

Abstract
Cognitive dysfunction is common in critically ill patients, not only during the acute illness but also long after its resolution. A large number of pathophysiologic mechanisms are thought to underlie critical illness-associated cognitive dysfunction, including neuro-transmitter abnormalities and occult diffuse brain injury. Markers that could be used to evaluate the influence of specific mechanisms in individual patients include serum anticholinergic activity, certain brain proteins, and tissue sodium concentration determination via high-resolution three-dimensional magnetic resonance imaging. Although recent therapeutic advances in this area are exciting, they are still too immature to influence patient care. Additional research is needed if we are to understand better the relative contributions of specific mechanisms to the development of critical illness-associated cognitive dysfunction and to determine whether these mechanisms might be amenable to treatment or prevention.
Introduction
Since its advent more than 40 years ago, the specialty of critical care has made remarkable advances in the care of severely ill patients. Mortality rates for many commonly encountered critical illnesses such as severe sepsis [1] and acute respiratory distress syndrome (ARDS) [2] have declined sharply over the past 2 decades. As greater numbers of patients survive intensive care, it is becoming increasingly evident that quality of life after critical illness is not always optimal. For instance, nearly half of ARDS survivors manifest neurocognitive sequelae 2 years after their illness, falling to below the 6th percentile of the normal distribution of cognitive function [3]. Considering that 89% of Americans would not wish to be kept alive if they had severe, irreversible neurologic damage [4], these findings are quite concerning.
Cognitive dysfunction (CD) is quite common in critically ill patients, not only during the acute illness but also long after the acute illness resolves [5]. Delirium, a form of acute CD that manifests as a fluctuating change in mental status, with inattention and altered level of consciousness, occurs in as many as 80% of mechanically ventilated intensive care unit (ICU) patients [6]. Most clinicians consider ICU delirium to be expected, iatrogenic, and without consequence. However, recent data associate delirium with increased duration of mechanical ventilation and ICU stay [7], worse 6-month mortality [8], and higher costs [9]. Chronically, critical illness-associated CD manifests as difficulties with memory, attention, executive function, mental processing speed, spatial abilities, and general intelligence. Interestingly, patients who develop acute CD often go on to develop chronic CD after hospital discharge [10-13], suggesting that the two entities may share a common etiology.
Although there are clearly defined risk factors for critical illness-associated CD, there is little understanding of the underlying pathophysiology. The precise mechanisms are unknown and there are likely to be multiple mechanisms at work in any given patient (Figure 1) [5,14,15]. We have chosen to focus on two mechanisms that appear to have the greatest merit: neurotransmitter abnormalities and occult diffuse brain injury. In this bench-to-bedside review, we discuss the evidence supporting these mechanisms, potential markers that could be used to evaluate each mechanism in individual patients, and emerging therapies that may prevent or mitigate critical illness-associated CD.
Figure 1.
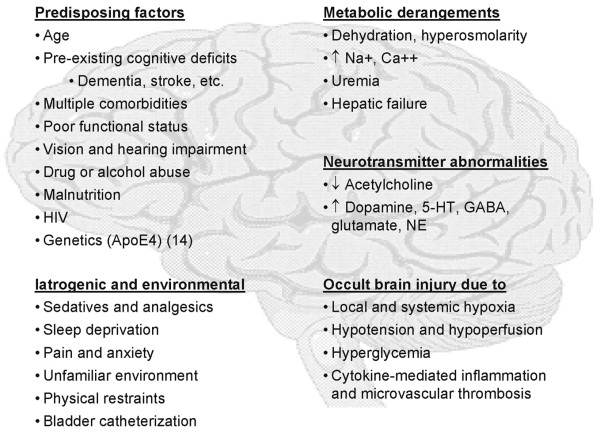
Pathophysiologic mechanisms and predisposing factors thought to underlie critical illness-associated cognitive dysfunction [5, 14, 15]. Apo, apolipoprotein; HIV, human immunodeficiiency virus; 5-HT, serotonin (5-hydroxytryptamine); GABA, γ-aminobutyric acid; NE, norepinephrine (noradrenaline).
Neurotransmitter abnormalities
For more than a century, clinicians have recognized that anticholinergic medications are a cause of both acute and chronic CD [16]. The mechanism is thought to be a direct reduction in central cholinergic activity [17], leading to relative dopamine excess in the central nervous system (CNS). Antipsychotics such as haloperidol, which antagonize central dopamine receptors, can counteract the cognitive effects of anticholinergic medications, further supporting the anticholinergic hypothesis.
Drugs with potent central anticholinergic effects, such as tricyclic antidepressants and antihistamines, are particularly likely to cause delirium. Many medications that are commonly used in the ICU yet not generally considered to be anti-cholinergic, such as H2 blockers, opiates, furosemide, digoxin, glucocorticoids, and benzodiazepines, were recently shown to have central anticholinergic properties [16,17]. Volatile anesthetics, such as sevoflurane, and intravenous anesthetics, such as propofol, also have anticholinergic effects and may be responsible not only for postoperative delirium but also for the more complex phenomena of postoperative cognitive dysfunction [18]. Acute illness itself may be associated with production of endogenous anticholinergic substances [19]. In one study, 8 out of 10 elderly medical inpatients had had detectable anticholinergic activity in their serum, even though no medication used by these individuals had anticholinergic activity. Characterization of such substances might improve our understanding of delirium and lead to useful intervention strategies. Considering that activation of specific cholinergic pathways can inhibit pro-inflammatory cytokine synthesis and protect against endo-toxemia and ischemia-reperfusion injury [20], it is tempting to speculate that inhibition of these pathways, whether exogenous or endogenous, might contribute not only to CD but also to other outcomes of critical illness.
In assessing the overall risk for developing CD posed by medications with central anticholinergic activity in a given patient, individual differences in drug pharmacokinetics make the dose received a poor estimate of a patient's overall anticholinergic burden [21,22]. However, we can objectively measure anticholinergic burden in individual patients using an assay referred to as serum anticholinergic activity (SAA) [16]. First described by Tune and Coyle [23], SAA measures the ability of a individual's serum to block central muscarinic receptors using a rat forebrain preparation. Elevated SAA levels are associated with cognitive impairment in studies of medical ward inpatients and community dwelling seniors [16,24-27]. Only a single, small study has used this assay to investigate CD in ICU patients. Golinger and colleagues [28] examined SAA levels in surgical ICU patients and found the mean SAA level drawn 4 hours after mental status change was significantly greater in delirious patients (n = 9) than in those without delirium (n = 16; 4.67 ng/ml versus 0.81 ng/ml; P = 0.007). Whether these results apply to all critically ill patients is uncertain because no study has examined SAA across a broad range of ICU admitting diagnoses or in medical ICU settings. Furthermore, because SAA measurement requires fresh rat brain preparations, its use is likely to remain limited to research settings for the foreseeable future.
Other neurotransmitter systems such as dopamine, serotonin, γ-aminobutyric acid (GABA), norepinephrine (noradrenaline), and glutamate are also thought to contribute to critical illness-associated CD. Dopaminergic hyperfunction is thought to underlie the cognitive symptoms of schizophrenia, and dopamine administration itself may be a risk factor for delirium [29]. Serotonin syndrome, a consequence of excess seroto-nergic agonism, can be seen not only with selective serotonin reuptake inhibitors but also with a variety of drugs and drug combinations [30]. Even a single therapeutic dose of an selective serotonin reuptake inhibitor can cause the syndrome, which manifests as mental status changes, autonomic hyperactivity, and neuromuscular abnormalities.
