

Abstract
New onset thrombocytopenia and multiple organ failure (TAMOF) presages poor outcome in critical illness. Patients who resolve thrombocytopenia by day 14 are more likely to survive than those who do not. Patients with TAMOF have a spectrum of microangiopathic disorders that includes thrombotic thrombocytopenic purpura (TTP), disseminated intravascular coagulation (DIC) and secondary thrombotic microanigiopathy (TMA). Activated protein C is effective in resolving fibrin-mediated thrombosis (DIC); however, daily plasma exchange is the therapy of choice for removing ADAMTS 13 inhibitors and replenishing ADAMTS 13 activity which in turn resolves platelet: von Willebrand Factor mediated thrombosis (TTP/secondary TMA).
Thrombocytopenia-associated multiple organ failure: what is it?
New onset thrombocytopenia in the critically ill patient has been established as an important independent risk factor for the development of multiple organ failure. Intensive care unit non-survivors commonly have thrombocytopenia out to 14 days whereas survivors do not [1-8]. It has long been established that thrombocytopenia at admission to the intensive care unit is a risk factor for mortality; however, this observation supports the concept that ongoing thrombocytopenia over time can be associated with pathological consequences similar to, for example, ongoing hypotension over time.
Laboratory and clinical studies have now confirmed that thrombocytopenia-associated multiple organ failure (TAMOF) is a thrombotic microangiopathic syndrome that can be defined by a spectrum of pathology that includes thrombotic thrombocytopenic purpura (TTP), secondary thrombotic microangiopathy (TMA), and disseminated intravascular coagulation (DIC). All three of these pathophysiological states have been reported in critically ill patients who developed endotheliopathy caused by exposure to cardiopulmonary bypass, infection, transplantation, radiation, chemotherapy, auto-immune disease, and transplantation medications. The preponderance of clinical evidence to date suggests that the use of plasma exchange for TTP and secondary TMA, and anticoagulant protein therapies, such as activated protein C, for DIC results in reversal of TAMOF and improved survival [9-51].
Understanding pathological coagulation and systemic endotheliopathy
Pro-thrombotic and anti-fibrinolytic responses, which are helpful during focal injury, may be injurious in the setting of systemic endothelial injury and are manifested by thrombocytopenia, systemic thrombosis, and multiple organ failure. Critically ill patients develop systemic endothelial microangiopathic disease after many types of systemic insults (Table 1). The pathophysiology of these thrombotic microangiopathies caused by systemic endothelial inury can be characterized as part of a spectrum of three phenotypes, TTP (Figure 1), consumptive DIC (Figure 2), and non-consumptive secondary TMA (Figure 3) [30-34].
Table 1.
Conditions associated with thrombocytopenia-associated multiple organ failure
| Cancer |
| Transplantation |
| Cardiovascular surgery/cardiopulmonary bypass |
| Autoimmune disease |
| Systemic infection |
| Vasculitis |
| Toxins |
| Cyclosporine A |
| FK 506 |
| Chemotherapy |
| Radiation |
| Ticlopidine |
| Hemolytic Uremic Syndrome variant syndromes |
Figure 1.
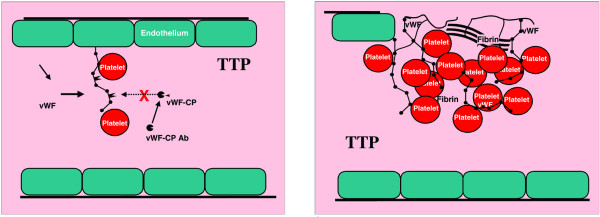
Systemic inflammation results in systemic coagulation. Thrombotic thrombocytopenuc purpura (TTP) is a microangiopathy phenotype characterized by ADAMTS 13 deficiency. Left: Platelets attach to ultra large vWF multimers. Because vWF-CP (ADAMTS 13) is inhibited this leads to massive vWF:platelet thrombosis (right). Ab, antibody; CP, cleaving protease; vWF, von Willebrand factor.
Figure 2.
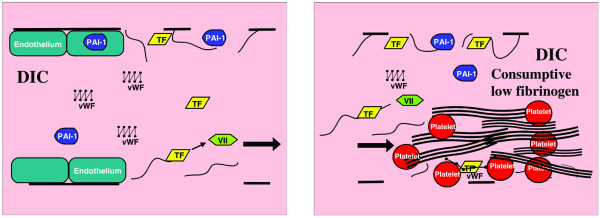
Disseminated intravascular coagulation (DIC) is a microangiopathy phenotype characterized by increased tissue factor (TF) and plasminogen activator inhibitor type I (PAI-1), unopposed by the anticoagulant proteins TFPI, protein C, antithrombin III, and prostacyclin. The severest forms also have an ADAMTS 13 deficiency. Tissue factor activates factor VII (left), leading to massive consumptive fibrin thrombosis (right). VII, factor VII; vWF, von Willebrand factor.
Figure 3.
