

Short abstract
How can protein-interaction networks can be made more complete?
Abstract
We estimate the full yeast protein-protein interaction network to contain 37,800-75,500 interactions and the human network 154,000-369,000, but owing to a high false-positive rate, current maps are roughly only 50% and 10% complete, respectively. Paradoxically, releasing raw, unfiltered assay data might help separate true from false interactions.
Networks are invaluable models for bettering our understanding of biological systems. Whether its constituent parts are molecules, cells, or living organisms, a network provides an organizing framework amenable to modeling the complex events that emerge from interactions among the parts. In functional genomics, concerted efforts over the past decade or so have produced rudimentary maps of the networks of genes, proteins, and metabolites controlling cells and, with these maps, have offered the promise of predictive, rather than just descriptive, models of molecular biology. Already, the network of physical interactions (the 'interactome') among yeast proteins, generated through a succession of experimental and algorithmic reconstructions, has proved its usefulness for discovering protein function [1,2], predicting cellular behavior [3,4], and the analysis of complex gene regulation [5-7]. Similar efforts for protein-interaction networks for Caenorhabditis elegans and Drosophila melanogaster are ongoing. We expect the human protein-interaction network to be equally informative; like the sequencing of the human genome, the construction of this map will represent a major step along the path towards understanding the functions of our genes.
Even in its current incomplete state, with interactions compiled from the literature, focused screens, and first-generation high-throughput interaction maps, the human protein-protein interaction network should be able to provide information about gene function and relevance to human health. For example, the emergent properties of proteins that are revealed in networks, as opposed to considering each protein in isolation, may identify genes and proteins critical to disease. Such a trend has been observed in yeast: a yeast gene's tendency to be essential correlates with the count of the encoded protein's interaction partners (the 'degree') [8]. Although not without its critics [9,10], this correlation would be exciting if present in animals. We have examined the current human protein-interaction network and find that this trend does indeed hold in humans (Figure 1). Among many other contributions, the human protein-interaction network will therefore focus attention on important hub proteins. Such proteins are likely to be essential to cell function and their disruption will often be lethal. Likewise, the network may focus attention on particularly important interactions: not all interactions are equally critical to the cell, and we might expect the network context of interactions (such as their centrality or association with essential proteins) to allow essential interactions to be identified.
Figure 1.
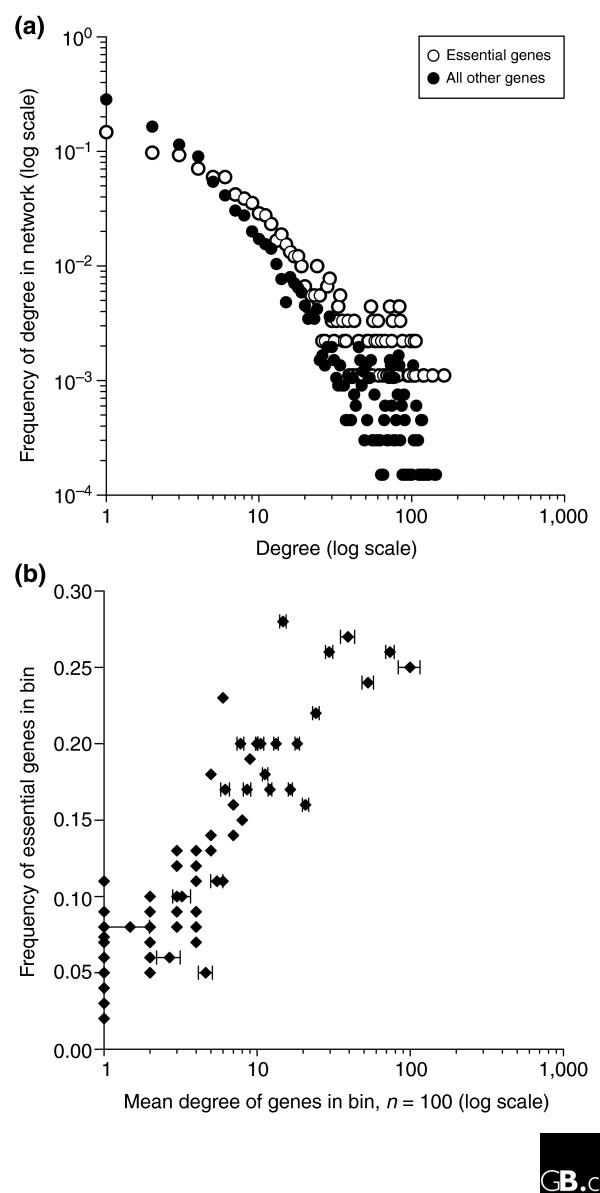
The tendency for a human gene to be essential correlates well with the number of its protein-interaction partners, suggesting that essential human genes can be identified directly from protein-interaction networks. (a) For a set of around 31,000 human protein interactions [49], the number of interactions per protein (the 'degree') is plotted for 907 essential vertebrate proteins known from mouse knockouts [50], human small interfering (si)RNA screens [51,52], and zebrafish random mutagenesis [53] and for the remaining 6,661 proteins in the network, considering only the largest connected network component. (b) The likelihood of being essential increases with increasing degree. Proteins were sorted by degree and divided into bins of 100 proteins each (filled diamonds). The observed frequency of essential genes in the bin is plotted against the average degree of the proteins in the bin, showing high correlation (R2 = 0.78) between degree and essentiality.
Although maps of both the yeast and human protein-interaction networks are well under way, their completion poses many problems, not least because of the anticipated scale of the human network, which could require multiple testing of all possible pairs of around 20,000-25,000 human proteins - roughly 200 million to 300 million pairs. The scale of this effort raises many questions. How do we even measure completion? The network is, after all, unknown. How close are we to completing the networks? How do we assess errors in the maps? Would maps obtained using only a single technique suffice?
