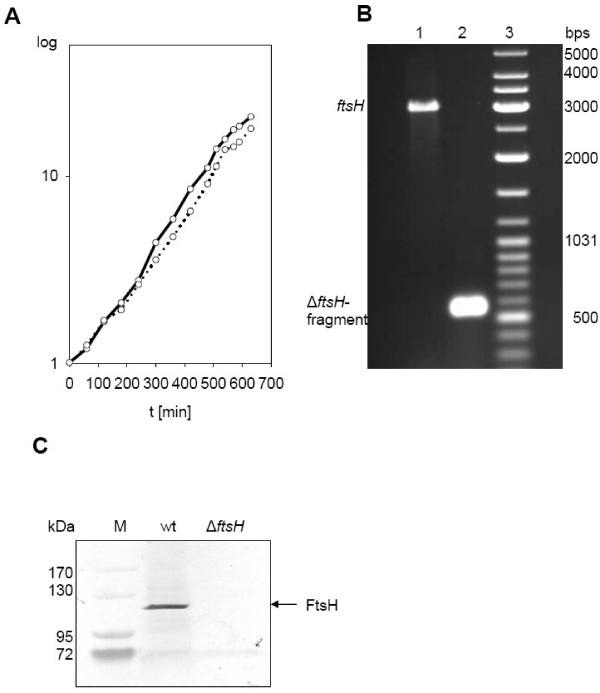
(A) Growth of the wild type strain ATCC 13032 (black line) and the deletion mutant ATCC 13032 ΔftsH (dotted line). Doubling times of the wild type and the mutant strain were very similar (2 h 18 min versus 2 h 38 min). (B) Control of ftsH deletion by PCR. Primers were designed to anneal approx. 214 bps up and down stream of ftsH gene (2562 bps). The PCR product comprised 2990 bps in the wild type strain (lane 1) and 530 bps in the deletion mutant (lane 2), lane 3 contains marker DNA. (C) Control of ftsH deletion by Western blotting. 25 μg of membrane protein of the wild type and ΔftsH were applied per lane. No signal was obtained with cytoplasmic proteins (data not shown).
Differences in the membrane proteome of wild type and ftsH deletion strain
FtsH is a membrane-bound AAA+ protease and therefore we started our investigations with an analysis of membrane proteins. While the separation of C. glutamicum membrane proteins by 2-D PAGE is restricted to those with two or less transmembrane helices [21,22], recently, a technique was established to separate highly hydrophobic proteins of the membrane fraction by anion exchange chromatography and 1-D SDS-PAGE [23]. This technique was applied for the comparison of membrane proteins from wild type and corresponding ftsH deletion strain.
The FtsH protease was identified for the wild type in a faint gel band in all three biological replicates, while this band was absent in the deletion strain (Figure 2). Compared to the wild type, five different proteins showed an increased abundance in the ftsH mutant strain (Table 1), namely all subunits of the succinate dehydrogenase complex (sdhA, sdhB and sdhCD), glutamate binding protein (gluB) and homocysteine methyltransferase (metE). Upregulation of protein concentration did not exceed a factor of five. While the succinate dehydrogenase complex could be a direct substrate of FtsH, GluB is a lipid-anchored glutamate binding protein [24], which is located at the outer face of the cytoplasmic membrane. Therefore, GluB must be an indirect target or processed by FtsH before secretion to the external site of the cell. C. glutamicum contains two homocysteine methyltransferares (metE and metH) catalyzing the final reaction of methionine synthesis, yet only metH requires vitamin B12 (cobalamin) as a cofactor [25]. Upregulation of metE could indicate that more of this enzyme is required for methionine synthesis, maybe due to reduced import of cobalamin in the ftsH deletion mutant, though further experiments are needed to verify this hypothesis. Interestingly, the ftsH deletion also influenced the abundance of ClpC, the ATPase component of the ClpCP protease complex. This protein was down-regulated in the mutant compared to the wild type by a factor of 0.4. However, the significance of this putative cross-talk between AAA+ proteases in C. glutamicum needs further investigation.
Figure 2.
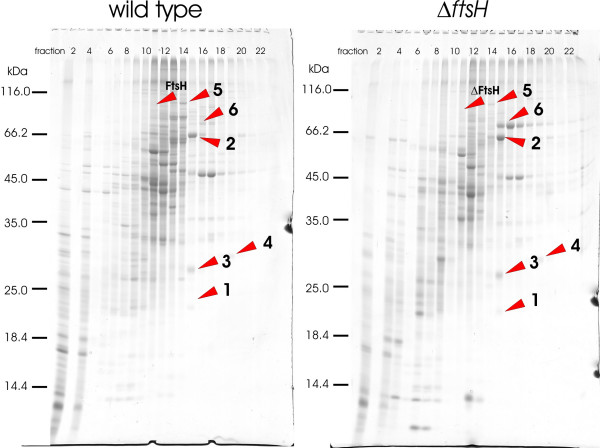
Coomassie-stained 1-D gels after ion exchange chromatography of wild type and ftsH deletion mutant membrane fraction. Protein spots which appear to be regulated are marked by red arrow heads and are listed in Table 1.
Table 1.
