

Abstract
Polycyclic aromatic hydrocarbons (PAHs) are ubiquitous environmental pollutants that have been linked to certain human cancers. The fjord region PAH dibenzo[a,l]pyrene exhibits the highest levels of carcinogenic activity of all PAH as yet tested in rodent tumor models. Another hexacyclic aromatic hydrocarbon, dibenzo[c,p]chrysene (DBC), is a unique PAH that possesses one bay region and two fjord regions within the same molecule. Due to its structure, which is a merger of the fjord region PAHs benzo[c]phenanthrene, benzo[c]chrysene, and benzo[g]chrysene, DBC is of considerable research interest. In order to investigate the pathway of regioselective metabolism we have studied the cytotoxicity, metabolic activation and DNA adduct formation of DBC in human mammary carcinoma MCF-7 cells in culture. The cytotoxicity assay indicated undisturbed cell proliferation even at concentrations as high as 4.5 μM (1.5 μg/ml) DBC. Concurrently, DNA adducts were detected in MCF-7 cells treated with DBC only in low amounts (0.6 pmol adducts/mg DNA). On the contrary, exposure to anti-DBC-1,2-diol-3,4-epoxide and anti-DBC-11,12-diol-13,14-epoxide, two putatively genotoxic metabolites of DBC, resulted in high levels of DNA adducts (33 and 51 pmol adducts/mg DNA, respectively). Although DBC was not efficiently transformed into DNA-reactive metabolites in MCF-7 cells in culture, the results from our study indicate that the two fjord region diol-epoxide derivatives of DBC may serve as ultimate genotoxic metabolites once they are enzymatically generated under certain circumstances in vitro or in vivo.
Keywords: Abbreviations: PAH polycyclic aromatic hydrocarbon, BP benzo [a] pyrene, DBP dibenzo [a l] pyrene, DBC dibenzo[c p] chrysene, BcC benzo[c]chrysene, BgC benzo[g]chrysene, BPh benzo[c]phenanthrene, DBA dibenz[a h]anthracene, MTT methylthiazolyldiphenyl-tetrazolium bromide, EROD ethoxyresorufin O-deethylase, DMSO dimethylsulfoxide, HPLC high performance liquid chromatography, CYP cytochrome P450
1. Introduction
Polycyclic aromatic hydrocarbons (PAHs) constitute an important class of environmental contaminants and it is well established that exposure to carcinogenic mixtures of PAHs causes several types of cancer in humans [1]. The sources of PAHs are either occupational (e.g., coal-tar processing in steel or aluminum industries) or environmental (e.g., combustion processes). Benzo[a]pyrene (BP) and dibenzo[a,l]pyrene (DBP) have been detected in cigarette smoke condensate [2], or in particulate matter formed during combustion of smoky coal [3] (Fig. 1). These potent carcinogens are metabolically activated through cytochrome P450 (CYP) enzymes to ultimate carcinogenic diol-epoxide derivatives which then may undergo covalent reaction with DNA, resulting in PAH-DNA adducts and subsequent induction of mutations. In fact, cancer patients with high levels of PAH-DNA adducts in lung tissue have been related to an early onset of lung cancer [4,5]. In addition, several studies have detected various DNA adducts in human breast tissue [6,7], indicating that PAH-DNA adduct formation might contribute in breast cancer development [8]. On the other hand, recent data again challenge this view and demonstrate that the role of PAHs in cancer etiology is still a matter of debate [9,10].
Fig. 1.