In this article, we discuss the techniques used up to now, describe strategies for recognizing network completion, and estimate our progress towards finished yeast and human protein-interaction maps. Even though large numbers of interactions have been mapped, we argue that assay false-positive rates are so high that only about half of the expected yeast network has been defined to date, and considerably less for the human one. Like whole-genome shotgun sequencing [11], interaction networks will require multiple-fold coverage for completion. We argue that raw interaction data should be released, pooled, and analyzed as a set, as was the case for the human genome sequence. Coverage is low enough and errors common enough in individual datasets to mean that the human interactome will only be fully mapped through integration of repeated analyses from many groups.
Current interaction mapping strategies and their potential for scaling
The primary approach to mapping human protein interactions is the same one that initiated the yeast interactome -the yeast two-hybrid assay [12,13]. This classic assay involves the creation of two fusion proteins, the 'bait' protein fused to a DNA-binding domain and the 'prey' protein fused to a transcriptional activator domain. An interaction between bait and prey reconstitutes a complete transcription factor, detected by transcription of a reporter gene. This approach has already identified more than 5,000 interactions between human proteins [14,15].
The second major approach is affinity purification followed by mass spectrometry [16,17]. Here, epitope-tagged proteins are purified by affinity chromatography, and their co-purified interaction partners are identified by mass spectrometry. This assay excels at identifying in vivo protein complexes in yeast and other systems [18], particularly when used with tandem affinity purification (TAP) [19] and genomic knock-in of tags [16] rather than overexpression of transgenes. Most importantly, this technique bypasses exhaustive trials of all binary protein pairs and may scale up well to the size of the human interactome. On the downside, the assay may be biased toward abundant proteins [20]. Also, human cells present more difficulties than yeast, especially in expressing tagged libraries of human genes and the need to grow large volumes of cells. Initial screens in human cells [21] have used transgenes, rather than genomic knock-ins, to simplify cloning.
The remaining main approaches to mapping yeast and human protein interactions are computational - inferring protein interactions by integrating evidence from comparative and functional genomics (see, for example [20,22-25]). Although these are in silico rather than in vivo or in vitro interaction assays, they use experimental data such as DNA microarrays or genome sequences to infer protein interactions, and are, therefore, ultimately based on experimental observations [26]. As large amounts of data are available, these data-mining methods scale-up easily and offer both in vivo relevance and the ability to detect stable and transient interactions. Disadvantages include the importance of measuring associated error rates and the need for independent validation to verify error rates.
Although the approaches described above are complementary, the differences between them have caused some confusion within the scientific community. The term 'protein-protein interaction' carries two meanings: direct physical binding or membership of the same multiprotein complex. The latter usage is common in the field at large: for example, both major efforts to map protein complexes in yeast describe "interactions" between co-complexed proteins [27,28]. Part of the ambiguity in usage arises from the fact that few biochemical assays, apart from in vitro binding assays, truly distinguish the two cases. Currently, only yeast two-hybrid assays are regarded as measuring direct physical interaction between proteins and, at least in principle, even these interactions might occasionally be mediated through other members of a nuclear protein complex. Protein co-immunoprecipitation, often considered a definitive test of direct physical interactions, more typically measures co-complex interactions, much like the closely related affinity purification/mass spectrometry interaction assays. In addition, for the mass spectrometry interactions, one can consider the bait-prey interactions (the 'spoke' model [29]) as well as the prey-prey interactions (the 'matrix' model), with the latter typically of lower accuracy.
Estimating the scale of the yeast and human protein interaction networks
Computational and experimental approaches have now mapped a great many yeast and human protein interactions, but how many interactions should we expect? We argue here that the sizes of the complete yeast and human protein interaction networks will be larger than most early estimates. We do not yet know the size of any complete protein-interaction network. We can, however, roughly estimate the expected sizes for the yeast network using two different approaches that agree reasonably well. These estimates are derived from considering the interactions shared between each pair of large-scale protein interaction assays published so far.
First, provided two large-scale assays sample the same portion of 'interaction space' (that is, they sample the same pairs of interacting proteins - usually a subset of the interactome), then the number of interactions detected by both assays should be distributed according to the hypergeometric distribution, well-approximated for large populations by the binomial distribution. Given two assays of size n1 and n2 interactions, respectively, with k in common, as well as estimates of the false-positive rates of the two assays (fpr1 and fpr2), the maximum likelihood estimate of the number of interactions, N, within that subspace is [n1(1 - fpr1) × n2(1 - fpr2)]/k, provided n1 and n2 are sufficiently large (n1(1 - fpr1) × n2(1 - fpr2) >>N; see Additional data file 1 for a derivation of the statistics). This intersection analysis (Figure 2) has a rich history in other fields, such as mark-recapture methods for estimating the size of an animal population [30], and has recently been applied to protein-interaction networks [31].
Figure 2.