GABA abnormalities are thought to contribute to hepatic encephalopathy, perhaps mediated by branched chain and aromatic amino acids acting as false neurotransmitters [31]. Excess GABA activity, such as that which occurs after withdrawal from chronic ethanol or benzodiazepine use, is a well known and quite dangerous cause of delirium [32]. Acutely, sedatives that stimulate GABA receptors, such as benzo-diazepines and (probably) propofol, impair cognitive function and are deliriogenic [8,33-35]. This raises the possibility that strategies to minimize sedative drug accumulation, such as daily interruption of sedative infusions [36], which have been shown to reduce duration of mechanical ventilation, and ICU and hospital length of stay, might also reduce the incidence or duration of delirium. Whether these sedative drugs lead to neurocognitive deficits long after their use is unknown, but this has been suggested in certain high-risk groups, such as the very old (>75 years) and those with pre-existing cognitive impairment [37,38].
Noradrenergic hyperfunction, as part of the 'fight or flight' response, can lead to panic attacks and delusions. Glutamate has been implicated in the 'Chinese food syndrome', in which food with high amounts of monosodium glutamate interferes with normal neurotransmission causing confusion [39]. For a more complete review of the other neurotransmitter abnormalities that may underlie delirium, the reader is referred elsewhere [40,41].
Occult diffuse brain injury
If critical illness-associated CD were solely due to acute medication effects, it would probably resolve after the exposure has ended. However, a significant percentage of individuals developing delirium in the hospital continue to demonstrate symptoms of CD after discharge [10-13]. These patients manifest decreased cerebral activity and increased cognitive deterioration, and are more likely to develop dementia than patients without delirium. Also, patients who develop delirium have a greater rate of decline on cognitive tests than do nondelirious patients [10-13]. Taken together, these observations raise the possibility that some degree of occult diffuse brain injury, as a consequence of the local hypoxia, hypoperfusion, cytokine-mediated inflammation and microvascular thrombosis that characterize the multisystem organ dysfunction of critical illness, might have occurred in these patients [42]. Given that every other organ system can be damaged by these forces, it seems implausible that the brain would be uniquely spared.
Many of the data supporting occult diffuse brain injury as a cause of critical illness-associated CD come from studies of sepsis and septic encephalopathy, a form of delirium. In animal models of sepsis, oxidative damage occurs early in the hippocampus, cerebellum, and cortex [43], and significant alterations in cerebral vascular hemodynamics and tissue acid-base balance indicate that cerebral ischemia and acidosis do occur [44-48]. Sharshar and colleagues completed several studies comparing brain pathology in small numbers of patients who died from septic shock with that in patients who died from other causes. Septic patients demonstrated diffuse severe ischemic and hemorrhagic CNS lesions [49], which correlated with persistent hypotension and severe coagulation disorders. Multiple microscopic foci of necrosis involving the white matter of the pons [50] were seen, as well as ischemia and apoptosis within the cerebral autonomic centers [51]. The white matter lesions were associated with elevated levels of proinflammatory cytokines, suggesting a possible role of inflammation and microvascular thrombosis in the genesis of CNS injury [52]. Although those studies demonstrated that ischemic brain injury occurs in sepsis, they did not determine whether delirium occurred.
Two studies attempted to examine the relationship of ischemic brain injury to delirium. In one study of 84 patients with severe sepsis and multiple organ dysfunction [53], severe hypotension was the only factor in multivariable analyses that was associated with delirium, suggesting that sepsis-related encephalo-pathy may be caused by ischemic damage rather than metabolic abnormalities. Another study examined cerebral blood flow and cerebral oxygen metabolic rates in patients with septic encephalopathy and multiple organ dysfunction [54], and it found that both were significantly lower than those in normal awake individuals. Although these studies support the idea of occult brain injury as a cause of delirium, the authors did not use a standardized Diagnostic and Statistical Manual of Mental Disorders (DSM)-IV based tool to diagnose delirium, such as the Confusion Assessment Method for the ICU [6].
Lending support to the hypothesis that acute inflammation leads to brain injury and subsequent development of delirium, a recent study found that delirium in postoperative hip-fractured patients was significantly associated with serum levels of C-reactive protein, an acute-phase protein that is a marker of acute inflammation [55]. Importantly, patients in the study were diagnosed with delirium using the Confusion Assessment Method (the ward-based predecessor to the Confusion Assessment Method for the ICU), providing the first DSM-IV based evidence that acute inflammation may be in the causative pathway of delirium.
The brain is a target for free radical damage because of its large lipid content, high rate of metabolism, and low anti-oxidant capacity. Free radical induced oxidative stress may play a role in the delirium seen after cardiopulmonary bypass. Karlidag and colleagues [56] noted that patients with low preoperative levels of catalase, a erythrocyte-based anti-oxidant enzyme, were more susceptible to delirium postoperatively. They suggested that preoperative catalase levels might some day be used to identify at-risk patients who could then be put on antioxidant treatment preoperatively. Whether this would reduce the incidence of delirium remains speculative.
Regional cerebral blood flow appears to be reduced in delirium. Using xenon-enhanced computed tomography (CT), Yakota and colleagues [57] demonstrated significant focal and global brain hypoperfusion in 10 ICU patients with hypoactive delirium. After recovery from delirium cerebral blood flow returned to normal, implying that cerebral hypo-perfusion may contribute to the development of delirium.
Studies of ARDS survivors suggest that a combination of acute hypoxia, hypoperfusion, and hyperglycemia plays an important role in the long-term cognitive sequelae of critical illness [3,58,59]. However, it has been difficult to demonstrate a clear relationship, given the lengthy interval between stimulus and effect and the great number of additional contributing variables that can obscure downstream effects. Among ARDS survivors, Hopkins and colleagues showed that the degree of CD at 1 year is significantly correlated with the durations of hypoxia [58] and mean arterial blood pressure less than 50 mmHg during the ICU stay [3]. In animals, hyperglycemia markedly enhances hypoxic-ischemic brain damage due to increased brain edema and disrupted cerebral metabolism [60]. In ARDS survivors, the duration of blood glucose greater than 180 mg/dl has been shown to correlate with worse visual spatial abilities, visual memory, processing speed, and executive function at 1 year [59]. Given the recent interest in maintaining tight glucose control during critical illness as a means of reducing mortality, it will be interesting to see whether patients managed using this technique have better cognitive outcomes. Clearly, such an approach will need to balance the benefits of tight glucose control with the known risks that hypoglycemia poses to the CNS.
One of the perceived difficulties with looking for evidence of occult brain injury in humans is the apparent need for CNS tissue specimens to prove that brain injury actually occurred. However, studies of stroke, trauma, and cardiopulmonary bypass-associated brain injury show that serum markers of brain injury correlate well with the extent of CNS damage. S-100β, neuron-specific enolase (NSE), and myelin basic protein (MBP) are three such markers that could be used to look for evidence of occult brain injury in critical illness-associated CD.
S-100 is a dimeric calcium-binding protein consisting of two subunits (α and β) [61]. The β unit (S-100β) is highly brain specific, located mainly in astrocytes. Circulating levels of S-100β are elevated in patients with cerebral ischemia [62], cardiopulmonary bypass-associated decline in explicit memory function [63,64], and traumatic brain injury (TBI) [65-67]. Even in mild head injury, serum levels of S-100β are correlated with clinical measures of injury severity, neuro-radiologic findings, and outcomes, including postconcussion symptoms [68]. Elevated serum S-100β levels were recently demonstrated in critically ill patients with respiratory failure [69] and in porcine models of endotoxic shock [70] and acute lung injury [71]. In this latter group, elevated S-100β levels were associated with hippocampal histopathologic changes, including basophilic shrunken neurons in the pyramidal cell layer [71]. Interestingly, S-100β may have both beneficial and detrimental effects, in that lower levels may have protective neurotrophic effects, yet higher levels can lead to exacerbation of neuroinflammation and neuronal dysfunction [72].