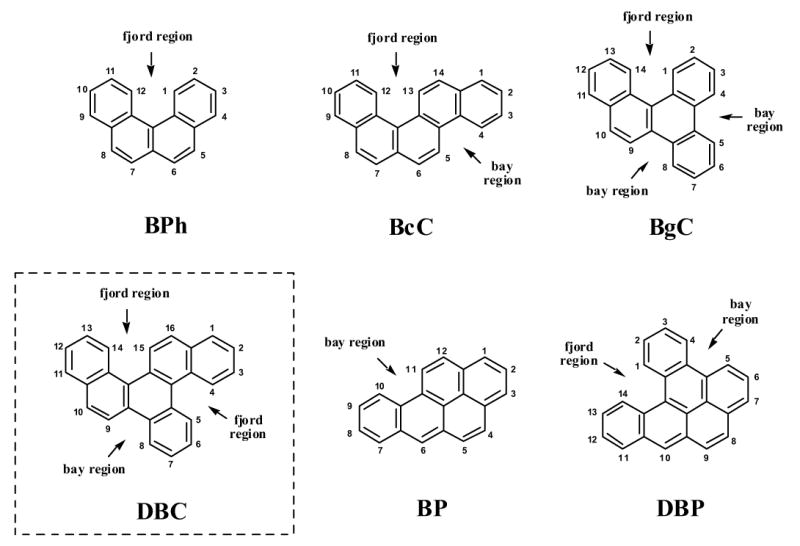
Structures of various PAHs containing the structural elements of sterically hindered bay and fjord regions. Depicted are the tetracyclic benzo[c]phenanthrene (BPh), the pentacyclic benzo[c]chrysene (BcC), benzo[g]chrysene (BgC), and benzo[a]pyrene (BP), as well as the hexacyclic dibenzo[c,p]pyrene (DBC) and dibenzo[a,l]pyrene (DBP). Among these compounds, BP, DBP and DBC have been applied in the present study.
Early studies demonstrated that the structure of a particular PAH can be an important determinant of its biological activity [11]. For a number of PAHs containing a bay region, including BP [12] and dibenz[a,h]anthracene (DBA) [13], their carcinogenic potency strongly depends on regioselective metabolic activation toward bay region anti-diol-epoxides and further oxidized metabolites [14,15]. In contrast, the situation is less clear for PAHs that contain the structural element of a fjord region, either alone or together with a bay region (Fig. 1). For instance, DBP that contains both a bay and a fjord region, has been found to be preferentially activated to fjord region syn- and anti-11,12-diol 13,14-epoxides. DBP is an exceptionally strong tumorigenic and toxic compound [16], and its diol-epoxide derivatives display extraordinary high mutagenicity in Chinese hamster V79 cells [17,18]. In general, the strong mutagenicity of PAH diol-epoxides has been attributed in part to a relatively high level of PAH-DNA adduct formation [18,19] and the repair resistance of those DNA lesions that have been formed [20].
Previous studies demonstrated that the orthologous forms of CYP1A1 and CYP1B1 from various animal species and humans are most important in activating a range of carcinogenic PAHs toward DNA-reactive bay or fjord region diol-epoxides [21,22]. Studies by Luch and colleagues [23,24] indicate that the metabolic precursor diols of DBP, benzo[c]chrysene (BcC) and benzo[g]chrysene (BgC) can be further activated by liver microsomes to the corresponding fjord region diol-epoxides. In addition, Amin and co-workers [25] recently demonstrated that both bay and fjord region diol-epoxides are formed as intermediates in the metabolism of BcC in vivo. From these metabolites, fjord region diol-epoxides are the more potent tumorigens as compared to bay region diol-epoxides in rat mammary tissue [25].
In the present study we have investigated the regioselectivity in the metabolism of DBC (Fig. 1), a high-molecular PAH with MW 328. Although nothing is known at the moment about its environmental presence and tumorigenic potency, this compound is of great research interest due to its structural features [26]. Given the constant improvement of preparative chemistry and analytical methods it can be expected, however, that current limits of detection will be resolved and environmental samples that typically contain complex mixtures of PAHs including constituents of molecular weights far beyond 300 will be better characterized in the near future [27]. Here we examined the CYP-mediated activation and DNA adduct formation of DBC or of its putatively proximate and ultimate metabolites in human mammary carcinoma-derived MCF-7 cells in culture. The following derivatives were included in the study: racemic DBC-1,2-diol, DBC-11,12-diol, anti-DBC-1,2-diol-3,4-epoxide, and anti-DBC-11,12-diol-13,14-epoxide, all of which have been synthesized and physico-chemically characterized previously (Fig. 2) [26].