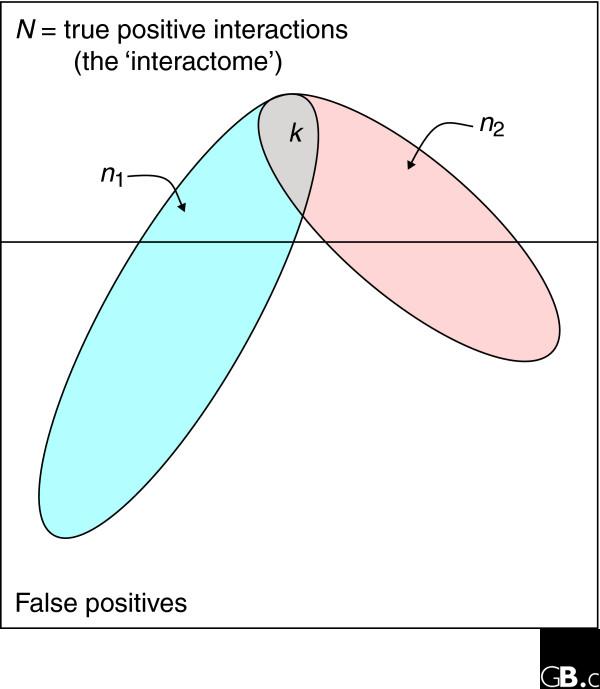
The method of intersection analysis for estimating interactome size. In an interactome, or subspace of an interactome, of N true interactions, two independent assays of n1 and n2 interactions are expected, under the hypergeometric distribution, to share k interactions by random chance. As described in the text, we can use the observed intersection of interaction assays to estimate N.
In order to use this method, the datasets must be corrected for their error rates. One method for estimating the false-positive rates of large-scale assays, described by D'haeseleer and Church [32], involves comparing the two datasets to each other and to a reference dataset. The method does not require a gold-standard reference; only that the reference not be biased toward either of the samples being measured. This requirement is met by comparing two similar assays: that is, either two mass spectrometry or two two-hybrid datasets. The method, described in Figure 3a, uses the ratio of the intersections of the three datasets to estimate the number of true positives in each sample. An example using the interactions derived from the two recent genome-scale TAP/mass spectrometry assays published by Gavin et al. [27] and Krogan et al. [28], compared to the Munich Information Center for Protein Sequences (MIPS) reference set [33], is presented in Figure 3b. In this and all subsequent analyses, the interaction data were used as for Krogan et al. [28] bait-prey pairs; for Gavin et al. [27] bait-prey pairs derived from lists of prey associated with each bait.
Figure 3.
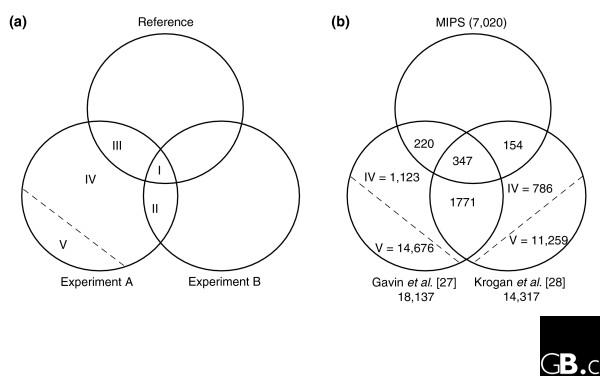
Estimating false-positive rates of large-scale assays. (a) As described by D'haeseleer and Church [32], the number of true positives in an interaction dataset can be estimated by examining the ratio of intersections of two similar datasets (A and B) and a reference dataset. If intersections contain all true positives, then the ratio of areas I and II is equal to the ratio of areas III and IV, where IV contains true positives (and V false positives, not shown to scale). The number of false positives can then be determined by simple subtraction, repeating the calculation for the other dataset. (b) Calculation of false-positive rates for the most recent yeast mass spectrometry assays of Gavin et al. [27] and Krogan et al. [28] within the interactome subspace sampled by both experiments (1,243 baits) and using MIPS as the reference sample [33]. Intersections (regions I, II, II) were determined by examining the data, and true- and false-positive populations (regions IV and V) were calculated as described in (a).
To estimate the interactome size by intersection analysis, we first take the interactions in each dataset that are derived from the common sample space of the two assays. (Figure 3b shows only the interactions in this common sample space.) Each group purified around 2,000 TAP-tagged strains for mass spectrometry, with the common set of baits numbering 1,243, of which 1,128 yielded at least one identical interaction. While a true 'apples-to-apples' comparison of these results is difficult given the data that these two groups have published, as discussed by Goll and Uetz [34], we tried to extract the interactions derived from these common baits for this analysis from the published filtered datasets. After calculating error rates and subtracting false positives from the two datasets, their intersection was used to predict the number of interactions within the subspace they sample. That prediction was then scaled up to the size of the whole interactome (around 5,8002/2) to estimate the total number of protein-protein interactions in the organism.
The error estimates for Gavin et al. [27] and Krogan et al. [28], as well as those for other large-scale yeast interaction datasets, are shown in Table 1. The false-positive rate of the computationally derived Jansen dataset [22] was determined by comparing it to Gavin et al. [27] and Krogan et al. [28] individually, although these comparisons may violate the no-bias requirement for the reference dataset. Table 2 shows the interactome size predictions derived from these pairs of mass spectrometry assays, which give an average interactome size of about 53,000 interactions, although the Gavin-Krogan pairwise estimate has the largest intersection and is, therefore, likely to be the most accurate estimate of the three. The two-hybrid assays [35,36] share too few interactions to give a meaningful estimate of interactome size.
Table 1.
Yeast protein-interaction assay false-positive rates: yeast datasets
| Dataset | Number of interactions | Derived false-positive rate* (%) | Published false-positive rate (%) | Average false-positive rate (%) |
| Uetz et al. [35] | 854 | 46 [32] | 32 [24]†,47 [44], 50 [37], 51 [42] | 45 |
| Ito [36] | 4,393 | 89 [32] | 71 [24]†, 78 [41], 85 [37], 91 [44] | 83 |
| Gavin et al. [16] | 3,180 | 68 [32] | 14 [24]†, 22 [4], <72 (upper bound [20]) | 35 |
| Ho et al. [17] | 3,618 | 83 [32], 81, 82, 80 | 55 [24]†, <97 (upper bound [20]) | 76 |
| Jansen et al. [22] | 15,922 | 81, 79 | - | 80 |
| Gavin et al. [27] | 18,137 | 78, 82, 86‡ | - | 82 |
| Krogan et al. [28] | 14,317 (7,123 core) | 75, 79, 66‡ (59, 65, 37‡ core) | - | 73 (54 core) |
| Overall | 51,419 | 72 |
*This interaction assay false-positive rate is taken from D'haeseleer and Church [32] or derived using the method therein. Multiple values derive from choosing either the GRID [2] or MIPS [33] reference sets. ‡This interaction assay false-positive rate is calculated with the EPR server of Deane et al. [42]. †The mean of four values estimated from Table S3 of Lee et al. [24] by fitting the interaction set as a linear combination of true-positive (small scale interactions) and false-positive (random pairs) interactions.