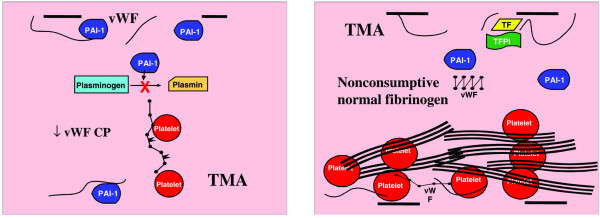
Secondary thrombotic microangiopathy (TMA) has a phenotype characterized by decreased ADAMTS 13, and increased plasminogen activator inhibitor type I (PAI-1) and von Willebrand factor (vWF) levels with normal or high fibrinogen levels. Platelets attach to increased large vWF multimers and form thrombi in the presence of decreased PAI-I activity (left), leading to systemic platelet thrombi with delayed fibrinolysis (right). CP, cleaving protease; TF, tissue factor; TFPI, tissue factor pathway inhibitor; vWF-CP, ADAMTS 13.
Thrombotic thrombocytopenic purpura
TTP has been described in two forms, acute and chronic relapsing (Table 2). It is described clinically as the constellation of fever, thrombocytopenia, abnormal mental status and or seizures, renal dysfunction, and microangiopathic hemolysis indicated by an elevated lactate dehydrogenase (LDH). There has been significant improvement in understanding of this disease in recent years. The acute form, which accounts for the majority of cases, occurs when antibody production against the von Willebrand factor (vWF)-cleaving proteinase (also called ADAMTS 13) destroys vWF cleaving proteinase activity (Figure 1). These patients have <10% of normal ADAMTS 13 activity. This leads to an inability to cleave unusually large and large multimers to their smaller, less thrombogenic multimers. Because these antibodies are produced in the presence of disease states associated with increased shear stress, the circulating large vWF multimers open and participate with near 100% efficiency in deposition of platelet thrombi. Because shear stress is greatest in the brain and kidney, these organs are most involved, although multiple organs are involved as well [9-16]. The less common but chronic relapsing form of TTP occurs in patients with a deficiency in ADAMTS 13 activity. These patients become ill during periods of systemic illness associated with increased microvascular shear stress. Fibrin thrombosis is involved as well. There is also a reduction in tissue factor pathway inhibitor (TFPI) levels without an increase in tissue factor levels, and an increase in plasminogen activator inhibitor type I (PAI-1) levels as well.
Table 2.
Diagnosing the pathophysiology of thrombocytopenia-associated multiple organ failure
| Diagnostic criteria | Treatment | |
| TTP | Fever | Steroids for 24 hours |
| Thrombocytopenia | Within 30 hours perform 1 1/2 volume plasma exchange then 1 volume daily until resolution of thrombocytopenia (median 18 days [18]) | |
| Increased LDH | ||
| Schistocytes >5% | If recalcitrant use cryopreserved supernatant | |
| Neurological and renal dysfunction | If continues at 28 days use vincristine | |
| DIC | Thrombocytopenia | Reverse shock and underlying disease (increase flow with fluids and consider vasodilators – nitroglycerin, milrinone, pentoxyfilline) |
| Decreased factors V and X, and fibrinogen | ||
| Decreased antithrombin III and protein C | Replace clotting factors with FFP, cryoprecipitate and platelets via plasma infusion or plasma exchange | |
| Increased D-dimers | ||
| Prolonged PT/aPTT | Anticoagulate with heparin, protein C, activated protein C, antithrombin III, or prostacyclin | |
| Use fibrinolytics for life or limb threatening thrombosis. Remember to keep PT/aPTT and platelets normal when giving fibrinolytics | ||
| Give anti-fibrinolytics if life threatening bleeding (rarely needed when PT/aPTT and platelet counts are maintained) | ||
| Secondary TMA | Thrombocytopenia | Remove source of secondary TMA |
| Increased LDH | Activated protein C for adult severe sepsis [26] | |
| Normal or elevated fibrinogen | TTP based plasma exchange (median 9 days [51]; median 12 days for children (Nguyen, 2006, submitted) | |
| <5% schistocytes | ||
| Multiple organ failure |
aPTT, activated partial thromboplastin time; DIC, disseminated intravascular coagulation; FFP, fresh frozen plasma; LDH, lactate dehydrogenase; PT, prothrombin time; TMA, thrombotic microangiopathy; TTP, thrombotic thrombocytopenic purpura.
Disseminated intravascular coagulation
DIC is a consumptive syndrome (consuming pro-coagulant factors such as fibrinogen, Table 2) that is represented in its most severe form by purpura fulminans and in its least severe form by abnormalities in platelet count and prothrombin time (PT)/activated partial thromboplastin time (aPTT). It is described clinically as the constellation of thrombocytopenia, decreased factors V and X, decreased fibrinogen and increased D-dimers. The depletion of factors and fibrinogen explains the common association with prolonged PT/aPTT.
There has been a significant improvement in understanding of thrombosis in patients with DIC syndrome in recent years. When observing and diagnosing the thrombotic process, it is important to understand how increased coagulation is occurring despite prolongation of PT/aPTT. We have been trained to think that prolonged PT/aPTT and reduced platelet count are indicative of a greater tendency to bleeding. How can prolonged PT/aPTT occur when a patient is in a pro-coagulant rather than an anti-coagulant state? How can investigators recommend heparin therapy for patients with DIC when the patient has thrombocytopenia and a prolonged PT/aPTT? PT and aPTT are dependent on coagulation factors and fibrinogen; PT and aPTT increase when these proteins are reduced and decrease when these proteins are increased.