Fig. 2.
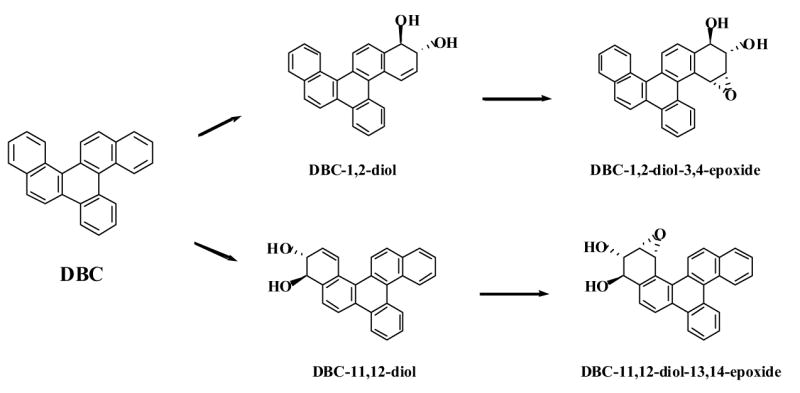
Schematic representation of the putative metabolic activation pathways at the two different fjord regions of DBC that lead to proximate diols and ultimate diol-epoxide metabolites. DBC and its racemic derivatives depicted were used in the present study.
2. Material and methods
2.1. Cell culture and treatment
The human mammary carcinoma-derived MCF-7 cell line was obtained from the Michigan Cancer Foundation (Karmanos Cancer Center, Detroit, MI). The cells were cultured in 75 cm2 flasks (Corning, Corning, NY) in a 1:1 mixture of F-12 Nutrient Mixture (Gibco-Invitrogen, Carlsbad, CA) and Dulbecco’s Modified Eagle’s Medium (DMEM) (Gibco-Invitrogen). The medium was supplemented with 10% fetal bovine serum (FBS) (Intergen, Purchase, NY), containing 15 mM HEPES buffer and antibiotics (200 units/ml penicillin, 200 μg/ml streptomycin, and 25 μg/ml ampicillin) at 37°C with 5% CO2. Cell cultures were subcultured at a ratio of 1:4 when the cells covered the entire surface of the flask.
Approximately 1 x 106 cells were treated in each flask in 20 ml of media with BP (2 μM), DBP (0.02 μM), DBC (1.5 μg/ml, 4.5 μM), DBC-1,2-diol or DBC-11,12-diol (1 μg/ml, 2.8 μM), and anti-DBC-1,2-diol-3,4-epoxide or anti-DBC-11,12-diol-13,14-epoxide (1 μg/ml, 2.6 μM), respectively. Dimethylsulfoxide (DMSO) was used as vehicle control. The treated cells were harvested after 48 h of continuous exposure, washed with phosphate-buffered saline (PBS), and the cell pellet was stored at −80°C.
2.2. Cytotoxicity assay
The methylthiazolyldiphenyl-tetrazolium bromide (MTT) cell proliferation assay was conducted as per manufacturer’s protocol (ATCC, Manassas, VA). Briefly, 35,000 cells were counted using a coulter counter (Coulter electronics, Hileah, FL) and plated in a 96-well plate. After 24 h the cells were treated with varying doses (0.75-4.5 μM) of DBC. Cells were also treated with highly cytotoxic doses of BP (6 μM) and DBP (5 μM) for comparison. The cells were incubated for 48 h at 37°C, after which 10 μl of MTT reagent [3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide] was added to each well, and incubated at 37°C for 3 h. Viable cells are capable of reducing this reagent to a purple formazan derivative. As soon as an intracellular purple precipitate was visible, the cells were washed with 100 μl of provided detergent reagent and incubated at room temperature for 2 h. Cell viability was determined by measuring the absorbance in each well using a Spectra MAX 250 plate reader (Molecular Devices, Sunnyvale, CA) at an absorbance wavelength of 570 nm. Each sample was assayed in triplicates and the results were compared to those of DMSO (solvent control) and cell-free media controls.