Table 2.
Prediction of the size of the yeast interactome
| Dataset pair | Common baits | Estimated interactions in common search space | Projected interactome size (95% CI) |
| Gavin-Krogan (core) [27,28] | 1,128 | 3,642 | 38,600 (37,800-39,500) |
| Ho-Gavin [16,17] | 241 | 718 | 50,000 (47,700-53,000) |
| Ho-Krogan (core) [17,28] | 282 | 1,109 | 69,000 (63,300-75,500) |
| Mean | 52,500 (37,800-75,500)* |
*The range of interactome sizes is the minimum and maximum from the confidence intervals (CI) generated from pairwise estimates.
These projected interactome sizes agree with those generated by a simple, very approximate, scaling argument: we observe approximately 5-10 unique interactions per yeast protein in current networks; multiplying these values by around 5,800 yeast genes gives estimates of approximately 29,000-58,000 interactions. These values are somewhat larger than previous estimates of 10,000-30,000 total yeast interactions [20,29,31,37-39].
Unfortunately, applying these techniques to high-throughput assays of human protein-protein interactions is still problematic. The two large-scale yeast two-hybrid screens published recently [14,15] share only six interactions, too small an intersection to generate reliable error rate or inter-actome size estimates; similarly, data from Stelzl et al. [15] share only 5 and 13 interactions with orthology-transferred interactions from Lehner and Fraser [40] and the computationally derived set of Rhodes et al. [23], ruling out these comparisons for estimating interactome size. However, comparison of the Rual et al. [14] data with those of Lehner and Fraser [40] and Rhodes et al. [23] yielded consistent false-positive estimates, suggesting that reference bias is minimal (Table 3). The human interactome estimates generated from these pairs of datasets are shown in Table 4. These projections, while consistent with the estimate of approximately 260,000 interactions offered by Rual et al. [14], still stem from small intersections and limited information about sample space, and should be considered very rough estimates.
Table 3.
Human protein-interaction assay false-positive rates: human datasets
| Dataset | Number of unique interactions | Derived false-positive rates* (%) | Published false-positive rates (%) | Average false-positive rates (%) |
| Lehner and Fraser [40] | 58,700 (9,396 core) | 96, 94, 93 (86, 81, 69 core) | - | 94 (79 core) |
| Rhodes et al. [23] | 38,379 | 87, 86, 83 | - | 85 |
| Stelzl et al. [15] | 3,150 (902 core) | 98, 98 (94,95 core) | 70 [45] | 98 (86 core) |
| Rual et al. [14] | 2,611 | 87, 93 | 8-66 [14]†, 54 [45] | 58 |
| Overall | 100,242 | 90 |
*This interaction assay false-positive rate is derived using the method of D'haeseleer and Church [32] and a reference set of 20,296 unique interactions from HPRD [54], BIND [55], Reactome [56], and Ramani et al. [49]. Multiple values derive from different choices of comparison sets. †A range of six values (mean 48%) estimated from Table 1 of Rual et al. [14] by fitting the interaction set CCSB-HI1 as a linear combination of true positive (LCI-core) and false positive (all possible) interactions.
Table 4.
Prediction of the size of the human interactome
| Dataset pair | Interactions in both datasets | Estimated interactions in common search space | Projected interactome size (95% CI) |
| Rual-Lehner (core) [14,40] | 35 | 28,200 | 261,000 (191,000-369,000) |
| Rual-Rhodes [14,23] | 59 | 20,200 | 189,000 (154,000-239,000) |
| Mean | 225,000 (154,000-369,000)* |
*The range of interactome sizes is the minimum and maximum from the confidence intervals (CI) generated from pairwise estimates.
The critical importance of measuring error rates
This analysis, with many others [20,32,37,41-45], only reinforces the importance of measuring error rates when mapping protein interactions. Observing an interaction experimentally (for example, as in a yeast two-hybrid assay) does not guarantee a true positive interaction; that is, one that occurs in vivo under native conditions during the life of the organism. All assays, experimental and computational, show errors and should be accompanied by measures of confidence. Many published methods exist for estimating interaction assay error rates [20,22,37,41-45] and for scoring individual protein-protein interactions. These latter scores either exploit assay-specific features [46] or use simple, but surprisingly effective, statistical criteria for separating true from false-positive interactions [43,47,48]. For example, although the full Ito et al. [36] yeast two-hybrid set has a measured false-positive rate of around 80%, a statistical measure based on the hypergeometric distribution can select a subset of around 45% of the interactions whose false-positive rate is only around 30% [43].
These high error rates underscore the difficulty in evaluating progress towards complete interactomes. Given these false-positive rates, and the resulting relatively small number of interactions detected in multiple assays, how far have we actually progressed towards the complete protein-interaction networks of yeast and humans?
How do we know when we're done?
As we can only approximate true interactome sizes, we have few sure measures of interactome completion beyond simply testing for coverage of confident interactions from the literature [2,20]. However, two empirical methods, assay saturation and dead reckoning, suggest that we are far from finished with either the yeast or human interactomes.