The tissue factor-factor VII pathway, not the factor XII pathway, is responsible for thrombosis in patients with DIC caused by systemic bacterial infection. When released into the circulation by monocyte micro-vesicles or exposed by injured endothelium, tissue factor forms a complex with factor VII and initiates thrombosis (Figure 2). If tissue factor promotes consumption of clotting factors to the point that factors V and X and fibrinogen are depleted, then the patient develops a prolonged PT/aPTT. The endogenous anticoagulant system is also reduced and, paradoxically, contributory to thrombosis in DIC. Protein C, protein S, and antithrombin III are significantly reduced in patients with DIC. Newborns with a congenital absence of protein C, protein S, or antithrombin III can develop spontaneous purpura fulminans, which is fatal if not treated with fresh frozen plasma infusion to replace the anti-coagulant proteins [35,42-44,47,48]. An increased anti-fibrinolytic system also contributes to sustained thrombosis in patients with DIC. Tissue plasminogen activator levels initially increase; however, within 12 to 24 hours the patients develop increased plasminogen activator inhibitor-1 antigen levels and a decrease in plasminα2-anti-plasmin production, indicative of a hypo-fibrinolytic state [10].
Non-consumptive secondary thrombotic microangiopathy
Non-consumptive secondary TMA occurs in critically ill patients with secondary TTP/Hemolytic Uremic Syndrome-like syndromes (Tables 1 and 2). It is identified clinically by the constellation of clinical criteria present with the primary form (TTP) with the exception of one; there is little evidence of hemolysis on peripheral smear [19,20,22-24]. The majority of patients with TMA have thrombocytopenia associated multiple organ failure with a normal or mildly elevated PT/aPTT. These patients have increased or normal levels of factors V, VIII, and X and fibrinogen but also have increased D-dimers. They also have very thrombogenic ultra-large vWF multimers, decreased ADAMTS 13 activity (<57% but rarely <10% as is seen in TTP), ADAMTS 13 inhibitors, and increased PAI-1 activity but normal TFPI activity and absent tissue factor activity (Figure 3). The systemic endothelium is in a platelet pro-coagulant and fibrin anti-fibrinolytic state but, unlike DIC, it is not in a fibrin pro-coagulant state. Thus, consumption of pro-coagulant factors is not observed to the degree noted during DIC.
Choosing a therapy to treat thrombocytopenia-associated multiple organ failure
There is an array of non-specific and specific therapies available to the intensivist for management of the critically ill patient with TAMOF (Figure 4, Table 3). TTP mortality was close to 100% before Bell and colleagues [17] demonstrated that the use of steroids and plasma exchange therapy reduced mortality to 10%. Interestingly, many of the patients treated in this way had evidence of DIC, and histology that showed fibrin and inflammatory cell lesions, and not only platelet-vWF thrombi, in the microvascular thrombi. These patients were defined as having TTP/HUS by a process of elimination when no other cause(s) (for example, infection, toxin, disease, and so on) could be found to explain the underlying microangiopathy. Rock and colleagues [18] also demonstrated that a median of 18 days of plasma exchange was superior to plasma infusion in improving survival in a cohort of patients with TTP and a normal PT/aPTT.
Figure 4.
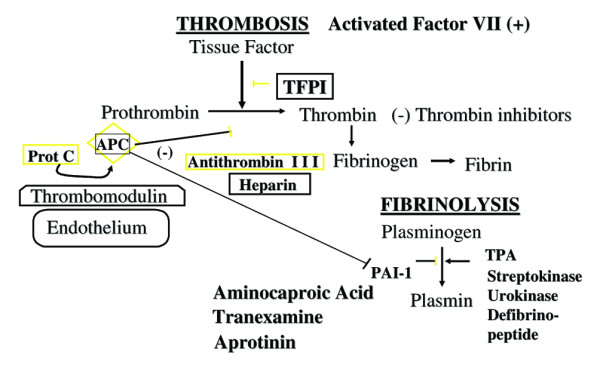
Specific therapies used to reverse or promote thrombosis and promote or stop fibrinolysis. Therapies used to reverse thrombosis include protein C concentrate (prot C), activated protein C (APC), tissue factor pathway inhibitor (TFPI), antithrombin III, heparin, and thrombin inhibitors such as argatroban and hyarudin. Therapies used to promote thrombosis include activated factor VII. Therapies used to promote fibrinolysis include tissue plasminogen activator (TPA), streptokinase, urokinase, and defibrinopeptide. Therapies used to stop fibrinolysis include aminocaproic acid, tranexamine, and aprotinin. PAI, plasminogen activator inhibitor type I.
Table 3.
Effect of non-specific therapy on coagulation and fibrinolysis
| Plasma infusion | Plasma exchange |
| Restores procoagulant factors | Restores procoagulant factor homeostasis |
| Restores anticoagulant factors (protein C, antithrombin III, TFPI) | Restores anticoagulant factor homeostasis (protein C, antithrombin III, TFPI) |
| Restores prostacyclin | Restores prostacyclin homeostasis |
| Restores tPA | Restores tPA homeostasis |
| Restores ADAMTS 13 | Restores ADAMTS 13 homeostasis |
| Removes ADAMTS 13 inhibitors | |
| Removes ultra-large vWF multimers | |
| Removes tissue factor | |
| Removes excess PAI-1 |
PAI, plasminogen activator inhibitor type I; TFPI, tissue factor pathway inhibitor; tPA, tissue plasminogen activator; vWF, von Willebrand factor.