Protein pattern of the membrane fraction of the wild type ATCC13032 and ftsH deletion mutant.
| Spot # | NCgl # | Protein (Gene) | ΔftsH/wildtype ratio | S.D. | p-value |
| 1 | 0359 | succinate dehydrogenase CD (sdhCD) | 1.6 | 0.4 | 0.0804 |
| 2 | 0360 | succinate dehydrogenase A (sdhA) | 4.5 | 0.2 | 0.0003 |
| 3 | 0361 | succinate dehydrogenase B (sdhB) | 2.4 | 0.9 | 0.0916 |
| 4 | 1876 | glutamate binding protein (gluB) | 2.2 | 0.7 | 0.0734 |
| 5 | 2585 | ATP-dependent protease (clpC) | 0.4 | 0.03 | 0.0045 |
| 6 | 1094 | homocysteine methyltransferase (metE) | 4.9 | 1.4 | 0.0147 |
| FtsH | 2603 | cell division protein | - |
Additionally, a role of FtsH in the response to nitrogen starvation and to improving nitrogen conditions after a starvation period was tested. Compared to the wild type, NADH dehydrogenase (ndh), a putative integral membrane protein (cg0952) and the ATPase component of an ABC-type sugar transport system (msiK) were down-regulated by a factor of 0.5 (data not shown).
Comparison of cytoplasmic protein profiles
In addition to the membrane proteome also the cytoplasmic protein fraction of wild type and ftsH deletion was analyzed by two-dimensional gel electrophoresis (2-D PAGE, Figure 3). By this approach, six proteins were found in increasing amounts, namely the biotin carboxylase/biotin carrier protein (accBC), glyceraldehyde-3-phosphate dehydrogenase (gap), malate synthase (aceB), isocitrate lyase (aceA), a conserved hypothetical protein (NCgl1985) and homocysteine methyltransferase (metE), which was also identified as an upregulated protein in the membrane fraction. AccBC was upregulated more than 50-fold, while GAP-DH was upregulated by a factor of four. 37 different protein spots showed a decreased abundance in the mutant. Almost one third of the proteins identified (presented in Table 2) is clearly involved in carbon and energy metabolism. These include the maltooligosyl trehalose synthase (treY), the 1,4-alpha-glucan branching enzyme (glgB), fumarate hydratase (fum), a putative L-lactate dehydrogenase (lldA), glyceraldehyde-3-phosphate dehydrogenase (gap), phosphoenolpyruvate carboxylase (ppc), pyruvate dehydrogenase E1 component (aceE), an acyl-CoA synthetase (fadD4), succinate dehydrogenase A (sdhA) and transaldolase (tal). Interestingly, GAPDH is present in two spots, which differ in their approximate pI, an upregulated one (see above) spot number 2 in Figure 4 and a downregulated spot (number 30), indicating a posttranslational modification of the protein. Furthermore in the ftsH deletion strain succinate dehydrogenase A (sdhA) is less present in the cytoplasm but enriched in the membrane fraction. This indicates that FtsH is involved, either directly or indirectly, in the release of this succinate dehydrogenase subunit from the membrane into the cytoplasm. Since FtsH lacks a robust unfolding activity, a cleavage of SdhA and release of the protein from the complex by FtsH is rather unlikely. Other identified proteins with decreased abundance were parts of amino acid metabolism such as glutamine synthetase (glnA), aspartate-ammonium-lyase (aspA), succinyl-diaminopimelate desuccinylase (dapE), and dihydroxy-acid dehydratase (ilvD) (Fig. 4). As in the case of the membrane proteome, the influence of FtsH on the cytoplasmic protein profile in dependence of the nitrogen status of the cell was analyzed. For unknown reasons, 2-D gels of proteins isolated from nitrogen-starved ΔftsH cells revealed in contrast to the wild type (see also [26]) reproducibly strong horizontal streaking. The reason for this FtsH-specific effect, which made comparisons impossible, is unknown and could not be prevented by alternative gel loading techniques such as cup loading (C. Lück, personal communication).
Figure 3.
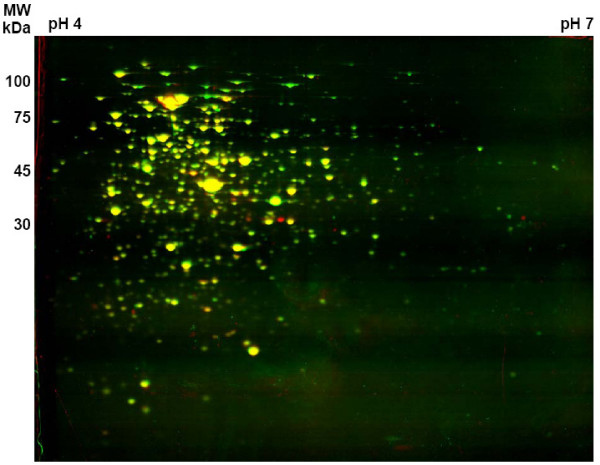
Comparison of cytoplasmic proteins of wild type strain ATCC 13032 and strain ΔftsH. Overlay of 2-D gels and false colour presentation: wild type proteins were stained in green, proteins of the deletion mutant in red. Spots present in both protein profiles appear in yellow. Molecular mass and pH range are indicated.