Assay saturation captures the notion that, early in interaction-network mapping, each new interaction assay largely discovers novel interactions, as was observed for the first two large-scale yeast two-hybrid assays [36]. Provided false-positive rates are well controlled, later assays should reveal proportionally fewer novel interactions, with the new interaction discovery rate dropping as interaction saturation approaches 100%. At this time, the portion of the interactome accessible to these assays will be complete, although this approach says nothing about how well this accessible portion covers the entire interactome. The saturation can be revealed by plotting, for each additional assay, the total interactions mapped versus the novel interactions mapped. Early assays fall along the diagonal (all interactions are new); later assays provide fewer new interactions, with the slope of the line decreasing, ultimately approaching zero for error-free, completely redundant assays.
We tested for assay saturation in yeast and humans (Figure 4). Not surprisingly, we detect no evidence of saturation in humans. In yeast, however, there appears to be some: the most recent yeast dataset from Krogan et al. [28] discovers 66% new interactions, on a par with the estimated false-positive rate of the dataset. As with previous screens [20], both recent large-scale mass spectrometry assays may be biased toward interactions between abundant proteins, and saturation is likely to be confined to interactions of abundant proteins. Nevertheless, achieving this level of completeness for a major fraction of yeast proteins is a worthy accomplishment, and serves as a guide for future large-scale assays exploring the rest of the yeast interactome.
Figure 4.
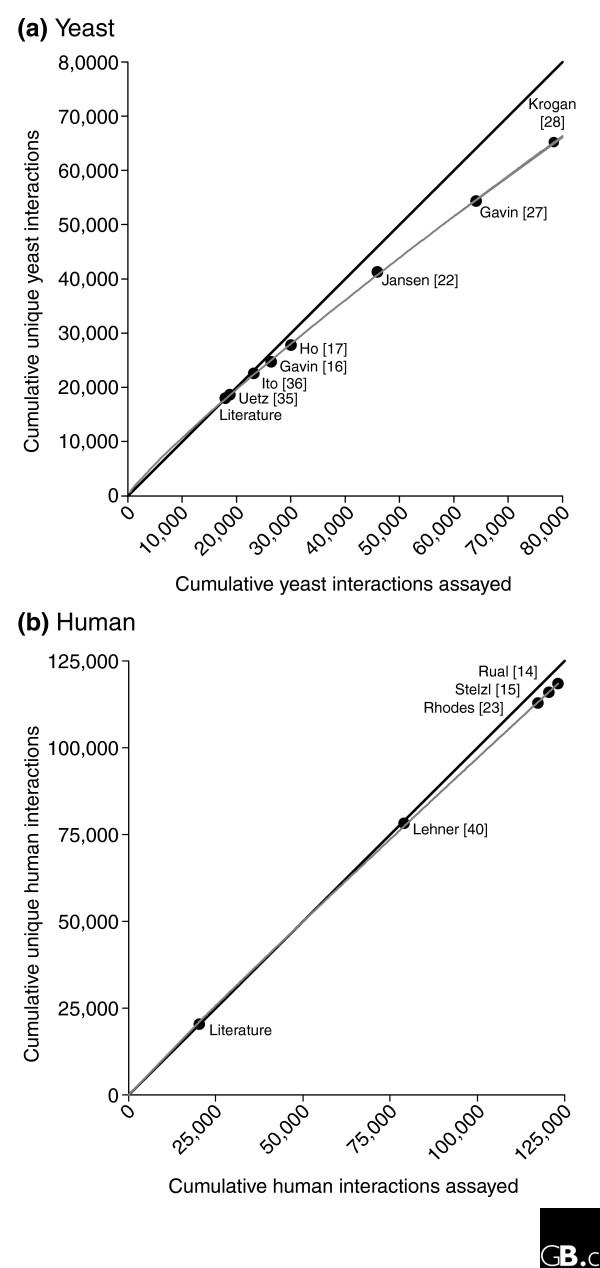
Comparison of the degree of completion of the yeast and human protein-interaction networks. Neither (a) the yeast nor (b) the human protein-interaction network is near completion as judged by the extent of assay saturation for the studies indicated here, although the yeast network shows higher saturation. With repeating assays on a finite set of interactions, we expect the rate of discovery of new interactions (gray line) to fall below 100% (black diagonal line) and asymptotically to approach the false-positive discovery rate. If false-positive rates are properly controlled, the rate of new interactions should level out, indicating the complete network assayable by these methods. In yeast, the most recent mass spectrometry study of Krogan et al. [28] (core set) shows 66% new interactions, suggesting initial saturation. Human protein interactions are under-sampled; the most recent study, Rual et al. [14], assayed 95% new interactions.
The method of dead reckoning measures total interactome completion from the number of interactions assayed and their associated false-positive rates, just as sailors on the high seas estimated distances from the ship's speed and the time traveled. For this approach, we assume all interactions observed by more than one assay are true positives. When assays are uncorrelated, this assumption holds for about 99.9% of the time for both yeast and human, given our estimates of interactome size. The number of additional true positives contributed by an assay of size n is n(1 - fpr) - x, where x is the number of interactions already observed in previous assays. By this measure, the yeast experiments in Tables 1 and 2 plus the comprehensive literature databases have contributed 24,800 true-positive interactions, or around 50% of the estimated interactome. Of this total, nearly 18,000 interactions come from curated literature databases [2,33], and 5,800 were detected in more than one high-throughput assay. Human protein-interaction assays have similarly covered about 25,000 true-positive interactions, or around 11% of the estimated interactome, with over 80% coming from sources based on literature mining. Note that these estimates assume that the literature sources are error-free, which is certainly not the case [14].