Whereas S-100β is a marker of astrocyte damage, NSE and MPB are markers of neuron and white matter (myelin) damage, respectively. NSE is protein-based enzyme that is found primarily in neurons. Serum levels of NSE are elevated after TBI, exhibiting a close relationship with outcome in severe head injury [73,74] and with volume of contusion in minor head injuries [75]. Interestingly, elevated NSE levels were recently shown to predict death in one small study (n = 29) of patients with severe sepsis [76], even though these patients had no acute CNS disorders, such as stroke or neurotrauma. MBP is the major protein component of myelin. Serum levels of MBP are elevated in diseases in which there is myelin breakdown. Studies of patients with TBI have shown that MBP levels correlate with clinical measures of severity and may allow early prediction of outcomes [74,77,78].
New developments in neuroimaging, such as functional magnetic resonance imaging (MRI) and positron emission tomography, have revolutionized our understanding of abnormal brain function in many disease states, including schizophrenia, Parkinson's disease, and post-traumatic stress disorder. To study further whether critical illness-associated CD is associated with occult brain injury in humans, it would be useful to have an imaging test that can detect subtle evidence of brain injury. Unfortunately, traditional CT scans and MRI do not appear to be sensitive enough to pick up the microscopic cellular changes that may underlie CD [42]. Two small studies assessed brain CT findings in critically ill patients with sepsis [79,80]. Neither study demonstrated any CT abnormalities, although brain pathology in nonsurvivors was consistent with the previously cited findings of Sharshar and colleagues [49-52]. A recent study of ARDS survivors (n = 15) [81] found that many of these individuals exhibited signs of significant brain atrophy and ventricular enlargement on head CTs obtained during their acute illness, but there were no significant correlations between these abnormalities and subsequent neurocognitive scores.
A new MRI technique may prove useful for identifying occult brain injury in critically ill patients. Specifically, high-resolution, three-dimensional MRI can be used to assess noninvasively differences in brain tissue sodium concentration, which is a highly sensitive marker of tissue viability that highlights areas that traditional MRI can miss [82-86]. The method is based on sodium ion homeostasis, which is tightly regulated in the body and is a major energy consuming process. Any event that perturbs the energy level of the cell enough to disrupt the sodium ion gradient, such as ischemia, has an important impact on cell viability. Although tissue sodium concentration MRI has been successfully used to evaluate the CNS, including nonhuman primate studies and clinical studies of stroke and reversible focal brain ischemia [87-89], it has not been used to assess patients with either acute or chronic critical illness-associated CD.
Emerging therapeutics
There are several recent developments that, although preliminary, are of interest because of their potential to prevent or mitigate critical illness-associated CD.
Haloperidol
Haloperidol has been used for many years to manage agitation in mechanically ventilated ICU patients, and it is the recommended drug for treatment of ICU delirium [90]. Kalisvaart and colleagues [91] compared the effect of halo-peridol prophylaxis (1.5 mg/day preoperatively and up to 3 days postoperatively) with that of placebo in 430 elderly hip surgery patients at risk for delirium. Although there was no difference in the incidence of postoperative delirium between treatment and control groups, those in the haloperidol group had significantly reduced severity and duration of delirium (5.4 days versus 11.8 days; P < 0.001). Haloperiodol also appeared to reduce the length of hospital stay among those who developed delirium (17.1 days versus 22.6 days; P < 0.001). A recent retrospective cohort study examined haloperidol use in 989 patients who were mechanically ventilated for longer than 48 hours [92]. Despite similar baseline characteristics, patients treated with haloperidol had significantly lower hospital mortality than did those who never received the drug (20.5% versus 36.1%; P = 0.004), an association that persisted after adjusting for potential confounders. Because of the observational nature of the study and the potential risks associated with haloperidol use, these findings require confirmation in a randomized, controlled trial before they may be applied to routine patient care.
Gabapentin
Leung and colleagues [93] tested the hypothesis that using gabapentin as an add-on agent for treating postoperative pain reduces the occurrence of postoperative delirium. Patients aged 45 years or older undergoing spine surgery were randomly assigned to gabapentin 900 mg or placebo by mouth 1 to 2 hours before surgery and continued for the first 3 days postoperatively. Postoperative delirium occurred in 0% (0/9) of gabapentin-treated patients and 42% (5/12) of placebo patients (P = 0.045). Reduction in delirium appeared to be due to the opioid-sparing effect of gabapentin. Given the small size of the study, these results require confirmation.
Donepezil
Donepezil, a cholinesterase inhibitor that increases synaptic availability of acetylcholine, improves cognitive function in Alzheimer's disease. Sampson and colleagues [94] randomly assigned 33 elderly patients undergoing elective total hip replacement to donepezil 5 mg or placebo immediately following surgery and every 24 hours for 3 days. Donepezil was well tolerated with no serious adverse events. Although the drug did not significantly reduce the incidence of delirium (9.5% versus 35.7%; P = 0.08) or length of hospital stay (mean ± standard error: 9.9 ± 0.73 days versus 12.1 ± 1.09 days; P = 0.09), both outcomes showed a consistent trend suggesting possible benefit. The authors project that a sample size of 95 patients would be required for a definitive trial.
Dexmedetomidine
Dexmedetomidine's sedative effects are due to selective stimulation of α2-adrenoreceptors in the locus ceruleus of the CNS. Because it does not have anticholinergic or GABA-stimulating effects, it has the potential to be a delirium-sparing sedative. In preliminary results presented in abstract form [95], cardiac surgery patients (n = 55) randomly assigned to dexmedetomidine for postoperative sedation had a nonsignificantly lower incidence of postoperative delirium as compared with those sedated with propofol or a combination of fentanyl and midazolam (5% versus 54% versus 46%). The authors of that report plan to enroll a total of 90 patient in the study; perhaps these impressive differences will be statistically significant with a greater number of patients.
Recombinant human erythropoietin
Recombinant human erythropoietin (rHuEPO) has received considerable attention as a potential transfusion sparing strategy in the ICU. Interestingly, EPO and its receptor are both expressed by the nervous system, and systemically administered rHuEPO can reach sites within the brain. In preclinical studies, rHuEPO reduced neuronal injury produced by focal ischemia, TBI, spinal cord injury, and subarachnoid hemorrhage [96-98]. Enthusiasm regarding its use as a general neuroprotectant in the ICU has been tempered by potential risks such as thromboembolism and the considerable cost of the drug. Concerns over safety may be at least partially addressed by the recent finding of erythropoietin derivatives with tissue protective but not hematopoietic properties [99].
Xenon
Xenon is a chemically inert gas that has been used as an anesthetic agent and for contrast enhancement in CT scans. In rats xenon appears to protect the brain from the neurologic damage associated with the use of cardiopulmonary bypass, an effect that is potentially related to N-methyl-D-aspartate receptor antagonism [100]. However, its tendency to expand gaseous bubbles, such as bypass-associated cerebral air emboli, could abolish any beneficial effect or even worsen cerebral outcome [101].
Other potentially therapeutic agents
In the setting of ischemic stroke or TBI, there are a variety of compounds with the potential to improve neurologic outcomes. For example, NXY-059, a free radical trapping agent, reduced disability at 90 days when given within 6 hours of stroke onset [102]. In a pilot randomized trial in 56 patients, simvastatin given up to 12 hours after stroke onset significantly improved neurologic functioning (National Institutes of Health Stroke Scale score) at 90 days [103]. Ethyl pyruvate, a pyruvate derivative that prevents mortality in murine sepsis models, reduced motor impairments, neurologic deficits, and infarct volume in a rat stroke model when given as late as 12 hours after middle cerebral artery occlusion [104]. In rodent models of TBI, cyclosporin A reduced acute motor deficits and improved cognitive performance, even when given after the traumatic insult [105]. A phase II dose escalation trial is currently underway in humans.