Table 2.
Cytoplasmic protein pattern of wild type strain ATCC13032 and ftsH deletion mutant. The listed proteins differ in their abundance of a factor of at least two.
| Spot # | NCgl # | Protein (Gene) | ΔftsH/wild type ratio | MW kDa | pI |
| 1 | 0670 | biotin carboxylase/biotin carboxyl carrier protein (accBC) | 53.48 | 63.5 | 5.02 |
| 2 | 1526 | glyceraldehyde-3-phosphate dehydrogenase (gap) | 4.24 | 36.2 | 5.16 |
| 3 | 2247 | malate synthase (aceB) | 2.95 | 82.5 | 5.0 |
| 4 | 2248 | isocitrate lyase (aceA) | 2.65 | 47.2 | 4.92 |
| 5 | 1985 | conserved hypothetical protein | 2.31 | 122.8 | 4.85 |
| 6 | 1094 | homocysteine methyltransferase (metE) | 2.16 | 81.3 | 4.78 |
| 7 | 2037 | maltooligosyl trehalose synthase (treY) | 0.48 | 90.5 | 5.03 |
| 8 | 1177 | 1,4-alpha-glucan branching enzyme (glgB) | 0.48 | 82.6 | 4.99 |
| 9 | 1023 | putative nicotinate-nucleotide pyrophosphorylase | 0.47 | 29.4 | 5.22 |
| 10 | 2431 | nicotinic acid phosphoribosyltransferase | 0.47 | 48.0 | 5.22 |
| 11 | 0187 | L-gulonolactone oxidase | 0.47 | 53.0 | 5.68 |
| 12 | 0578 | inositol-monophosphate dehydrogenase (guaB2) | 0.47 | 53.4 | 5.99 |
| 13 | 0094 | AMP nucleosidase (amn) | 0.46 | 53.7 | 5.23 |
| 14 | 0358 | transcriptional regulator, MerR family (ramB) | 0.46 | 53.9 | 6.29 |
| 15 | 0704 | putative DNA helicase | 0.46 | 84.1 | 5.35 |
| 16 | 2718 | sulfite reductase (hemoprotein) (cysI) | 0.43 | 63.0 | 5.53 |
| 17 | 0251 | catalase (katA) | 0.42 | 58.7 | 5.18 |
| 18 | 0200 | quinone oxidoreductase | 0.41 | 33.2 | 4.99 |
| 19 | 2133 | glutamine synthetase (glnA) | 0.41 | 53.3 | 4.90 |
| 20 | 0471 | DNA-directed RNA polymerase beta chain (rpoB) | 0.41 | 128.8 | 4.86 |
| 21 | 1446 | aspartate ammonia-lyase (aspartase) (aspA) | 0.4 | 57.6 | 5.69 |
| 22 | 1440 | ATPases of the AAA+ class | 0.4 | 58 | 4.91 |
| 23 | 1835 | polyphosphate glucokinase (ppgK) | 0.4 | 26.7 | 4.97 |
| 24 | 0371 | probable formyltetrahydrofolate deformylase protein (purU) | 0.39 | 34.3 | 5.68 |
| 25 | 2986 | N-acetymuramyl-L-alanine amidase (cwlM) | 0.38 | 44.5 | 5.63 |
| 26 | 0967 | fumarate hydratase (fum) | 0.37 | 49.8 | 5.06 |
| 27 | 1442 | aspartyl aminopeptidase (pepC) | 0.36 | 44.9 | 5.10 |
| 28 | 2817 | putative L-lactate dehydrogenase (lldA) | 0.34 | 45.7 | 5.72 |
| 29 | 2126 | dihydrolipoamide succinyltransferase (sucB) | 0.34 | 70.9 | 4.26 |
| 30 | 1526 | glyceraldehyde-3-phosphate dehydrogenase (gap) | 0.34 | 36.0 | 5.16 |
| 31 | 1523 | phosphoenolpyruvate carboxylase (ppc) | 0.33 | 103.2 | 4.92 |
| 32 | 0251 | catalase (katA) | 0.29 | 58.7 | 5.18 |
| 33 | 1064 | succinyl-diaminopimelate desuccinylase (dapE) | 0.29 | 40.0 | 4.84 |
| 34 | 2586 | inositol-monophosphate dehydrogenase (guaB1) | 0.28 | 50.8 | 6.39 |
| 35 | 2487 | GCN5-related N-acetyltransferase | 0.27 | 32.1 | 5.86 |
| 36 | 2167 | pyruvate dehydrogenase E1 component (aceE) | 0.27 | 102.8 | 5.26 |
| 37 | 1151 | acyl-CoA synthetase (fadD4) | 0.26 | 63.7 | 5.08 |
| 38 | 0360 | succinate dehydrogenase A (sdhA) | 0.25 | 74.7 | 5.37 |
| 39 | 0570 | predicted carbohydrate kinase | 0.19 | 60.0 | 5.08 |
| 40 | 0707 | superfamily II DNA/RNA helicase, SNF2 family | 0.16 | 106.9 | 5.65 |
| 41 | 1219 | dihydroxy-acid dehydratase (ilvD) | 0.16 | 64.2 | 5.18 |
| 42 | 1513 | transaldolase (tal) | 0.15 | 38.3 | 4.47 |
| 43 | 2602 | GTP cyclohydrolase (folE) | 0.08 | 22.0 | 6.08 |
Figure 4.