For both organisms, a number of factors could extend the current datasets to cover more of the interactome, such as considering the matrix model of interactions discovered by mass spectrometry [29]. Although this increases the false positives, statistical scores can identify true positives [43], increasing the overall quality and number of interactions.
Raw data release could be the way forward
High error rates in large-scale assays dictate that the community must oversample the interactome in order to approach completion. Whole-proteome interactome mapping is, therefore, analogous to whole-genome shotgun sequencing [11]: each assay reveals a subset of the interactions (sequence), requiring multiple-fold coverage of the interactome (genome) for completion of the true-positive set. In shotgun sequencing, assembly of sequencing reads is the algorithmically difficult step. By contrast, controlling and measuring error rates is currently the more challenging step in 'shotgun' interactome mapping. With false-positive rates exceeding 50%, and false-negative rates (the proportion of true interactions missed) for two-hybrid assays in particular approaching 90%, it is clear that each subspace must be sampled many times to provide complete coverage - and the problem remains of separating the true interactome from the false positives.
This last problem has made it clear that many alternative approaches will be required to complete the network. Comparing results from different approaches will continue to be crucial for validating interactions and estimating error rates, as the biases of one technique are easily overcome by integrating interactions from other methods. To this end, we strongly encourage all participants in interactome mapping to make public their raw data as well as their analyzed and filtered high-confidence interactions, as weak signals detected across multiple assays can be integrated to help distinguish real from spurious interactions. To further this discussion, many of the primary groups mapping the human protein interaction network met last August at the Joint Cold Spring Habor/Wellcome Trust Conference on Interactome Networks in Hinxton, UK, to compare results and coordinate efforts and announced plans to meet again next August. This effort may yet coalesce into a collaborative consortium like the human genome sequencing consortium, and an open forum now exists as the mapping proceeds.
Additional data files
Additional data on the statistics used are available online as Additional data file 1.
Supplementary Material
Estimating interactome size: the maximum likelihood approach.
References
- Bandyopadhyay S, Sharan R, Ideker T. Systematic identification of functional orthologs based on protein network comparison. Genome Res. 2006;16:428–435. doi: 10.1101/gr.4526006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reguly T, Breitkreutz A, Boucher L, Breitkreutz BJ, Hon GC, Myers CL, Parsons A, Friesen H, Oughtred R, Tong A, et al. Comprehensive curation and analysis of global interaction networks in Saccharomyces cerevisiae. J Biol. 2006;5:11. doi: 10.1186/jbiol36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almaas E, Kovacs B, Vicsek T, Oltvai ZN, Barabasi AL. Global organization of metabolic fluxes in the bacterium Escherichia coli. Nature. 2004;427:839–843. doi: 10.1038/nature02289. [DOI] [PubMed] [Google Scholar]
- Herrgard MJ, Lee BS, Portnoy V, Palsson BO. Integrated analysis of regulatory and metabolic networks reveals novel regulatory mechanisms in Saccharomyces cerevisiae. Genome Res. 2006;16:627–635. doi: 10.1101/gr.4083206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharchenko P, Church GM, Vitkup D. Expression dynamics of a cellular metabolic network. Mol Syst Biol. 2005 doi: 10.1038/msb4100023. doi:10.1038/-msb4100023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maciag K, Altschuler SJ, Slack MD, Krogan NJ, Emili A, Greenblatt JF, Maniatis T, Wu LF. Systems-level analyses identify extensive coupling among gene expression machines. Mol Syst Biol. 2006 doi: 10.1038/msb4100045. doi:10.1038/msb4100045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guido NJ, Wang X, Adalsteinsson D, McMillen D, Hasty J, Cantor CR, Elston TC, Collins JJ. A bottom-up approach to gene regulation. Nature. 2006;439:856–860. doi: 10.1038/nature04473. [DOI] [PubMed] [Google Scholar]
- Jeong H, Mason SP, Barabasi AL, Oltvai ZN. Lethality and centrality in protein networks. Nature. 2001;411:41–42. doi: 10.1038/35075138. [DOI] [PubMed] [Google Scholar]
- Gandhi TK, Zhong J, Mathivanan S, Karthick L, Chandrika KN, Mohan SS, Sharma S, Pinkert S, Nagaraju S, Periaswamy B, et al. Analysis of the human protein interactome and comparison with yeast, worm and fly interaction datasets. Nat Genet. 2006;38:285–293. doi: 10.1038/ng1747. [DOI] [PubMed] [Google Scholar]
- Coulomb S, Bauer M, Bernard D, Marsolier-Kergoat MC. Gene essentiality and the topology of protein interaction networks. Proc Biol Sci. 2005;272:1721–1725. doi: 10.1098/rspb.2005.3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber JL, Myers EW. Human whole-genome shotgun sequencing. Genome Res. 1997;7:401–409. doi: 10.1101/gr.7.5.401. [DOI] [PubMed] [Google Scholar]
- Fields S, Song O. A novel genetic system to detect protein-protein interactions. Nature. 1989;340:245–246. doi: 10.1038/340245a0. [DOI] [PubMed] [Google Scholar]