Acute TTP is treated successfully as follows. Because the process can be mediated by antibodies to vWF-cleaving proteinase, a trial of steroid therapy is reasonable as a first step. Daily plasma exchange should be used if resolution is not attained within 24 hours of steroid therapy. Plasma exchange is more effective than plasma infusion because antibodies can be removed from the recipient, and ADAMTS 13 can be replaced by the donor plasma. In patients who are recalcitrant to fresh frozen plasma, some recommend use of cryo-preserved supernatant (fresh frozen plasma minus cryoprecipitate) or solvent detergent treated (soluble detergent) plasma because these plasma products are poor in large vWF multimers. Plasma exchange therapy is most effective when implemented within the first 24 hours of disease, and is required for an average of 15.8 days to restore platelet counts without recrudescence of thrombocytopenia. The endpoint of therapy is resolution of thrombocytopenia (attainment of a platelet count greater than 150,000) and no further deterioration of neurological status. Vincristine is recommended to stop antibody production in patients who are recalcitrant to 28 days of plasma exchange therapy. Chronic relapsing TTP, though much less common, requires chronic plasma infusion therapy after resolution of the acute episode. Plasma infusions may be required on a monthly basis. The benefits of these therapies are considerable. The short-term risks associated with plasma exchange therapy include the need for a large bore intravenous catheter, hypocalcemia secondary to citrate requiring calcium replacement, hypotension requiring inotropes or vasopressors in patients with shock, awakening requiring increased use of sedation in some patients, and secondary catheter-related infections. The long-term risks include blood borne virus exposure.
DIC is a primary determinant of outcome in critically ill patients. The most important determinant of outcome is aggressive fluid resuscitation, restoration of normal or hyperdynamic circulation, and removal of any nidus of infection. With this approach, DIC is now the least common manifestation of organ failure in patients with MOF. However, despite reversal of shock, there are still patients who have DIC and coagulopathy is a predictor of mortality if it persists.
The present mainstay of therapy for DIC is replacement of plasma until PT/aPTT is corrected. This approach could be theoretically counterproductive in some patients. Although PT/aPTT can improve as antithrombin III, protein C and protein S are replaced, some have wondered whether concomitant replacement of coagulation factors in fresh frozen plasma is 'fueling the fire'. For this reason, many investigators who use plasma infusion recommend concomitant heparin infusion to allow ongoing anti-coagulation. In countries where antithrombin III or protein C concentrate are available, physicians may use these concentrates in place of, or in combination with, plasma infusion. Both approaches have been shown to be effective in reversing DIC. An international multicenter study in adults comparing use of activated protein C to standard therapies found a reduction in 28 day mortality from 30.8% to 26.3% in adults with severe sepsis [26]. Although patients with platelet counts less than 30,000/mm3 were excluded from this study, the greatest benefit was found in patients with platelets counts <100,000 and elevated thrombin-antithrombin complexes diagnostic of DIC.
TFPI concentrate is also effective in reversing DIC but not approved for use. Several other infusion therapies have been promoted by various centers. Many use heparin to prevent ongoing thrombosis; however, heparin is a co-factor for antithrombin III and, therefore, does not prevent clotting efficiently if antithrombin III levels are low. Also, combined use of heparin and antithrombin III concentrate can cause an increased tendency to bleeding and actually increase mortality. Prostacyclin infusion can improve microcirculatory flow and decrease platelet thromboses. Other infusion therapies with similar effects include nitroglycerin, nitroprusside, milrinone, amrinone, and pentoxyfilline. Several investigators have reported that fibrinolytic therapy with tissue plasminogen activator, urokinase, or streptokinase leads to remarkable restoration of limb perfusion and unexpected survival with purpura fulminans. Continued use of urokinase requires intermittent plasma infusion to replace depleted plasminogen. The untoward complication of continued use of fibrinolytic therapies can be bleeding if exogenous plasminogen activator activity is far greater than endogenous plasminogen activator inhibitor activity. It is likely prudent to maintain higher platelet counts and pro-coagulant factor levels (for example, platelet, fresh frozen plasma, and cryoprecipitate infusion) when using fibrinolytic therapies. If patients develop life-threatening bleeding from these therapies, then one can consider anti-fibrinolytic therapies, including aminocaproic acid, tranexamine, and aprotinin.
Recently, Ono and colleagues [24] reported that the degree of ADAMTS 13 deficiency in DIC patients is associated with both the degree of renal failure and the likelihood of resolution of renal failure. Plasma exchange is a non-specific therapy that has been reported by several centers to be effective for reversal of DIC. The theory behind this therapy is straightforward. If DIC is caused by increased circulating tissue factor and plasminogen activator inhibitor activity, reduced antithrombin III, protein C, protein S, prostacyclin activity, and ADAMTS 13 activity, then why not simultaneously correct each of the abnormalities without causing fluid overload? Plasma exchange is performed using 1 1/2 volume exchange, which replaces approximately 78% of host plasma. An aPTT >50 seconds predicts poor outcome in meningococcemia. Plasma exchange reversed coagulopathy and resulted in survival in seven out of nine children with meningococcus-associated purpura fulminans who had a predicted mortality of greater than 90% based on prolonged PTT [36]. Interestingly, aPTT was corrected because factor II, V, VII, and VIII levels were restored, but protein C and antithrombin III levels were only minimally increased by plasma exchange. These authors did not measure the effect of plasma exchange on ADAMTS 13 levels. Attainment of protein C levels of 0.25 IU/ml is associated with normalization of coagulation in neonates with congenital purpura fulminans. Supplementation of plasma exchange with protein C and antithrombin III might be efficacious in patients with consumptive microangiopathy.