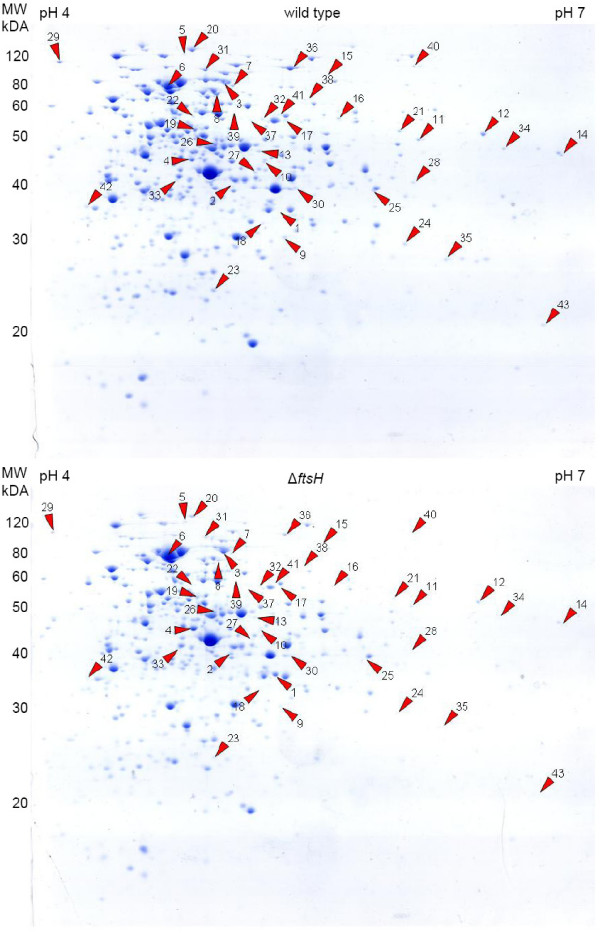
Coomassie-stained 2-D gels of wild type ATCC 13032 and corresponding ftsH deletion mutant. Protein spots which appear to be regulated are marked by red arrow heads and are listed in Table 2. Molecular mass and pH range are indicated.
Discussion
Data which hint to an involvement of FtsH in GlnK signal transduction protein degradation [13] prompted us to investigate the influence of this AAA+ protease on membrane and cytoplasmic protein profiles in C. glutamicum. Using a combination of anion exchange chromatography and SDS-PAGE for membrane protein analysis and 2-D PAGE for cytoplasmic proteins, we were able to show that FtsH regulates only a few proteins under the growth conditions tested. However, since the applied method only covers about 10% of the C. glutamicum membrane proteome [23], some FtsH targets may have been missed due to technical limitations. For example, the FtsH target GlnK is degraded depending on FtsH but proteolysis is also influenced by ClpCP and ClpXP [13]. In contrast to the situation in E. coli (for recent review, see [27]), we found that deletion of the ftsH gene is tolerated by C. glutamicum cells very well, although this gene is conserved in all other corynebacterial genome sequences published so far, i. e. in the C. diphtheriae [28], C. efficiens [29] and Corynebacterium jeikeium [30] genome, and although no obvious paralog of the ftsH gene is encoded in the C. glutamicum genome. Obvious detrimental effects of an ftsH deletion were not observed. In this respect the C. glutamicum results resemble the situation in B. subtilis and C. crescentus. Also for these organisms, a less severe effect of ftsH mutation compared to an E. coli mutant was shown. In B. subtilis, FtsH is involved in sporulation, stress adaptation and protein secretion [18,19], and the effect of its deletion on the cytosolic proteome has been studied [31]. FtsH deletion resulted in increased levels of an arginase, a protein similar to a quinone oxidoreductase, and penicillin binding protein, but for the latter direct proteolytic action could be excluded and for the other two proteins it was not verified. FtsH of M. tuberculosis, which is phylogenetically closely related to C. glutamicum, was heterologously expressed in E. coli, and proteolytic activity against the known E. coli substrates heat shock transcription factor σ32 protein, protein translocation subunit SecY, and bacteriophage λCII repressor protein was observed [32]. For M. tuberculosis no experimental verification exists if SecY is indeed a target of FtsH, and our data for C. glutamicumdoes not support this hypothesis, but it does not completely rule this out, too. For C. crescentus an involvement of FtsH in development, stress response and heat shock control was shown [20]. The ftsH gene is expressed transiently after temperature upshift and in stationary phase in this organism, while during normal growth conditions FtsH is dispensable. In C. crescentus a mutation of ftsH influences chaperones, DnaK is derepressed under normal temperature compared to the wild type, while an influence on GroEL abundance was not observed. In contrast, the ftsH deletion in C. glutamicum had no influence on DnaK and even less GroEL was observed compared to the wild type. Further differences besides chaperone activation are sporulation and cell cycle proteins, processes which are absent in C. glutamicum. The majority of proteins identified to be differentially synthesized in dependence of FtsH C. glutamicum seem to be involved in carbon and energy metabolism.