Hypothermia
Mounting evidence suggests that mild-to-moderate hypothermia can mitigate neurologic injury. Shankaran and colleagues [106] found that whole-body hypothermia (33.5°C for 72 hours) reduced the risk for death or disability in infants with moderate or severe hypoxic-ischemic encephalopathy. In adults successfully resuscitated after cardiac arrest, moderate hypothermia (32–34°C for 12 to 24 hours) increased rates of favorable neurologic outcomes and reduced mortality [107,108]. A practical limitation of therapeutic hypothermia is that reaching target temperatures takes at least 2 hours using the fastest currently available cooling techniques. However, Polderman and colleagues [109] demonstrated that hypothermia could be induced safely and quickly (about 60 min) by means of ice-cold intravenous fluid combined with ice-water cooling blankets.
Cognitive rehabilitation
Cognitive rehabilitation involves the teaching of skills and strategies to target specific problems in perception, memory, thinking and problem solving, with the goal of improving function and compensating for deficits. The benefits of cognitive rehabilitation are well known to those that care for patients with stroke, anoxia, or TBI. Predicting who will benefit and how much has proven challenging, but even severely disabled patients sometimes make dramatic neuro-cognitive recoveries [110]. Although there are no studies evaluating the effectiveness of cognitive rehabilitation in patients recovering from non-neurologic critical illness, it stands to reason that such patients could benefit when they are found to be cognitively impaired. Because cognitive impairments in critically ill patients appear to be under-recognized by ICU and physical rehabilitation providers [111], few patients are referred for cognitive rehabilitation therapy [3]. Education regarding the cognitive sequelae of critical illness is needed to enhance referrals for rehabilitation, not only for weakness and physical debilitation but also for cognitive impairments.
Conclusion
Cognitive function is an important and relatively understudied outcome of critical illness. Evidence suggests that neuro-transmitter abnormalities and occult diffuse brain injury are important pathophysiologic mechanisms that underlie critical illness-associated CD. Markers that could be used to evaluate the influence of these mechanisms in individual patients include the following: SAA, certain brain proteins (S-100β, NSE, and MPB), and MRI tissue sodium concentration. Although recent advances in this area are exciting, they are still too immature to influence patient care. Additional research is needed if we are to understand better the relative contributions of specific mechanisms to the development of critical illness-associated cognitive dysfunction and to determine whether these mechanisms might be amenable to treatment or prevention.
Abbreviations
ARDS = acute respiratory distress syndrome; CD = cognitive dysfunction; CNS = central nervous system; CT = computed tomography; DSM = Diagnostic and Statistical Manual of Mental Disorders; GABA = γ-aminobutyric acid; ICU = intensive care unit; MBP = myelin basic protein; MRI = magnetic resonance imaging; NSE = neuron-specific enolase; rHuEPO = recombinant human erythropoietin; SAA = serum anticholinergic activity; TBI = traumatic brain injury.
Competing interests
The authors declare that they have no competing interests.
Note
This article is part of a thematic series on Translational research, edited by John Kellum.
Other articles in the series can be found online at http://ccforum.com/articles/theme-series.asp?series=CC_Trans
Acknowledgments
Acknowledgements
This work was performed at the University of Pittsburgh School of Medicine, Pittsburgh, Philadelphia, USA.
References
- Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348:1546–1554. doi: 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]
- Milberg JA, Davis DR, Steinberg KP, Hudson LD. Improved survival of patients with acute respiratory distress syndrome (ARDS): 1983–1993. JAMA. 1995;273:306–309. doi: 10.1001/jama.273.4.306. [DOI] [PubMed] [Google Scholar]
- Hopkins RO, Weaver LK, Collingridge D, Parkinson RB, Chan KJ, Orme JF., Jr Two-year cognitive, emotional, and quality-of-life outcomes in acute respiratory distress syndrome. Am J Respir Crit Care Med. 2005;171:340–347. doi: 10.1164/rccm.200406-763OC. [DOI] [PubMed] [Google Scholar]
- Fried TR, Bradley EH, Towle VR, Allore H. Understanding the treatment preferences of seriously ill patients. N Engl J Med. 2002;346:1061–1066. doi: 10.1056/NEJMsa012528. [DOI] [PubMed] [Google Scholar]
- Milbrandt EB, Angus DC. Potential mechanisms and markers of critical illness-associated cognitive dysfunction. Curr Opin Crit Care. 2005;11:355–359. doi: 10.1097/01.ccx.0000170508.63067.04. [DOI] [PubMed] [Google Scholar]
- Ely EW, Inouye SK, Bernard GR, Gordon S, Francis J, May L, Truman B, Speroff T, Gautam S, Margolin R, Hart RP, Dittus R. Delirium in mechanically ventilated patients: validity and reliability of the confusion assessment method for the intensive care unit (CAM-ICU) Jama. 2001;286:2703–10. doi: 10.1001/jama.286.21.2703. [DOI] [PubMed] [Google Scholar]
- Ely W, Gautam S, Margolin R, Francis J, May L, Speroff T, Truman B, Dittus R, Bernard R, Inouye K. The impact of delirium in the intensive care unit on hospital length of stay. Intensive Care Med. 2001;27:1892–1900. doi: 10.1007/s00134-001-1132-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ely EW, Shintani A, Truman B, Speroff T, Gordon SM, Harrell FE, Jr, Inouye SK, Bernard GR, Dittus RS. Delirium as a predictor of mortality in mechanically ventilated patients in the intensive care unit. Jama. 2004;291:1753–1762. doi: 10.1001/jama.291.14.1753. [DOI] [PubMed] [Google Scholar]
- Milbrandt EB, Deppen S, Harrison PL, Shintani AK, Speroff T, Stiles RA, Truman B, Bernard GR, Dittus RS, Ely EW. Costs associated with delirium in mechanically ventilated patients. Crit Care Med. 2004;32:955–962. doi: 10.1097/01.CCM.0000119429.16055.92. [DOI] [PubMed] [Google Scholar]
- Kiely DK, Bergmann MA, Murphy KM, Jones RN, Orav EJ, Marcantonio ER. Delirium among newly admitted postacute facility patients: prevalence, symptoms, and severity. J Gerontol A Biol Sci Med Sci. 2003;58:M441–M445. doi: 10.1093/gerona/58.5.m441. [DOI] [PubMed] [Google Scholar]