Secondary TMA can be diagnosed in critically ill patients with new-onset thrombocytopenia, organ failure, and elevated LDH and an underlying predisposing condition (Table 1). Poor outcomes of these processes are well documented. Favorable responses of adults and children with secondary TMA have been found with the use of the TTP-based plasma exchange therapy protocol. The biological plausibility for positive effects of plasma exchange in patients with TTP or DIC has been discussed; the biological plausibility for therapeutic effect in patients with secondary TMA is similar. Plasma exchange normalizes plasminogen activator inhibitor activity allowing endogenous tissue plasminogen activator to lyse fibrin thrombi in a controlled and progressive fashion without bleeding. Plasma exchange also has a beneficial effect on vWF pathophysiology. It removes ADAMTS 13 inhibitors and ultra-large vWF multimers, restores ADAMTS 13 activity, and improves organ function.
Because protein C is an inhibitor of plasminogen activator type 1 activity, its use could also have a role in children with TAMOF with and without prolonged PT/aPTT. Darmon and colleagues [51] recently reported that plasma exchange for a median of 9 days reduced multiple organ failure and improved survival in critically ill patients with TAMOF caused by secondary TMA compared to plasma infusion therapy alone. In this regard, a single center study in adults with severe sepsis using plasma exchange therapy for a median of 3 days showed a reduction in mortality from 54% to 33%, with an absolute relative risk reduction of 20.5% and a number of patients needed to treat to save one patient equal to 4.9 [25].
Interpreting the literature on therapy for TAMOF
The medical literature on therapy for patients with TAMOF is growing. Activated protein C studies in adults and children show it has the best effect in patients with severe sepsis and DIC. The bleeding risk can be minimized by correcting thrombocytopenia (maintaining platelet counts >30,000/m3) with platelet transfusion and prolonged PT/PTT with FFP infusion, before administering the drug. Clinical studies testing plasma exchange have consistently shown positive results in patients with TAMOF (TTP, secondary TMA) but varied results in those with severe sepsis. Its use for treatment of TTP is universally accepted; however, it is important to note that therapy is continued until restoration of platelet count, usually after 18 days of therapy. Darmon and colleagues [51] demonstrated improved outcomes (reduction in mortality from 40% to 0%) when comparing plasma exchange for a median of 9 days compared to plasma infusion. Similar to the experience in the TTP trials [18], these authors found that recrudescence was common when plasma exchange was attempted for more abbreviated periods of time. Reeves and colleagues [49] performed a clinical trial of continuous plasma filtration without full plasma replacement for 36 hours in adults and children with severe sepsis and found no benefit. The authors did not state whether their patients had TAMOF; however, one would not expect a benefit if TTP-like pathophysiology was the target (this needs up to 18 days of treatment), nor if DIC pathophysiology was the target (this needs full plasma replacement to replace deficient anti-coagulant proteins). Interestingly, Busund and colleagues [25] performed a trial of daily centrifugation-based full plasma exchange for three days in patients with severe sepsis and showed improved survival. Stegmayr and colleagues [50] also reported improved outcome with one to three treatments of centrifugation-based plasma exchange in severe sepsis. Improvement was less likely to be from reversal of TTP-like pathophysiology (due to short duration) and more likely to be from reversal of DIC pathophysiology.
We interpret these findings as follows. Activated protein C (four day infusion) should be used to treat adult severe sepsis with greatest benefit expected in the DIC population [26]. Plasma exchange should be performed on a daily basis for patients with TTP [51] or secondary TMA [18] until resolution of thrombocytopenia (a median 9 to 16 days) and recrudescence of thrombocytopenia should be treated with resumption of daily plasma exchange therapy.
Conclusion
A consensus is developing that reversal of microvascular thrombosis is a therapeutic target in patients with TAMOF defined by the clinical triad of new onset thrombocytopenia, multiple organ failure, and elevated LDH levels. As with all therapeutic targets, the underlying cause of disease must be removed for the therapy to have long-term effects. Microvascular thrombosis is associated with systemic insults, including shock, infection, drugs, toxins, and radiation. For therapies directed at microangiopathy to be beneficial, shock must be reversed, infection eradicated and removed, and precipitating drugs, toxins, and radiation stopped. Anti-thrombotic/fibrinolytic therapies can only be expected to have beneficial effects on outcome if and when these tasks have been accomplished.
New-onset thrombocytopenia is a clinical indicator of TMA in patients with MOF, and resolution of thrombocytopenia is an indicator of resolving TMA. Therefore, resolution of thrombocytopenia is the goal for directed use of therapy. Activated protein C use is associated with improved outcomes in children and adults with severe sepsis and DIC; however, activated protein C does not address the deficiency of ADAMTS 13 in the severest forms of DIC, nor in TTP or secondary TMA. Thus, development of human recombinant ADAMTS 13 could be an important drug discovery. There is also an important need to develop clinical laboratory testing that allows bedside determination of ADAMTS 13 activity. At this time, clinical trials support the use of steroids and intensive daily centrifugation-based plasma exchange therapy to reverse TTP/DIC/secondary TMA, and improve survival for patients with TAMOF [18,25,51].