Conclusion
The data obtained in this study, indicate that C. glutamicum AAA+ metalloprotease FtsH is not involved in the cellular response to heat or osmotic stress as shown in other bacteria. An astonishingly small amount of membrane and cytoplasmic proteins is affected by an ftsH deletion. From these data an involvement of FtsH in regulation of energy and carbon metabolism as well as in amino acid biosynthesis is indicated.
Methods
Strains and growth conditions
C. glutamicum type strain ATCC 13032 [33] and ftsH deletion mutant [13] were routinely grown on a rotary shaker at 30°C. A fresh C. glutamicum culture in BHI medium was used to inoculate minimal medium (per litre 42 g MOPS, 20 g (NH4)2SO4, 5 g urea, 0.5 g K2HPO4 × 3 H2O, 0.5 g KH2PO4, 0.25 g MgSO4 × 7 H2O, 0.01 g CaCl2, 50 g glucose, 0.2 mg biotin, 10 mg FeSO4, 10 mg MnSO4, 1 mg ZnSO4, 0.2 mg CuSO4, 0.02 mg NiCl2 × 6 H2O, 0.09 mg H3BO3, 0.06 mg CoCl2 × 6 H2O, 0.009 mg NaMoO4 × 2 H2O; pH adjusted to pH 7.0 using NaOH; [34]) for overnight growth. This culture, with an overnight OD600 of approximately 25 to 30, was used to inoculate fresh minimal medium to an OD600 of approximately 1, and cells were grown for 4 to 6 hours until the exponential growth phase was reached (OD600 approximately 4–5). To induce nitrogen starvation, cells were harvested by centrifugation and the pellet was suspended in and transferred to pre-warmed minimal medium without nitrogen source. The nitrogen-deprived cells were incubated at 30°C under aeration.
Polymerase chain reaction
To verify the deletion of the ftsH gene, PCR experiments were carried out. Primers were designed to anneal approx. 214 bps up and down stream of ftsH gene (2562 bps) (ftsH+200up fw: 5'-GTG GGC TAC GGA CTT GAT TTC G-3'; ftsH+200down rv: 5'-GAA CCA ACT CTT CAT GGC CCT C-3'). Chromosomal DNA prepared by phenol-chloroform extraction was used as template. PCR was performed using Taq polymerase and the following program: 95°C 5 min; 30 cycles (95°C 30 s; 64°C 30 s; 72°C 3 min) followed by 72°C 10 min and cooling down to 4°C. PCR products were analyzed by agarose gel electrophoresis [35].
SDS-PAGE and Western blotting
To demonstrate deletion of ftsH on protein level, cells were disrupted band fractionated as described below or 2-D PAGE. Cytoplasmic proteins and membrane fraction of the wild type ATCC13032 and the deletion strain were separated by Tricine-buffered 9.5% SDS gels as described [36]. After SDS-PAGE proteins were transferred onto a polyvinylidene difluoride membrane (PVDF, Immobilon-P, pore size 0.45 μm, Millipore, Bedford, MA, USA) by semi-dry electroblotting. Immunodetection of FtsH was performed with antibodies against peptide fragments of E. coli FtsH, produced in rabbit. Antibody binding was visualised by using appropriate anti-antibodies coupled to alkaline phosphatase (Sigma-Aldrich, Traufkirchen, Germany) and the BCIP/NBT alkaline phosphatase substrate (Sigma-Aldrich, Traufkirchen, Germany).
Membrane proteomics
For analysis of membrane proteins, a combination of anion exchange chromatography and SDS-PAGE was applied as described previously [23]. For this method, cells were disrupted by French Press treatment; the membrane fraction was separated from cell debris and cytoplasm by differential (ultra)centrifugation and washed with 2.5 M NaBr to remove membrane-associated proteins. Membrane proteins were subsequently solubilised in buffer containing 2% ASB-14 and separated by anion exchange chromatography. After TCA precipitation and SDS-PAGE, gels were scanned and analyzed using the LabScan software package (Amersham Biosciences, Freiburg, Germany). The scanner was calibrated with a greyscale marker (Kodak) and the same settings applied for all gels. Scanning was carried out at 300 dpi and 8 bit greyscale. Gel bands were quantified relative to each other by densitometry using the software scion image (version beta 4.0.2; Scion Corporation). Proteins from three independent experiments (biological replicates) were regarded as regulated if a p-value < 0.1 was calculated for a Student's t-test (paired, two-tailed).