- Levkoff SE, Evans DA, Liptzin B, Cleary PD, Lipsitz LA, Wetle TT, Reilly CH, Pilgrim DM, Schor J, Rowe J. Delirium. The occurrence and persistence of symptoms among elderly hospitalized patients. Arch Intern Med. 1992;152:334–40. doi: 10.1001/archinte.152.2.334. [DOI] [PubMed] [Google Scholar]
- Marcantonio ER, Simon SE, Bergmann MA, Jones RN, Murphy KM, Morris JN. Delirium symptoms in post-acute care: prevalent, persistent, and associated with poor functional recovery. J Am Geriatr Soc. 2003;51:4–9. doi: 10.1034/j.1601-5215.2002.51002.x. [DOI] [PubMed] [Google Scholar]
- McNicoll L, Pisani MA, Zhang Y, Ely EW, Siegel MD, Inouye SK. Delirium in the intensive care unit: occurrence and clinical course in older patients. J Am Geriatr Soc. 2003;51:591–598. doi: 10.1034/j.1600-0579.2003.00201.x. [DOI] [PubMed] [Google Scholar]
- Girard T, Jackson JC, Pun BT, Ely EW, Shintani A, Renfrew JW, Dunn J, Laskowitz DT. Apolipoprotein E4 polymorphism as a genetic predisposition to delirium in humans. Crit Care Med. 2006;33:A22. doi: 10.1097/00003246-200512002-00086. [DOI] [PubMed] [Google Scholar]
- Inouye SK. Delirium in older persons. N Engl J Med. 2006;354:1157–1165. doi: 10.1056/NEJMra052321. [DOI] [PubMed] [Google Scholar]
- Mulsant BH, Pollock BG, Kirshner M, Shen C, Dodge H, Ganguli M. Serum anticholinergic activity in a community-based sample of older adults: relationship with cognitive performance. Arch Gen Psychiatry. 2003;60:198–203. doi: 10.1001/archpsyc.60.2.198. [DOI] [PubMed] [Google Scholar]
- Carnahan RM, Lund BC, Perry PJ, Pollock BG. A critical appraisal of the utility of the serum anticholinergic activity assay in research and clinical practice. Psychopharmacol Bull. 2002;36:24–39. [PubMed] [Google Scholar]
- Pratico C, Quattrone D, Lucanto T, Amato A, Penna O, Roscitano C, Fodale V. Drugs of anesthesia acting on central cholinergic system may cause post-operative cognitive dysfunction and delirium. Med Hypotheses. 2005;65:972–982. doi: 10.1016/j.mehy.2005.05.037. [DOI] [PubMed] [Google Scholar]
- Flacker JM, Wei JY. Endogenous anticholinergic substancesmay exist during acute illness in elderly medical patients. J Gerontol A Biol Sci Med Sci. 2001;56:M353–M355. doi: 10.1093/gerona/56.6.m353. [DOI] [PubMed] [Google Scholar]
- Czura CJ, Friedman SG, Tracey KJ. Neural inhibition of inflammation: the cholinergic anti-inflammatory pathway. J Endotoxin Res. 2003;9:409–413. doi: 10.1179/096805103225002755. [DOI] [PubMed] [Google Scholar]
- Carnahan RM, Lund BC, Perry PJ, Culp KR, Pollock BG. The relationship of an anticholinergic rating scale with serum anti-cholinergic activity in elderly nursing home residents. Psychopharmacol Bull. 2002;36:14–19. [PubMed] [Google Scholar]
- Carnahan RM, Lund BC, Perry PJ, Culp KR, Pollock BG. Comparing models for estimating anticholinergic burden from medications using serum anticholinergic activity as the gold standard. Pharmacotherapy. 2006;25:1496–1497. [Google Scholar]
- Tune L, Coyle JT. Serum levels of anticholinergic drugs in treatment of acute extrapyramidal side effects. Arch Gen Psychiatry. 1980;37:293–297. doi: 10.1001/archpsyc.1980.01780160063007. [DOI] [PubMed] [Google Scholar]
- Tune LE, Damlouji NF, Holland A, Gardner TJ, Folstein MF, Coyle JT. Association of postoperative delirium with raised serum levels of anticholinergic drugs. Lancet. 1981;2:651–653. doi: 10.1016/S0140-6736(81)90994-6. [DOI] [PubMed] [Google Scholar]
- Flacker JM, Cummings V, Mach JR, Jr, Bettin K, Kiely DK, Wei J. The association of serum anticholinergic activity with delirium in elderly medical patients. Am J Geriatr Psychiatry. 1998;6:31–41. [PubMed] [Google Scholar]
- Inouye SK, van Dyck CH, Alessi CA, Balkin S, Siegal AP, Horwitz RI. Clarifying confusion: the confusion assessment method. A new method for detection of delirium. Ann Intern Med. 1990;113:941–8. doi: 10.7326/0003-4819-113-12-941. [DOI] [PubMed] [Google Scholar]
- Mussi C, Ferrari R, Ascari S, Salvioli G. Importance of serum anticholinergic activity in the assessment of elderly patients with delirium. J Geriatr Psychiatry Neurol. 1999;12:82–86. doi: 10.1177/089198879901200208. [DOI] [PubMed] [Google Scholar]
- Golinger RC, Peet T, Tune LE. Association of elevated plasma anticholinergic activity with delirium in surgical patients. Am J Psychiatry. 1987;144:1218–1220. doi: 10.1176/ajp.144.9.1218. [DOI] [PubMed] [Google Scholar]
- Sommer BR, Wise LC, Kraemer HC. Is dopamine administration possibly a risk factor for delirium? Crit Care Med. 2002;30:1508–1511. doi: 10.1097/00003246-200207000-00019. [DOI] [PubMed] [Google Scholar]
- Boyer EW, Shannon M. The serotonin syndrome. N Engl J Med. 2005;352:1112–1120. doi: 10.1056/NEJMra041867. [DOI] [PubMed] [Google Scholar]
- Fischer JE, Rosen HM, Ebeid AM, James JH, Keane JM, Soeters PB. The effect of normalization of plasma amino acids on hepatic encephalopathy in man. Surgery. 1976;80:77–91. [PubMed] [Google Scholar]
- Glue P, Nutt D. Overexcitement and disinhibition. Dynamic neurotransmitter interactions in alcohol withdrawal. Br J Psychiatry. 1990;157:491–499. doi: 10.1192/bjp.157.4.491. [DOI] [PubMed] [Google Scholar]
- Gaudreau JD, Gagnon P, Roy MA, Harel F, Tremblay A. Association between psychoactive medications and delirium in hospitalized patients: a critical review. Psychosomatics. 2005;46:302–316. doi: 10.1176/appi.psy.46.4.302. [DOI] [PubMed] [Google Scholar]
- Pandharipande P, Ely EW. Sedative and analgesicmedications: risk factors for delirium and sleep disturbances in the critically ill. Crit Care Clin. 2006;22:313–27. doi: 10.1016/j.ccc.2006.02.010. vii. [DOI] [PubMed] [Google Scholar]
- Pandharipande P, Shintani A, Peterson J, Pun BT, Wilkinson GR, Dittus RS, Bernard GR, Ely EW. Lorazepam is an independent risk factor for transitioning to delirium in intensive care unit patients. Anesthesiology. 2006;104:21–26. doi: 10.1097/00000542-200601000-00005. [DOI] [PubMed] [Google Scholar]
- Kress JP, Pohlman AS, O'Connor MF, Hall JB. Daily interruption of sedative infusions in critically ill patients undergoing mechanical ventilation. N Engl J Med. 2000;342:1471–7. doi: 10.1056/NEJM200005183422002. [DOI] [PubMed] [Google Scholar]