- Golemis EA, Brent R. Fused protein domains inhibit DNA binding by LexA. Mol Cell Biol. 1992;12:3006–3014. doi: 10.1128/mcb.12.7.3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rual JF, Venkatesan K, Hao T, Hirozane-Kishikawa T, Dricot A, Li N, Berriz GF, Gibbons FD, Dreze M, Ayivi-Guedehoussou N, et al. Towards a proteome-scale map of the human protein-protein interaction network. Nature. 2005;437:1173–1178. doi: 10.1038/nature04209. [DOI] [PubMed] [Google Scholar]
- Stelzl U, Worm U, Lalowski M, Haenig C, Brembeck FH, Goehler H, Stroedicke M, Zenkner M, Schoenherr A, Koeppen S, et al. A human protein-protein interaction network: a resource for annotating the proteome. Cell. 2005;122:957–968. doi: 10.1016/j.cell.2005.08.029. [DOI] [PubMed] [Google Scholar]
- Gavin AC, Bosche M, Krause R, Grandi P, Marzioch M, Bauer A, Schultz J, Rick JM, Michon AM, Cruciat CM, et al. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature. 2002;415:141–147. doi: 10.1038/415141a. [DOI] [PubMed] [Google Scholar]
- Ho Y, Gruhler A, Heilbut A, Bader GD, Moore L, Adams SL, Millar A, Taylor P, Bennett K, Boutilier K, et al. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature. 2002;415:180–183. doi: 10.1038/415180a. [DOI] [PubMed] [Google Scholar]
- Butland G, Peregrin-Alvarez JM, Li J, Yang W, Yang X, Canadien V, Starostine A, Richards D, Beattie B, Krogan N, et al. Interaction network containing conserved and essential protein complexes in Escherichia coli. Nature. 2005;433:531–537. doi: 10.1038/nature03239. [DOI] [PubMed] [Google Scholar]
- Rigaut G, Shevchenko A, Rutz B, Wilm M, Mann M, Seraphin B. A generic protein purification method for protein complex characterization and proteome exploration. Nat Biotechnol. 1999;17:1030–1032. doi: 10.1038/13732. [DOI] [PubMed] [Google Scholar]
- von Mering C, Krause R, Snel B, Cornell M, Oliver SG, Fields S, Bork P. Comparative assessment of large-scale data sets of protein-protein interactions. Nature. 2002;417:399–403. doi: 10.1038/nature750. [DOI] [PubMed] [Google Scholar]
- Bouwmeester T, Bauch A, Ruffner H, Angrand PO, Bergamini G, Croughton K, Cruciat C, Eberhard D, Gagneur J, Ghidelli S, et al. A physical and functional map of the human TNF-alpha/NF-kappa B signal transduction pathway. Nat Cell Biol. 2004;6:97–105. doi: 10.1038/ncb1086. [DOI] [PubMed] [Google Scholar]
- Jansen R, Yu H, Greenbaum D, Kluger Y, Krogan NJ, Chung S, Emili A, Snyder M, Greenblatt JF, Gerstein M. A Bayesian networks approach for predicting protein-protein interactions from genomic data. Science. 2003;302:449–453. doi: 10.1126/science.1087361. [DOI] [PubMed] [Google Scholar]
- Rhodes DR, Tomlins SA, Varambally S, Mahavisno V, Barrette T, Kalyana-Sundaram S, Ghosh D, Pandey A, Chinnaiyan AM. Probabilistic model of the human protein-protein interaction network. Nat Biotechnol. 2005;23:951–959. doi: 10.1038/nbt1103. [DOI] [PubMed] [Google Scholar]
- Lee I, Date SV, Adai AT, Marcotte EM. A probabilistic functional network of yeast genes. Science. 2004;306:1555–1558. doi: 10.1126/science.1099511. [DOI] [PubMed] [Google Scholar]
- Mellor JC, Yanai I, Clodfelter KH, Mintseris J, DeLisi C. Predictome: a database of putative functional links between proteins. Nucleic Acids Res. 2002;30:306–309. doi: 10.1093/nar/30.1.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser AG, Marcotte EM. A probabilistic view of gene function. Nat Genet. 2004;36:559–564. doi: 10.1038/ng1370. [DOI] [PubMed] [Google Scholar]
- Gavin AC, Aloy P, Grandi P, Krause R, Boesche M, Marzioch M, Rau C, Jensen LJ, Bastuck S, Dumpelfeld B, et al. Proteome survey reveals modularity of the yeast cell machinery. Nature. 2006;440:631–636. doi: 10.1038/nature04532. [DOI] [PubMed] [Google Scholar]
- Krogan NJ, Cagney G, Yu H, Zhong G, Guo X, Ignatchenko A, Li J, Pu S, Datta N, Tikuisis AP, et al. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature. 2006;440:637–643. doi: 10.1038/nature04670. [DOI] [PubMed] [Google Scholar]
- Bader GD, Hogue CW. Analyzing yeast protein-protein interaction data obtained from different sources. Nat Biotechnol. 2002;20:991–997. doi: 10.1038/nbt1002-991. [DOI] [PubMed] [Google Scholar]
- Seber GAF. The Estimation of Animal Abundance and Related Parameters. 2. Caldwell, NJ: Blackburn Press; 1982. [Google Scholar]
- Grigoriev A. On the number of protein-protein interactions in the yeast proteome. Nucleic Acids Res. 2003;31:4157–4161. doi: 10.1093/nar/gkg466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'haeseleer P, Church GM. Proc IEEE Comput Syst Bioinform Conf: August 16-19 2004; California. IEEE Computer Society; 2004. Estimating and improving protein interaction error rates. pp. 216–223. [DOI] [PubMed] [Google Scholar]
- Guldener U, Munsterkotter M, Oesterheld M, Pagel P, Ruepp A, Mewes HW, Stumpflen V. MPact: the MIPS protein interaction resource on yeast. Nucleic Acids Res. 2006;(34 Database):D436–D441. doi: 10.1093/nar/gkj003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goll J, Uetz P. The elusive yeast interactome. Genome Biol. 2006;7:223. doi: 10.1186/gb-2006-7-6-223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uetz P, Giot L, Cagney G, Mansfield TA, Judson RS, Knight JR, Lockshon D, Narayan V, Srinivasan M, Pochart P, et al. A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature. 2000;403:623–627. doi: 10.1038/35001009. [DOI] [PubMed] [Google Scholar]