Abbreviations
DIC = disseminated intravascular coagulation; PT = prothrombin time; TAMOF = thrombocytopenia-associated multiple organ failure; TF = tissue factor; TFPI = tissue factor pathway inhibitor; TMA = thrombotic microangiopathy; TTP = thrombotic thrombocytopenic purpura; vWF = von Wille-brand factor.
Competing interests
The authors declare that they have no competing interests.
Note
This article is part of a thematic series on Translational research, edited by John Kellum.
Other articles in the series can be found online at http://ccforum.com/articles/theme-series.asp?series=CC_Trans
References
- Akca S, Haji-Michael P, de Mendonca A, Suter P, Levi M, Vincent JL. Time course of platelet counts in critically ill patients. Crit Care Med. 2002;30:753–756. doi: 10.1097/00003246-200204000-00005. [DOI] [PubMed] [Google Scholar]
- Baughman RP, Lower EE, Flessa HC, Tollerud DJ. Thrombocytopenia in the intensive care unit. Chest. 1993;104:1243–1247. doi: 10.1378/chest.104.4.1243. [DOI] [PubMed] [Google Scholar]
- Brun-Buisson C, Doyon F, Carlet J, Dellamonica P, Gouin F, Lepoutre A, Mercier JC, Offenstadt G, Regnier B. Incidence, risk factors, and outcome of severe sepsis and septic shock in adults. A multicenter prospective study in intensive care units. French ICU Group for Severe Sepsis. JAMA. 1995;274:968–974. doi: 10.1001/jama.274.12.968. [DOI] [PubMed] [Google Scholar]
- Lee KH, Hui KP, Tan WC. Thrombocytopenia in sepsis: a predictor of mortality in the intensive care unit. Singapore Med J. 1993;34:245–246. [PubMed] [Google Scholar]
- Nguyen TC, Han YY, Watson S, Carcillo JA, Kiss J. Thrombocytopenia is associated with multiple organ failure and death in children. Crit Care Med. 2000;28S:A50. [Google Scholar]
- Sprung CL, Peduzzi PN, Shatney CH, Schein RM, Wilson MF, Sheagren JN, Hinshaw LB. Impact of encephalopathy on mortality in the sepsis syndrome. The Veterans Administration Systemic Sepsis Cooperative Study Group. Crit Care Med. 1990;18:801–806. doi: 10.1097/00003246-199001000-00034. [DOI] [PubMed] [Google Scholar]
- Stephan F, Hollande J, Richard O, Cheffi A, Maier-Redelsperger M, Flahault A. Thrombocytopenia in a surgical ICU. Chest. 1999;115:1363–1370. doi: 10.1378/chest.115.5.1363. [DOI] [PubMed] [Google Scholar]
- Vanderschueren S, De Weerdt A, Malbrain M, Vankersschaever D, Frans E, Wilmer A, Bobbaers H. Thrombocytopenia and prognosis in intensive care. Crit Care Med. 2000;28:1871–1876. doi: 10.1097/00003246-200006000-00031. [DOI] [PubMed] [Google Scholar]
- Furlan M, Robles R, Galbusera M, Remuzzi G, Kyrle PA, Brenner B, Krause M, Scharrer I, Aumann V, Mittler U, et al. von Willebrand factor-cleaving protease in thrombotic thrombocytopenic purpura and the hemolyticuremic syndrome. N Engl J Med. 1998;339:1578–1584. doi: 10.1056/NEJM199811263392202. [DOI] [PubMed] [Google Scholar]
- Green J, Doughty L, Kaplan SS, Carcillo JA. The tissue factor and plasminogen activator type 1 response ion children with sepsis induced multiple organ failure. Thromb Haemost. 2002;87:218–223. [PubMed] [Google Scholar]
- Moake JL. Thrombotic microangiopathies. N Engl J Med. 2002;347:589–600. doi: 10.1056/NEJMra020528. [DOI] [PubMed] [Google Scholar]
- Tsai HM, Lian EC. Antibodies to von Willebrand factor-cleaving protease in acute thrombotic thrombocytopenic purpura. N Engl J Med. 1998;339:1585–1594. doi: 10.1056/NEJM199811263392203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadler JE. Biochemistry and genetics of von Willebrand factor. Annu Rev Biochem. 1998;67:395–424. doi: 10.1146/annurev.biochem.67.1.395. [DOI] [PubMed] [Google Scholar]
- van Mourik JA, Boertjes R, Huisveld IA, Fijnvandraat K, Pajkrt D, van Genderen PJ, Fijnheer R. von Willebrand factor propeptide in vascular disorders: A tool to distinguish between acute and chronic endothelial cell perturbation. Blood. 1999;94:179–185. [PubMed] [Google Scholar]
- Levy GG, Nichols WC, Lian EC, Foroud T, McClintick JN, McGee BM, Yang AY, Siemieniak DR, Stark KR, Gruppo R, et al. Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature. 2001;413:488–494. doi: 10.1038/35097008. [DOI] [PubMed] [Google Scholar]
- Asada Y, Sumiyoshi A, Hayashi T, Suzumiya J, Kaketani K. Immunohistochemistry of vascular lesion in thrombotic thrombocytopenic purpura, with special reference to factor VIII related antigen. Thromb Res. 1985;38:469–479. doi: 10.1016/0049-3848(85)90180-X. [DOI] [PubMed] [Google Scholar]