- Ancelin ML, de Roquefeuil G, Ledesert B, Bonnel F, Cheminal JC, Ritchie K. Exposure to anaesthetic agents, cognitive functioning and depressive symptomatology in the elderly. Br J Psychiatry. 2001;178:360–366. doi: 10.1192/bjp.178.4.360. [DOI] [PubMed] [Google Scholar]
- Dodds C, Allison J. Postoperative cognitive deficit in the elderly surgical patient. Br J Anaesth. 1998;81:449–462. doi: 10.1093/bja/81.3.449. [DOI] [PubMed] [Google Scholar]
- Crippen D. Agitation in the ICU: part one – anatomical and physiologic basis for the agitated state. Crit Care (Lond) 1999;3:R35–R46. doi: 10.1186/cc348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flacker JM, Lipsitz LA. Neural mechanisms of delirium: current hypotheses and evolving concepts. J Gerontol A Biol Sci Med Sci. 1999;54:B239–B246. doi: 10.1093/gerona/54.6.b239. [DOI] [PubMed] [Google Scholar]
- van der Mast RC. Pathophysiology of delirium. J Geriatr Psychiatry Neurol. 1998;11:138–145. doi: 10.1177/089198879801100304. [DOI] [PubMed] [Google Scholar]
- Orlikowski D, Sharshar T, Annane D. The brain in sepsis. Adv Sepsis. 2003;3:8–14. [Google Scholar]
- Barichello T, Fortunato JJ, Vitali AM, Feier G, Reinke A, Moreira JC, Quevedo J, Dal Pizzol F. Oxidative variables in the rat brain after sepsis induced by cecal ligation and perforation. Crit Care Med. 2006;34:886–889. doi: 10.1097/01.CCM.0000201880.50116.12. [DOI] [PubMed] [Google Scholar]
- Christenson JT, Kuikka JT, Owunwanne A, Al-Sarraf AA. Cerebral circulation during endotoxic shock with special emphasis on the regional cerebral blood flow in vivo. Nucl Med Commun. 1986;7:531–540. doi: 10.1097/00006231-198607000-00007. [DOI] [PubMed] [Google Scholar]
- Ekstrom-Jodal B, Haggendal J, Larsson LE, Westerlind A. Cerebral hemodynamics, oxygen uptake and cerebral arteriove-nous differences of catecholamines following E. coli endotoxin in dogs. Acta Anaesthesiol Scand. 1982;26:446–452. doi: 10.1111/j.1399-6576.1982.tb01797.x. [DOI] [PubMed] [Google Scholar]
- Ekstrom-Jodal B, Haggendal E, Larsson LE. Cerebral blood flow and oxygen uptake in endotoxic shock. An experimental study in dogs. Acta Anaesthesiol Scand. 1982;26:163–170. doi: 10.1111/j.1399-6576.1982.tb01746.x. [DOI] [PubMed] [Google Scholar]
- Parker JL, Emerson TE., Jr Cerebral hemodynamics, vascular reactivity, and metabolism during canine endotoxin shock. Circ Shock. 1977;4:41–53. [PubMed] [Google Scholar]
- Rudinsky BF, Lozon M, Bell A, Hipps R, Meadow WL. Group B streptococcal sepsis impairs cerebral vascular reactivity to acute hypercarbia in piglets. Pediatr Res. 1996;39:55–63. doi: 10.1203/00006450-199601000-00008. [DOI] [PubMed] [Google Scholar]
- Gray F, Sharshar T, De La Grandmaison GL, Annane D. Neuropathology of septic shock. Neuropathol Appl Neurobiol. 2002;28:159. doi: 10.1046/j.1365-2990.2002.39286_30.x. [DOI] [Google Scholar]
- Sharshar T, Gray F, Poron F, Raphael JC, Gajdos P, Annane D. Multifocal necrotizing leukoencephalopathy in septic shock. Crit Care Med. 2002;30:2371–2375. doi: 10.1097/00003246-200210000-00031. [DOI] [PubMed] [Google Scholar]
- Sharshar T, Gray F, Lorin de la Grandmaison G, Hopkinson NS, Ross E, Dorandeu A, Orlikowski D, Raphael JC, Gajdos P, Annane D. Apoptosis of neurons in cardiovascular autonomic centres triggered by inducible nitric oxide synthase after death from septic shock. Lancet. 2003;362:1799–1805. doi: 10.1016/S0140-6736(03)14899-4. [DOI] [PubMed] [Google Scholar]
- Sharshar T, Annane D, De La Grandmaison GL, Brouland JP, Hopkinson NS, Francoise G. The neuropathology of septic shock. Brain Pathol. 2004;14:21–33. doi: 10.1111/j.1750-3639.2004.tb00494.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijdicks EF, Stevens M. The role of hypotension in septicencephalopathy following surgical procedures. Arch Neurol. 1992;49:653–656. doi: 10.1001/archneur.1992.00530300093015. [DOI] [PubMed] [Google Scholar]
- Maekawa T, Fujii Y, Sadamitsu D, Yokota K, Soejima Y, Ishikawa T, Miyauchi Y, Takeshita H. Cerebral circulation and metabolism in patients with septic encephalopathy. Am J Emerg Med. 1991;9:139–143. doi: 10.1016/0735-6757(91)90175-J. [DOI] [PubMed] [Google Scholar]
- Beloosesky Y, Grinblat J, Pirotsky A, Weiss A, Hendel D. Different C-reactive protein kinetics in post-operative hip-fractured geriatric patients with and without complications. Gerontology. 2004;50:216–222. doi: 10.1159/000078350. [DOI] [PubMed] [Google Scholar]
- Karlidag R, Unal S, Sezer OH, Bay KA, Battaloglu B, But A, Ozcan C. The role of oxidative stress in postoperative delirium. Gen Hosp Psychiatry. 2006;28:418–423. doi: 10.1016/j.genhosppsych.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Yokota H, Ogawa S, Kurokawa A, Yamamoto Y. Regional cerebral blood flow in delirium patients. Psychiatry Clin Neurosci. 2003;57:337–339. doi: 10.1046/j.1440-1819.2003.01126.x. [DOI] [PubMed] [Google Scholar]
- Hopkins RO, Weaver LK, Pope D, Orme JF, Bigler ED, Larson-LOHR V. Neuropsychological sequelae and impaired health status in survivors of severe acute respiratory distress syndrome. Am J Respir Crit Care Med. 1999;160:50–56. doi: 10.1164/ajrccm.160.1.9708059. [DOI] [PubMed] [Google Scholar]
- Hopkins RO, Suchyta MR, Jephson A, et al. Hyperglycemia and neurocognitive outcome in ARDS survivors [abstract] Am J Respir Crit Care Med. 2005;2:A36. [Google Scholar]
- Chew W, Kucharczyk J, Moseley M, Derugin N, Norman D. Hyperglycemia augments ischemic brain injury: in vivo MR imaging/spectroscopic study with nicardipine in cats with occluded middle cerebral arteries. AJNR Am J Neuroradiol. 1991;12:603–609. [PMC free article] [PubMed] [Google Scholar]
- Aberg T. Signs of brain cell injury during open heart operations: past and present. Ann Thorac Surg. 1995;59:1312–1315. doi: 10.1016/0003-4975(95)00194-P. [DOI] [PubMed] [Google Scholar]
- Herrmann M, Vos P, Wunderlich MT, de Bruijn CH, Lamers KJ. Release of glial tissue-specific proteins after acute stroke: a comparative analysis of serum concentrations of protein S-100B and glial fibrillary acidic protein. Stroke. 2000;31:2670–2677. doi: 10.1161/01.str.31.11.2670. [DOI] [PubMed] [Google Scholar]
- Svenmarker S, Sandstrom E, Karlsson T, Aberg T. Is there an association between release of protein S100B during car-diopulmonary bypass and memory disturbances? Scand Cardiovasc J. 2002;36:117–122. doi: 10.1080/140174302753675410. [DOI] [PubMed] [Google Scholar]