2-D PAGE, staining and protein analysis
For 2-D PAGE analyses C. glutamicum cells were disrupted using glass beads and a Q-BIOgene FastPrep FP120 instrument (Q-BIOgene, Heidelberg, Germany) by lyzing the cells four times for 30 sec and 6.5 m sec-1 in the presence of the proteinase inhibitor Complete as recommended by the supplier (Roche, Basel, Switzerland). Proteins were separated by ultracentrifugation in cytoplasmic and membrane-associated protein fractions [37,21]. For classical 2-D PAGE, only the cytoplasmic proteins were analyzed further. Protein concentrations were determined according to Dulley and Grieve [38]. For isoelectric focusing (IEF) 24 cm pre-cast IPG strips pI 4–7 and an IPGphor IEF unit (Amersham Biosiences, Freiburg, Germany) were used as described [39]. 100 μg and 200 μg of protein were loaded by rehydration for 24 h in a sample buffer containing 6 M urea, 2 M thiourea, 4% CHAPS, 0.5% Pharmalyte (3–10) and 0.4% DTT. The isoelectric focusing was performed for 48 000 Vh. The run for the second dimension was carried out using 12.5% polyacrylamide gels and an Ettan Dalt II system (Amersham Biosiences, Freiburg, Germany). After electrophoresis 2-D gels were stained with Coomassie brilliant Blue [35]. The Coomassie-stained gels were aligned using the Delta2D software, version 3.3 (Decodon, Greifswald, Germany). All samples were separated at least twice by 2-D PAGE to minimize irregularities (technical replicates). To validate the results, each comparison of interest was performed using samples from at least three independent experiments (biological replicates). The Delta2D software (version 3.3) was also used for spot quantification. Proteins were regarded as regulated if (i) the corresponding ratios referring to the relative volumes of the spots changed more than two-fold and if (ii) this regulation pattern was found in all biological and technical replicates. All other proteins were classified as "not regulated". Pearson coefficients for wild type gels were higher than 0.9962, for ftsH gels 0.9929, and for the comparisons of wild type and ftsH mutant 0.9510.
Protein identification
Protein spots or bands with significantly altered abundance in the ftsH mutant compared to the wild type were analyzed by trypric in-gel digest and MALDI-ToF-MS as described earlier [26].
Authors' contributions
AL carried out growth experiments and 2-D PAGE, RK supported the project by discussions, AB supervised the experiments and was responsible for the draft of the manuscript, DS analyzed the membrane proteome and was supervised by AP. AP additionally wrote the final version of the manuscript. All authors read and approved the final manuscript.
Acknowledgments
Acknowledgements
FtsH-specific antibodies were kindly provided by Teru Ogura (Kumamoto University, Japan). The authors wish to thank C. Lück (Technical University Munich), U. Meyer and S. Morbach (University of Cologne) for providing unpublished data. The financial support of the Deutsche Forschungsgemeinschaft (Sonderforschungsbereich 635, TP17) and the Bundesministerium für Bildung und Forschung (Neue Methoden zur Proteomanalyse: Anwendung und Verknüpfung mit Metabolomanalysen am Beispiel von Corynebacterium glutamicum) is gratefully acknowledged.
Contributor Information
Alja Lüdke, Email: alja.luedke@uni-koeln.de.
Reinhard Krämer, Email: r.kraemer@uni-koeln.de.
Andreas Burkovski, Email: aburkov@biologie.uni-erlangen.de.
Daniela Schluesener, Email: daniela.schluesener@rub.de.
Ansgar Poetsch, Email: ansgar.poetsch@rub.de.
References
- Hermann T. Industrial production of amino acids by coryneform bacteria. J Biotechnol. 2003;104:155–172. doi: 10.1016/S0168-1656(03)00149-4. [DOI] [PubMed] [Google Scholar]
- Leuchtenberger W. Amino acids – technical production and use. In: Rehm H, Reed G, editor. Products of primary metabolism Biotechnology. Vol. 6. VCH Verlagsgesellschaft, Weinheim, Germany; 1996. pp. 465–502. [Google Scholar]
- Stackebrandt E, Rainey FA, Ward-Rainey NL. Proposal for a new hierachic classification system, Actinobacteria classis nov. Int J System Bacteriol. 1997;47:97–102. [Google Scholar]
- Ikeda M, Nakagawa S. The Corynebacterium glutamicum genome: features and impacts on biotechnological processes. Appl Microbiol Biotechnol. 2003;62:99–109. doi: 10.1007/s00253-003-1328-1. [DOI] [PubMed] [Google Scholar]
- Kalinowski J, Bathe B, Bischoff N, Bott M, Burkovski A, Dusch N, Eggeling L, Eikmanns BJ, Gaigalat L, Goesmann A, Hartmann M, Huthmacher K, Krämer R, Linke B, McHardy AC, Meyer F, Möckel B, Pfefferle W, Pühler A, Rey D, Rückert C, Sahm H, Wendisch VF, Wiegräbe I, Tauch A. The complete Corynebacterium glutamicum ATCC 13032 genome sequence and its impact on the production of L-aspartate-derived amino acids and vitamins. J Biotechnol. 2003;104:5–25. doi: 10.1016/S0168-1656(03)00154-8. [DOI] [PubMed] [Google Scholar]