- Ito T, Chiba T, Ozawa R, Yoshida M, Hattori M, Sakaki Y. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc Natl Acad Sci USA. 2001;98:4569–4574. doi: 10.1073/pnas.061034498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprinzak E, Sattath S, Margalit H. How reliable are experimental protein-protein interaction data? J Mol Biol. 2003;327:919–923. doi: 10.1016/S0022-2836(03)00239-0. [DOI] [PubMed] [Google Scholar]
- Tucker CL, Gera JF, Uetz P. Towards an understanding of complex protein networks. Trends Cell Biol. 2001;11:102–106. doi: 10.1016/S0962-8924(00)01902-4. [DOI] [PubMed] [Google Scholar]
- Legrain P, Wojcik J, Gauthier JM. Protein-protein interaction maps: a lead towards cellular functions. Trends Genet. 2001;17:346–352. doi: 10.1016/S0168-9525(01)02323-X. [DOI] [PubMed] [Google Scholar]
- Lehner B, Fraser AG. A first-draft human protein-interaction map. Genome Biol. 2004;5:R63. doi: 10.1186/gb-2004-5-9-r63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader JS, Chaudhuri A, Rothberg JM, Chant J. Gaining confidence in high-throughput protein interaction networks. Nat Biotechnol. 2004;22:78–85. doi: 10.1038/nbt924. [DOI] [PubMed] [Google Scholar]
- Deane CM, Salwinski L, Xenarios I, Eisenberg D. Protein interactions: two methods for assessment of the reliability of high throughput observations. Mol Cell Proteomics. 2002;1:349–356. doi: 10.1074/mcp.M100037-MCP200. [DOI] [PubMed] [Google Scholar]
- Lee I, Narayanaswamy R, Marcotte EM. Bioinformatic prediction of yeast gene function. In: Stansfield I, Stark M, editor. Yeast Gene Analysis. Amsterdam: Elsevier Press; 2007. Preprint at http://polaris.icmb.utexas.edu/publications.html. [Google Scholar]
- Mrowka R, Patzak A, Herzel H. Is there a bias in proteome research? Genome Res. 2001;11:1971–1973. doi: 10.1101/gr.206701. [DOI] [PubMed] [Google Scholar]
- Patil A, Nakamura H. Filtering high-throughput protein-protein interaction data using a combination of genomic features. BMC Bioinformatics. 2005;6:100. doi: 10.1186/1471-2105-6-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giot L, Bader JS, Brouwer C, Chaudhuri A, Kuang B, Li Y, Hao YL, Ooi CE, Godwin B, Vitols E, et al. A protein interaction map of Drosophila melanogaster. Science. 2003;302:1727–1736. doi: 10.1126/science.1090289. [DOI] [PubMed] [Google Scholar]
- Samanta MP, Liang S. Predicting protein functions from redundancies in large-scale protein interaction networks. Proc Natl Acad Sci USA. 2003;100:12579–12583. doi: 10.1073/pnas.2132527100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlitt T, Palin K, Rung J, Dietmann S, Lappe M, Ukkonen E, Brazma A. From gene networks to gene function. Genome Res. 2003;13:2568–2576. doi: 10.1101/gr.1111403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramani AK, Bunescu RC, Mooney RJ, Marcotte EM. Consolidating the set of known human protein-protein interactions in preparation for large-scale mapping of the human interactome. Genome Biol. 2005;6:R40. doi: 10.1186/gb-2005-6-5-r40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eppig JT, Bult CJ, Kadin JA, Richardson JE, Blake JA, Anagnostopoulos A, Baldarelli RM, Baya M, Beal JS, Bello SM, et al. The Mouse Genome Database (MGD): from genes to mice-a community resource for mouse biology. Nucleic Acids Res. 2005;(33 Database):D471–D475. doi: 10.1093/nar/gki113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harborth J, Elbashir SM, Bechert K, Tuschl T, Weber K. Identification of essential genes in cultured mammalian cells using small interfering RNAs. J Cell Sci. 2001;114:4557–4565. doi: 10.1242/jcs.114.24.4557. [DOI] [PubMed] [Google Scholar]
- Kittler R, Putz G, Pelletier L, Poser I, Heninger AK, Drechsel D, Fischer S, Konstantinova I, Habermann B, Grabner H, et al. An endoribonuclease-prepared siRNA screen in human cells identifies genes essential for cell division. Nature. 2004;432:1036–1040. doi: 10.1038/nature03159. [DOI] [PubMed] [Google Scholar]
- Amsterdam A, Nissen RM, Sun Z, Swindell EC, Farrington S, Hopkins N. Identification of 315 genes essential for early zebrafish development. Proc Natl Acad Sci USA. 2004;101:12792–12797. doi: 10.1073/pnas.0403929101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra GR, Suresh M, Kumaran K, Kannabiran N, Suresh S, Bala P, Shivakumar K, Anuradha N, Reddy R, Raghavan TM, et al. Human protein reference database - 2006 update. Nucleic Acids Res. 2006;(34 Database):D411–D414. doi: 10.1093/nar/gkj141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfarano C, Andrade CE, Anthony K, Bahroos N, Bajec M, Bantoft K, Betel D, Bobechko B, Boutilier K, Burgess E, et al. The Biomolecular Interaction Network Database and related tools 2005 update. Nucleic Acids Res. 2005;(33 Database):D418–D424. doi: 10.1093/nar/gki051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi-Tope G, Gillespie M, Vastrik I, D'Eustachio P, Schmidt E, de Bono B, Jassal B, Gopinath GR, Wu GR, Matthews L, et al. Reactome: a knowledgebase of biological pathways. Nucleic Acids Res. 2005;(33 Database):D428–D432. doi: 10.1093/nar/gki072. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Estimating interactome size: the maximum likelihood approach.