- Jonsson H, Johnsson P, Alling C, Westaby S, Blomquist S. Significance of serum S100 release after coronary artery bypass grafting. Ann Thorac Surg. 1998;65:1639–1644. doi: 10.1016/S0003-4975(98)00229-X. [DOI] [PubMed] [Google Scholar]
- Hardemark HG, Ericsson N, Kotwica Z, Rundstrom G, Mendel-Hartvig I, Olsson Y, Pahlman S, Persson L. S-100 protein and neuron-specific enolase in CSF after experimental traumatic or focal ischemic brain damage. J Neurosurg. 1989;71:727–731. doi: 10.3171/jns.1989.71.5.0727. [DOI] [PubMed] [Google Scholar]
- Raabe A, Grolms C, Keller M, Dohnert J, Sorge O, Seifert V. Correlation of computed tomography findings and serum brain damage markers following severe head injury. Acta Neurochir (Wien) 1998;140:787–791. doi: 10.1007/s007010050180. [DOI] [PubMed] [Google Scholar]
- Raabe A, Grolms C, Sorge O, Zimmermann M, Seifert V. Serum S-100B protein in severe head injury. Neurosurgery. 1999;45:477–483. doi: 10.1097/00006123-199909000-00012. [DOI] [PubMed] [Google Scholar]
- Ingebrigtsen T, Romner B. Biochemical serum markers for brain damage: a short review with emphasis on clinical utility in mild head injury. Restor Neurol Neurosci. 2003;21:171–176. [PubMed] [Google Scholar]
- Routsi C, Stamataki E, Nanas S, Psachoulia C, Zervou M, Koro-neos A, Stathopoulos A, Roussos C. Increased levels of serum S100B protein in critically ill patients without brain injury [poster presentation] Crit Care. 2005;(Suppl 1):P289. doi: 10.1186/cc3352. [DOI] [PubMed] [Google Scholar]
- Lipcsey M, Larsson A, Sjolin J, Hansson L, Eriksson M. Blood-brain barrier damage is an early event in porcine endotoxemic shock [poster presentation] Crit Care. 2005;(Suppl 1):P278. doi: 10.1186/cc3341. [DOI] [Google Scholar]
- Fries M, Bickenbach J, Henzler D, Beckers S, Dembinski R, Sell-haus B, Rossaint R, Kuhlen R. S-100 protein and neuro-histopathologic changes in a porcine model of acute lung injury. Anesthesiology. 2005;102:761–767. doi: 10.1097/00000542-200504000-00011. [DOI] [PubMed] [Google Scholar]
- Van Eldik LJ, Wainwright MS. The Janus face of glial-derived S100B: beneficial and detrimental functions in the brain. Restor Neurol Neurosci. 2003;21:97–108. [PubMed] [Google Scholar]
- Dauberschmidt R, Marangos PJ, Zinsmeyer J, Bender V, Klages G, Gross J. Severe head trauma and the changes of concentration of neuron-specific enolase in plasma and in cere-brospinal fluid. Clin Chim Acta. 1983;131:165–170. doi: 10.1016/0009-8981(83)90085-2. [DOI] [PubMed] [Google Scholar]
- Yamazaki Y, Yada K, Morii S, Kitahara T, Ohwada T. Diagnostic significance of serum neuron-specific enolase and myelin basic protein assay in patients with acute head injury. Surg Neurol. 1995;43:267–270. doi: 10.1016/0090-3019(95)80012-6. [DOI] [PubMed] [Google Scholar]
- Skogseid IM, Nordby HK, Urdal P, Paus E, Lilleaas F. Increased serum creatine kinase BB and neuron specific enolase following head injury indicates brain damage. Acta Neurochir (Wien) 1992;115:106–111. doi: 10.1007/BF01406367. [DOI] [PubMed] [Google Scholar]
- Weigand MA, Volkmann M, Schmidt H, Martin E, Bohrer H, Bar-denheuer HJ. Neuron-specific enolase as a marker of fatal outcome in patients with severe sepsis or septic shock. Anesthesiology. 2000;92:905–907. doi: 10.1097/00000542-200003000-00057. [DOI] [PubMed] [Google Scholar]
- Thomas DG, Palfreyman JW, Ratcliffe JG. Serum-myelin-basic-protein assay in diagnosis and prognosis of patients with head injury. Lancet. 1978;1:113–115. doi: 10.1016/S0140-6736(78)90415-4. [DOI] [PubMed] [Google Scholar]
- Thomas DG, Rabow L, Teasdale G. Serum myelin basic protein, clinical responsiveness, and outcome of severe head injury. Acta Neurochir Suppl (Wien) 1979;28:93–95. doi: 10.1007/978-3-7091-4088-8_20. [DOI] [PubMed] [Google Scholar]
- Jackson AC, Gilbert JJ, Young GB, Bolton CF. The encephalopathy of sepsis. Can J Neurol Sci. 1985;12:303–307. doi: 10.1017/s0317167100035381. [DOI] [PubMed] [Google Scholar]
- Young B. Neurologic complications of systemic critical illness. Neurologic Clinics. 1995;13:645–658. [PubMed] [Google Scholar]
- Hopkins RO, Gale SD, Weaver LK. Brain atrophy and cognitive impairment in survivors of acute respiratory distress syndrome. Brain Inj. 2006;20:263–271. doi: 10.1080/02699050500488199. [DOI] [PubMed] [Google Scholar]
- Boada FE, Gillen JS, Shen GX, Chang SY, Thulborn KR. Fast three dimensional sodium imaging. Magn Reson Med. 1997;37:706–715. doi: 10.1002/mrm.1910370512. [DOI] [PubMed] [Google Scholar]
- Clayton DB, Lenkinski RE. MR imaging of sodium in thehuman brain with a fast three-dimensional gradient-recalled-echo sequence at 4 T. Acad Radiol. 2003;10:358–365. doi: 10.1016/S1076-6332(03)80023-5. [DOI] [PubMed] [Google Scholar]
- Constantinides CD, Kraitchman DL, O'Brien KO, Boada FE, Gillen J, Bottomley PA. Noninvasive quantification of total sodium concentrations in acute reperfused myocardial infarction using 23Na MRI. Magn Reson Med. 2001;46:1144–1151. doi: 10.1002/mrm.1311. [DOI] [PubMed] [Google Scholar]
- Hancu I, Boada FE, Shen GX. Three-dimensional triple-quantum-filtered (23)Na imaging of in vivo human brain. Magn Reson Med. 1999;42:1146–1154. doi: 10.1002/(SICI)1522-2594(199912)42:6<1146::AID-MRM20>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Lin SP, Song SK, Miller JP, Ackerman JJ, Neil JJ. Direct, longitudinal comparison of (1)H and (23)Na MRI after transient focal cerebral ischemia. Stroke. 2001;32:925–932. doi: 10.1161/01.str.32.4.925. [DOI] [PubMed] [Google Scholar]
- LaVerde GC, Boada FE, Davis D, Nemoto E, Jungreis CA. International Society for Magnetic Resonance in Medicine 12th Scientific Meeting and Exhibition 2004, Abstract number 1695 Proceedings of the 12th Scientific Meeting of the International Society for Magnetic Resonance in Medicine 15–21 May 2004. Kyoto International Conference Hall, Kyoto, Japan. Mira Digital Publishing. St. Louis, MO; Sodium MRI of reversible focal brain inchemia in the monkey [abstract] [Google Scholar]
- Thulborn KR, Gindin TS, Davis D, Erb P. Comprehensive MRimaging protocol for stroke management: tissue sodium concentration as a measure of tissue viability in nonhuman primate studies and in clinical studies. Radiology. 1999;213:156–166. doi: 10.1148/radiology.213.1.r99se15156. [DOI] [PubMed] [Google Scholar]