- Wendisch VF. Genome-wide expression analysis in Corynebacterium glutamicum using DNA microarrays. J Biotechnol. 2003;104:273–285. doi: 10.1016/S0168-1656(03)00147-0. [DOI] [PubMed] [Google Scholar]
- Strelkov S, von Elstermann M, Schomburg D. Comprehensive analysis of metabolites in Corynebacterium glutamicum by gas chromatography/mass spectrometry. Biol Chem. 2004;385:853–861. doi: 10.1515/BC.2004.111. [DOI] [PubMed] [Google Scholar]
- Wittmann C, de Graaf AA. Metabolic flux analysis in Corynebacterium glutamicum. In: Bott M, Eggeling L, editor. Handbook of Corynebacterium glutamicum. CRC Press LLC, Boca Raton, FL; 2005. pp. 277–304. [Google Scholar]
- Schaffer S, Burkovski A. Genome-based approaches: proteomics. In: Bott M, Eggeling L, editor. Handbook of Corynebacterium glutamicum. CRC Press LLC, Boca Raton, FL; 2005. pp. 99–118. [Google Scholar]
- Burkovski A. Ammonium assimilation and nitrogen control in Corynebacterium glutamicum and its relatives: an example for new regulatory mechanisms in actinomycetes. FEMS Microbiol Rev. 2003;27:617–628. doi: 10.1016/S0168-6445(03)00067-6. [DOI] [PubMed] [Google Scholar]
- Burkovski A. I do it my way: Regulation of ammonium uptake and ammonium assimilation in Corynebacterium glutamicum. Arch Microbiol. 2003;179:83–88. doi: 10.1007/s00203-002-0505-4. [DOI] [PubMed] [Google Scholar]
- Burkovski A. Nitrogen metabolism and its regulation. In: Bott M, Eggeling L, editor. Handbook of Corynebacterium glutamicum. CRC Press LLC, Boca Raton, FL; 2005. pp. 333–349. [Google Scholar]
- Strösser J, Lüdke A, Schaffer S, Krämer R, Burkovski A. Regulation of GlnK activity: modification, membrane sequestration, and proteolysis as regulatory principles in the network of nitrogen control in Corynebacterium glutamicum. Mol Microbiol. 2004;54:132–147. doi: 10.1111/j.1365-2958.2004.04247.x. [DOI] [PubMed] [Google Scholar]
- Engels S, Schweitzer J, Ludwig C, Bott M, Schaffer S. clpC and clpP1P2 gene expression in Corynebacterium glutamicum is controlled by a regulatory network involving the transcriptional regulators ClgR and HspR as well as the ECF sigma factor σH. Mol Microbiol. 2004;52:285–302. doi: 10.1111/j.1365-2958.2003.03979.x. [DOI] [PubMed] [Google Scholar]
- Gottesman S. Proteolysis in bacterial regulatory circuits. Annu Rev Cell Dev Biol. 2003;19:565–587. doi: 10.1146/annurev.cellbio.19.110701.153228. [DOI] [PubMed] [Google Scholar]
- Sauer RT, Bolon DN, Burton BM, Burton RE, Flynn JM, Grant RA, Hersch GL, Joshi SA, Kenniston JA, Levchenko I, Neher SB, Oakes ESC, Siddiqui SM, Wah DA, Baker TA. Sculpting the proteome with AAA+ proteases and disassembly machines. Cell. 2004;119:9–18. doi: 10.1016/j.cell.2004.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begg KJ, Tomoyasu T, Donachie WD, Khattar M, Niki H, Yamanaka K, Hiraga S, Ogura T. Escherichia coli mutantY16 is a double mutant carrying thermosensitive ftsH and ftsI mutations. J Bacteriol. 1992;174:2516–2417. doi: 10.1128/jb.174.7.2416-2417.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deuerling E, Mogk A, Richter C, Purucker M, Schumann W. The ftsH gene of Bacillus subtilis is involved in major cellular processes such as sporulation, stress adaptation and secretion. Mol Microbiol. 1997;23:921–933. doi: 10.1046/j.1365-2958.1997.2721636.x. [DOI] [PubMed] [Google Scholar]
- Lysenko E, Ogura T, Cutting SM. Characterization of the ftsH gene of Bacillus subtilis. Microbiology. 1997;143:971–978. doi: 10.1099/00221287-143-3-971. [DOI] [PubMed] [Google Scholar]
- Fischer B, Rummel G, Aldrige P, Jenal U. The FtsH protease is involved in development, stress response and heat shock control in Caulobacter crescentus. Mol Microbiol. 2002;44:461–478. doi: 10.1046/j.1365-2958.2002.02887.x. [DOI] [PubMed] [Google Scholar]
- Hermann T, Finkemeier M, Pfefferle W, Wersch G, Krämer R, Burkovski A. Two-dimensional electrophoretic analysis of Corynebacterium glutamicum membrane fraction and surface proteins. Electrophoresis. 2000;21:654–659. doi: 10.1002/(SICI)1522-2683(20000201)21:3<654::AID-ELPS654>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Schaffer S, Weil B, Nguyen VD, Dongmann G, Günther K, Nickolaus M, Hermann T, Bott M. A high-resolution reference map for cytoplasmatic and membrane proteins of Corynebacterium glutamicum. Electrophoresis. 2001;22:4404–4422. doi: 10.1002/1522-2683(200112)22:20<4404::AID-ELPS4404>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Schluesener D, Fischer F, Kruip J, Rögner M, Poetsch A. Mapping the membrane proteome of Corynebacterium glutamicum. Proteomics. 2005;5:1317–1330. doi: 10.1002/pmic.200400993. [DOI] [PubMed] [Google Scholar]