- Bell WR, Braine HG, Ness PM, Kickler TS. Improved survival in thrombotic thrombocytopenic purpura-hemolytic uremic syndrome. Clinical experience in 108 patients. N Engl J Med. 1991;325:398–403. doi: 10.1056/NEJM199108083250605. [DOI] [PubMed] [Google Scholar]
- Rock GA, Shumak KH, Buskard NA, Blanchette VS, Kelton JG, Nair RC, Spasoff RA. Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. Canadian Apheresis Study Group. N Engl J Med. 1991;325:393–397. doi: 10.1056/NEJM199108083250604. [DOI] [PubMed] [Google Scholar]
- Brilliant SE, Lester PA, Ohno AK, Carlon MJ, Davis BJ, Cusher HM. Hemolytic uremic syndrome without evidence of microangiopathic hemolysis on peripheral blood smear. South Med J. 1996;89:342–345. doi: 10.1097/00007611-199603000-00018. [DOI] [PubMed] [Google Scholar]
- Fava S, Galizia AC. Thrombotic thrombocytopenic purpura-like syndrome in the absence of schistocytes. Br J Haematol. 1995;89:643–644. doi: 10.1111/j.1365-2141.1995.tb08379.x. [DOI] [PubMed] [Google Scholar]
- Tsai HM, Li A, Rock G. Inhibitors of von Willebrand factor-cleaving protease in thrombotic thrombocytopenic purpura. Clin Lab. 2001;47:387–392. [PubMed] [Google Scholar]
- Moore JC, Hayward CP, Warkentin TE, Kelton JG. Decreased von Willebrand factor protease activity associated with thrombocytopenic disorders. Blood. 2001;98:1842–1846. doi: 10.1182/blood.V98.6.1842. [DOI] [PubMed] [Google Scholar]
- Veyradier A, Obert B, Houllier A, Meyer D, Girma JP. Specific von Willebrand factor-cleaving protease in thrombotic microangiopathies: a study of 111 cases. Blood. 2001;98:1765–1772. doi: 10.1182/blood.V98.6.1765. [DOI] [PubMed] [Google Scholar]
- Ono T, Mimuro J, Madoiwa S, Soejima K, Kashiwakura Y, Ishiwata A, Takano K, Ohmori T, Sakata Y. Severe secondary deficiency of von Willebrand factor-cleaving protease (ADAMTS13) in patients with sepsis-induced disseminated intravascular coagulation: its correlation to development of renal failure. Blood. 2006;107:528–534. doi: 10.1182/blood-2005-03-1087. [DOI] [PubMed] [Google Scholar]
- Busund R, Koukline V, Utrobin U, Nedashkovsky E. Plasmapheresis in severe sepsis and septic shock: a prospective, randomised, controlled trial. Intensive Care Med. 2002;28:1434–1439. doi: 10.1007/s00134-002-1410-7. [DOI] [PubMed] [Google Scholar]
- Bernard GR, Vincent JL, Laterre PF, LaRosa JP, Dhainaut JF, Lopez-Rodriguez A, Steingrad JS, Garbo GE, Heltebrand JD, Ely W, Fisher CJ. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001;344:699–709. doi: 10.1056/NEJM200103083441001. [DOI] [PubMed] [Google Scholar]
- Chauhan AK, Motto DG, Lamb CB, Bergmeier W, Dockal M, Plaimauer B, Scheiflinger F, Ginsburg D, Wagner DD. The metalloprotease ADAMTS13 is a natural anti-thrombotic. J Exp Med. 2006;203:767–776. doi: 10.1084/jem.20051732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motto D, Zhang W, Zhu G. Additional environmental and/or genetic factors are required to trigger TTP in ADAMTS13-deficient mice (Abstract) Blood. 2004;104:258. doi: 10.1182/blood-2004-04-1438. [DOI] [Google Scholar]
- Motto DG, Chauhan AK, Zhu G, Homeister J, Lamb CB, Desch KC, Zhang W, Tsai HM, Wagner DD, Ginsburg D. Shigatoxin triggers thrombotic thrombocytopenic purpura in genetically susceptible ADAMTS13-deficient mice. J Clin Invest. 2005;115:2752–2761. doi: 10.1172/JCI26007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bick RL. Disseminated intravascular coagulation: Pathophysiologic mechanisms and manifestations. Semin Thromb Hemost. 1998;24:3–18. doi: 10.1055/s-2007-994971. [DOI] [PubMed] [Google Scholar]
- Bick RL. Disseminated intravascular coagulation: Objective clinical and laboratory diagnosis, treatment and assessment of therapeutic response. Semin Thromb Hemost. 1996;22:69–88. doi: 10.1055/s-2007-998993. [DOI] [PubMed] [Google Scholar]
- Kwan HC. Thrombotic microangiopathy. Semin Hematol. 1987;24:69–81. [PubMed] [Google Scholar]
- Kwan HC. Miscellaneous secondary thrombotic microangiopathy. Semin Hematol. 1987;24:141–147. [PubMed] [Google Scholar]
- Nguyen T, Hall Y, Fiedor M, Hasset A, Lopez-Plena I, Watson S, Lum L, Carcillo JA. Microvascular thrombosis in pediatric multiple organ failure: is it a therapeutic target? Pediatr Crit Care Med. 2001;2:187–196. doi: 10.1097/00130478-200107000-00001. [DOI] [PubMed] [Google Scholar]