- Lin SP, Song SK, Miller JP, Ackerman JJ, Neil JJ. Direct, longitudinal comparison of (1)H and (23)Na MRI after transient focal cerebral ischemia. Stroke. 2001;32:925–932. doi: 10.1161/01.str.32.4.925. [DOI] [PubMed] [Google Scholar]
- Jacobi J, Fraser GL, Coursin DB, Riker RR, Fontaine D, Wittbrodt ET, Chalfin DB, Masica MF, Bjerke HS, Coplin WM, et al. Clinical practice guidelines for the sustained use of sedatives and analgesics in the critically ill adult. Crit Care Med. 2002;30:119–41. doi: 10.1097/00003246-200201000-00020. [DOI] [PubMed] [Google Scholar]
- Kalisvaart KJ, de Jonghe JF, Bogaards MJ, Vreeswijk R, Egberts TC, Burger BJ, Eikelenboom P, van Gool WA. Haloperidol prophylaxis for elderly hip-surgery patients at risk for delirium: a randomized placebo-controlled study. J Am Geriatr Soc. 2005;53:1658–1666. doi: 10.1111/j.1532-5415.2005.53503.x. [DOI] [PubMed] [Google Scholar]
- Milbrandt EB, Kersten A, Kong L, Weissfeld LA, Clermont G, Dremsizov T, Fink MP, Angus DC. Haloperidol use in mechanically ventilated patients is associated with lower hospital mortality. Crit Care Med. 2005;33:226–229. doi: 10.1097/01.CCM.0000150743.16005.9A. [DOI] [PubMed] [Google Scholar]
- Leung JM, Sands LP, Rico M, Petersen KL, Rowbotham MC, Dahl JB, Ames C, Chou D, Weinstein P. Pilot clinical trial of gabapentin to decrease postoperative delirium in older patients. Neurology. 2006;67:1251–1253. doi: 10.1212/01.wnl.0000233831.87781.a9. [DOI] [PubMed] [Google Scholar]
- Sampson EL, Raven PR, Ndhlovu PN, Vallance A, Garlick N, Watts J, Blanchard MR, Bruce A, Blizard R, Ritchie CW. A randomized, double-blind, placebo-controlled trial of donepezil hydrochloride (Aricept) for reducing the incidence of postoperative delirium after elective total hip replacement. Int J Geriatr Psychiatry. 2006 doi: 10.1002/gps.1679. [DOI] [PubMed] [Google Scholar]
- Maldonado JR, van der Starre P, Wysong A, Block T. Dexmedetomidine: Can it reduce the incidence of ICU delirium in postcardiotomy patients? Psychosomatics. 2004;45:173. [Google Scholar]
- Bernaudin M, Marti HH, Roussel S, Divoux D, Nouvelot A, MacKenzie ET, Petit E. A potential role for erythropoietin in focal permanent cerebral ischemia in mice. J Cereb Blood Flow Metab. 1999;19:643–651. doi: 10.1097/00004647-199906000-00007. [DOI] [PubMed] [Google Scholar]
- Brines ML, Ghezzi P, Keenan S, Agnello D, de Lanerolle NC, Cerami C, Itri LM, Cerami A. Erythropoietin crosses the blood-brain barrier to protect against experimental brain injury. Proc Natl Acad Sci USA. 2000;97:10526–10531. doi: 10.1073/pnas.97.19.10526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasso G, Buemi M, Alafaci C, Sfacteria A, Passalacqua M, Sturiale A, Calapai G, De Vico G, Piedimonte G, Salpietro FM, et al. Beneficial effects of systemic administration of recombinant human erythropoietin in rabbits subjected to subarachnoid hemorrhage. Proc Natl Acad Sci USA. 2002;99:5627–5631. doi: 10.1073/pnas.082097299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leist M, Ghezzi P, Grasso G, Bianchi R, Villa P, Fratelli M, Savino C, Bianchi M, Nielsen J, Gerwien J, et al. Derivatives of erythropoietin that are tissue protective but not erythropoietic. Science. 2004;305:239–242. doi: 10.1126/science.1098313. [DOI] [PubMed] [Google Scholar]
- Ma D, Yang H, Lynch J, Franks NP, Maze M, Grocott HP. Xenon attenuates cardiopulmonary bypass-induced neurologic andneurocognitive dysfunction in the rat. Anesthesiology. 2003;98:690–698. doi: 10.1097/00000542-200303000-00017. [DOI] [PubMed] [Google Scholar]
- Jungwirth B, Gordan ML, Blobner M, Schmehl W, Kochs EF, Mackensen GB. Xenon impairs neurocognitive and histologic outcome after cardiopulmonary bypass combined with cerebral air embolism in rats. Anesthesiology. 2006;104:770–776. doi: 10.1097/00000542-200604000-00022. [DOI] [PubMed] [Google Scholar]
- Lees KR, Zivin JA, Ashwood T, Davalos A, Davis SM, Diener HC, Grotta J, Lyden P, Shuaib A, Hardemark HG, Wasiewski WW. NXY-059 for acute ischemic stroke. N Engl J Med. 2006;354:588–600. doi: 10.1056/NEJMoa052980. [DOI] [PubMed] [Google Scholar]
- Montaner J, Chacon P, Krupinski J, Rubio F, Millan M, Escudero D, Hereu P, Molina C, Quintana M, Alvarez-Sabin J. Safety and efficacy of statin in the acute phase of ischemic stroke: the MISTICS Trial [Abstract] Stroke. 2004;35:293. [Google Scholar]
- Yu YM, Kim JB, Lee KW, Kim SY, Han PL, Lee JK. Inhibition of the cerebral ischemic injury by ethyl pyruvate with a wide therapeutic window. Stroke. 2005;36:2238–2243. doi: 10.1161/01.STR.0000181779.83472.35. [DOI] [PubMed] [Google Scholar]
- Alessandri B, Rice AC, Levasseur J, DeFord M, Hamm RJ, Bullock MR. Cyclosporin A improves brain tissue oxygen consumption and learning/memory performance after lateral fluid percussion injury in rats. J Neurotrauma. 2002;19:829–841. doi: 10.1089/08977150260190429. [DOI] [PubMed] [Google Scholar]
- Shankaran S, Laptook AR, Ehrenkranz RA, Tyson JE, McDonald SA, Donovan EF, Fanaroff AA, Poole WK, Wright LL, Higgins RD, et al. Whole-body hypothermia for neonates with hypoxic-ischemic encephalopathy. N Engl J Med. 2005;353:1574–1584. doi: 10.1056/NEJMcps050929. [DOI] [PubMed] [Google Scholar]
- Hypothermia after Cardiac Arrest Study Group Mild therapeutic hypothermia to improve the neurologic outcome after cardiac arrest. N Engl J Med. 2002;346:549–556. doi: 10.1056/NEJMoa012689. [DOI] [PubMed] [Google Scholar]
- Bernard SA, Gray TW, Buist MD, Jones BM, Silvester W, Gutteridge G, Smith K. Treatment of comatose survivors of out-of-hospital cardiac arrest with induced hypothermia. N Engl J Med. 2002;346:557–563. doi: 10.1056/NEJMoa003289. [DOI] [PubMed] [Google Scholar]
- Polderman KH, Rijnsburger ER, Peerdeman SM, Girbes ARJ. Induction of hypothermia in patients with various types of neurologic injury with use of large volumes of ice-cold intravenous fluid. Crit Care Med. 2005;33:2744–2751. doi: 10.1097/01.CCM.0000190427.88735.19. [DOI] [PubMed] [Google Scholar]
- Kaplan CP. Anoxic-hypotensive brain injury: neuropsychological performance at 1 month as an indicator of recovery. Brain Inj. 1999;13:305–310. doi: 10.1080/026990599121674. [DOI] [PubMed] [Google Scholar]
- Hopkins RO, Brett S. Chronic neurocognitive effects of critical illness. Curr Opin Crit Care. 2005;11:369–375. doi: 10.1097/01.ccx.0000166399.88635.a5. [DOI] [PubMed] [Google Scholar]