- Kronemeyer W, Peekhaus N, Krämer R, Sahm H, Eggeling L. Structure of the gluABCD cluster encoding the glutamate uptake system of Corynebacterium glutamicum. J Bacteriol. 1995;177:1152–1158. doi: 10.1128/jb.177.5.1152-1158.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rückert C, Pühler A, Kalinowski J. Genome-wide analysis of the L-methionine biosynthetic pathway in Corynebacterium glutamicum by targeted gene deletion and homologous complementation. J Biotechnol. 2003;104:213–228. doi: 10.1016/S0168-1656(03)00158-5. [DOI] [PubMed] [Google Scholar]
- Silberbach M, Schäfer M, Hüser A, Kalinowski J, Pühler A, Krämer R, Burkovski A. Adaptation of Corynebacterium glutamicum to ammonium-limitation: a global analysis using transcriptome and proteome techniques. Appl Environ Microbiol. 2005;71:2391–2402. doi: 10.1128/AEM.71.5.2391-2402.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Akiyama Y. Cellular functions, mechanism of action, and regulation of FtsH protease. Annu Rev Microbiol. 2005;59:211–231. doi: 10.1146/annurev.micro.59.030804.121316. [DOI] [PubMed] [Google Scholar]
- Cerdeno-Tarraga AM, Efstratiou A, Dover LG, Holden MTG, Pallen M, Bentley SD, Besra GS, Churcher C, James KD, De Zoysa A, Chillingworth T, Cronin A, Dowd L, Feltwell T, Hamlin N, Holroyd S, Jagels K, Moule S, Quail MA, Rabbinowitch E, Rutherford KM, Thomson NR, Unwin L, Whitehead S, Barrell BG, Parkhill J. The complete genome sequence and analysis of Corynebacterium diphtheriae NCTC13129. Nucleic Acids Res. 2003;31:6516–6523. doi: 10.1093/nar/gkg874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fudou R, Jojima Y, Seto A, Yamada K, Rimura E, Nakamatsu T, Hirashi A, Yamanaka S. Corynebacterium efficiens sp. Nov., a glutamic-acid-producing species from soil and plant material. Int J Syst Evol Microbiol. 2002;52:1127–1131. doi: 10.1099/ijs.0.02086-0. [DOI] [PubMed] [Google Scholar]
- Tauch A, Kaiser O, Hain T, Goesmann A, Weisshaar B, Albersmeier A, Bekel T, Bischoff N, Brune I, Chakraborty T, Kalinowski J, Meyer F, Rupp O, Schneiker S, Viehoever P, Pühler A. Complete genome sequence and analysis of the multiresistant nosocomial pathogen Corynebacterium jeikeium K411, a lipid-requiring bacterium of the human skin flora. J Bacteriol. 2005;187:4671–4682. doi: 10.1128/JB.187.13.4671-4682.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zellmeier S, Zuber U, Schumann W, Wiegert T. The absence of FtsH metalloprotease activity causes overexpression of the sigmaW-controlled pbpE gene, resulting in filamentous growth of Bacillus subtilis. J Bacteriol. 2003;185:973–982. doi: 10.1128/JB.185.3.973-982.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan R, Anilkumar G, Rajeswari H, Ajitkumar P. Functional characterization of AAA family FtsH protease of Mycobacterium tuberculosis. FEMS Microbiol Lett. 2006;259:97–105. doi: 10.1111/j.1574-6968.2006.00251.x. [DOI] [PubMed] [Google Scholar]
- Abe S, Takayama K, Kinoshita S. Taxonomical studies on glutamic acid-producing bacteria. J Gen Microbiol. 1967;13:279–301. [Google Scholar]
- Keilhauer C, Eggeling L, Sahm H. Isoleucine synthesis in Corynebacterium glutamicum : molecular analysis of the ilvB-ilvN-ilvC operon. J Bacteriol. 1993;175:5595–5603. doi: 10.1128/jb.175.17.5595-5603.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2. Cold Spring Habor Laboratory Press, Cold Spring Habor, N.Y; 1989. [Google Scholar]
- Schägger H, von Jagow G. Tricine-sodium dodecyl sulfate-polyacrylamide gel eletrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal Biochem. 1987;166:368–379. doi: 10.1016/0003-2697(87)90587-2. [DOI] [PubMed] [Google Scholar]
- Hermann T, Wersch G, Uhlemann E-M, Schmid R, Burkovski A. Mapping and identification of Corynebacterium glutamicum proteins by two-dimensional gel electrophoresis and microsequencing. Electrophoresis. 1998;19:3217–3221. doi: 10.1002/elps.1150191827. [DOI] [PubMed] [Google Scholar]
- Dulley JR, Grieve PA. A simple technique for eliminating interference by detergent in the Lowry method of protein determination. Anal Biochem. 1975;64:136–141. doi: 10.1016/0003-2697(75)90415-7. [DOI] [PubMed] [Google Scholar]
- Büttner K, Bernhardt J, Scharf C, Schmid R, Mäder U, Eymann C, Antelmann H, Völker A, Völker U, Hecker M. A comprehensive two-dimensional map of cytosolic proteins of Bacillus subtilis. Electrophoresis. 2001;22:2908–2935. doi: 10.1002/1522-2683(200108)22:14<2908::AID-ELPS2908>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]