- Dreyfus M, Masterson M, David M, Rivard GE, Muller FM, Krenz W, Beeg T, Minard A, Allgrove J, Cohen JD, et al. Replacement therapy with a monoclonal Ab purified protein C concentrate in newborns with severe congenital protein C deficiency. Semin Thromb Hemost. 1995;21:371–381. doi: 10.1055/s-2007-1000658. [DOI] [PubMed] [Google Scholar]
- Churchwell KB, McManus ML, Kent P, Gorlin J, Galacki D, Humphreys D, Kevy SV. Intensive blood and plasma exchange for treatment of coagulopathy in meningococcemia. J Clin Apheresis. 1995;10:171–177. doi: 10.1002/jca.2920100403. [DOI] [PubMed] [Google Scholar]
- Fourrier F, Chopin C, Huart JJ, Runge I, Caron C, Goudemand J. Double blind placebo controlled trial of antithrombin III concentrate in septic shock with disseminated intravascular coagulation. Chest. 1993;104:882–888. doi: 10.1378/chest.104.3.882. [DOI] [PubMed] [Google Scholar]
- Gross SM, Kennan JJ. Whole blood transfusion for exsanguinating coagulopathy in a US field surgical hospital in Kosovo. J Trauma. 2000;49:145–148. doi: 10.1097/00005373-200007000-00022. [DOI] [PubMed] [Google Scholar]
- Harrison CN, Lawrie AS, Iqbal A, Hunter A, Machin SJ. Plasma exchange with solvent/detergent treated plasma of resistant thrombotic thrombocytopenic purpura. Br J Haematol. 1996;94:756–758. doi: 10.1046/j.1365-2141.1996.d01-1836.x. [DOI] [PubMed] [Google Scholar]
- Hattersley PG, Kuntel M. Cryoprecipitate as a souce of fibrinogen in treatment of disseminated intravascular coagulation. Transfusion. 1976;16:641–645. doi: 10.1046/j.1537-2995.1976.16677060249.x. [DOI] [PubMed] [Google Scholar]
- Leclerc F, Hazelzet JA, Jude B, Hofhuis W, Hue V, Martinot A, Van de Vort E. Protein C and S deficiency in severe infectious purpura of children: a collaborative study of 40 cases. Intensive Care Med. 1992;18:202–225. doi: 10.1007/BF01709832. [DOI] [PubMed] [Google Scholar]
- McManus ML, Churchwell KD. Coagulopathy as a predictor of outcome in meningococcal sepsis and systemic inflammatory response syndrome with purpura. Crit Care Med. 1993;21:706–711. doi: 10.1097/00003246-199305000-00014. [DOI] [PubMed] [Google Scholar]
- Riewald M, Reiss H. Treatment options for clinically recognized disseminated intravascular coagulation. Semin Thromb Hemost. 1998;24:53–59. doi: 10.1055/s-2007-995823. [DOI] [PubMed] [Google Scholar]
- Rintala E, Kauppila M, Seppala O, Voipio-Pulkk L, Pettila V, Rasi V, Kotilainen P. Protein C substitution in sepsis-associated purpura fulminans. Crit Care Med. 2000;28:2373–2378. doi: 10.1097/00003246-200007000-00032. [DOI] [PubMed] [Google Scholar]
- Rock G, Shumak KH, Sutton DM, Buskard NA, Nair RC. Cryosupernatant as replacement fluid for plasma exchange in thrombotic thrombocytopenic purpura. Members of the Canadian Apheresis Group. Br J Haematol. 1996;94:383–386. doi: 10.1046/j.1365-2141.1996.d01-1800.x. [DOI] [PubMed] [Google Scholar]
- Sagripanti A, Carpi A, Rosaia B, Morelli E, Innocenti M, D'Acunto G, Nicolini A. Iloprost in the treatment of thrombotic microangiopathy:report of thirteen cases. Biomed Pharmacother. 1996;50:350–356. doi: 10.1016/S0753-3322(96)89667-3. [DOI] [PubMed] [Google Scholar]
- Williams CK, Fernbach B, Cuttner J, Hallert JF, Essien ER. Management of leukemia associated disseminated intravascular coagulation. Haematologia (Budap) 1982;15:287–295. [PubMed] [Google Scholar]
- Zenz W, Bodo Z, Zobel G, Fanconi S, Rettenbacher A. Recombinant tissue plasminogen activator restores perfusion in meningococcal purpura fulminans. Crit Care Med. 1998;26:969–971. doi: 10.1097/00003246-199805000-00039. [DOI] [PubMed] [Google Scholar]
- Reeves JH. A review of plasma exchange in sepsis. Blood Purification. 2002;20:282–286. doi: 10.1159/000047021. [DOI] [PubMed] [Google Scholar]
- Stegmayr BG, Banga R, Berggren L, Norda R, Rydvall A, Vikerfors T. Plasma exchange as rescue therapy in multiple organ failure including acute renal failure. Crit Care Med. 2003;31:1730–1736. doi: 10.1097/01.CCM.0000064742.00981.14. [DOI] [PubMed] [Google Scholar]
- Darmon M, Azoulay E, Thiery G, Ciroldi M, Galicier L, Parquet N, Veyradier A, Le Gall JR, Oksenhendler E, Schlemmer B. Time course of organ dysfunction in thrombotic microanguiopathy patients receiveign either plasma perfusion or plasm exchange. Crit Care Med. 2006;34:2127–2133. doi: 10.1097/01.CCM.0000227659.14644.3E. [DOI] [PubMed] [Google Scholar]