2.3. DNA isolation
A standard DNA isolation protocol was used [22]. Briefly, cell pellets were homogenized in a glass homogenizer with EDTA, SDS buffer [10 mM Tris, 1 mM Na2EDTA, 1% SDS (w/v), pH 8]. The homogenates were treated with RNase, DNase-free (50 U/ml) (Boehringer-Mannheim, Indianapolis, IN) and RNase T1 (1000 U/ml) (Boehringer-Mannheim) at 37°C for 1 h, followed by treatment with proteinase K (500 μg/ml) (Sigma, St. Louis, MO) at 37°C for 1 h. The DNA was extracted with equal volumes of Tris-equilibrated phenol (Boehringer-Mannheim) followed by extraction with 1:1 volume of Tris-equilibrated phenol and chloroform:isoamyl alcohol (24:1) and then with equal volumes of chloroform:isoamyl alcohol (24:1). The aqueous layer was treated with 1/10 volume of 5 M NaCl and twice the volume of cold 100% ethanol to precipitate the DNA, which was then dissolved in double-distilled water. The concentration was determined by UV absorbance at 260 nm.
2.4. Microsome isolation
Microsomes were prepared as described previously [28] with minor modifications. Briefly, cell culture samples were homogenized with a steel homogenizer in microsomal homogenization buffer [0.25 M K2HPO4, 0.15 M KCl 10 mM EDTA, and 0.25 mM phenylmethylsulfonylfluoride (PMSF)] and were centrifuged at 15,000 g for 20 min at 4°C. The supernatant was centrifuged at 100,000 g for 90 min at 4°C, and the pellet was resuspended in microsome dilution buffer (0.1 M KH2PO4, 20% glycerol, 10 mM EDTA, 0.1 mM DTT and 0.25 mM PMSF). The protein concentration was spectrophotometrically determined at 562 nm using the Bicinchonic acid colorimetric assay (Pierce, Rockford, IL).
2.5. Ethoxyresorufin O-deethylase (EROD) assay
Fifty micrograms of microsomal protein were added to 1 μM 7-ethoxyresorufin (Sigma) in 200 μl of 0.1 M Tris HCl (pH 7.8) buffer per well in a black 96-well plate format (E&K Scientific, Campbell, CA). NADPH (Sigma) was added to each well and the fluorescence was measured in a Spectra MAX Gemini plate reader (Molecular Devices, Sunnyvale, CA). The excitation and emission wavelengths were 535 nm and 585 nm, respectively, and the kinetic assay was monitored over 10 min. The experiment was repeated three times and each sample was assayed in triplicates. The amount of resorufin produced was calculated from the fluorescence of a known concentration of resorufin.
2.6. 33P-Postlabeling of PAH-DNA adducts
Postlabeling and HPLC separation of PAH-DNA adducts was carried out as described previously [22,29]. The BP-DNA adducts were resolved by elution at 1 ml/min with 0.1 M ammonium phosphate, pH 5.5 (solvent A) and 100% HPLC grade methanol (solvent B). The elution gradient was as follows: 44–55% solvent B over 5 min, 55–60% solvent B over 5 min, 60–90% solvent B over 20 min, 90–100% solvent B over 5 min and 100–44% solvent B over 5 min. DBP-DNA adducts were resolved by elution at 1 ml/min with 0.1 M ammonium phosphate, pH 5.5 (solvent A) and 50% HPLC grade methanol/50% acetonitrile (solvent B). The elution gradient was as follows: 20–44% solvent B over 20 min, 44–60% solvent B over 40 min, 60–80% solvent B over 15 min and 80–20% solvent B over 1 min. DNA adducts formed from DBC diol or diol-epoxide derivatives were resolved by elution at 1 ml/min with water (solvent A) and 90% HPLC grade methanol/10% acetonitrile (solvent B). The elution gradient was as follows: 46–60% solvent B over 30 min, 60–90% solvent B over 30 min, 90–100% solvent B over 10 min and 100–46% solvent B over 1 min. The level of DNA binding was calculated based on the labeling efficiency of a [3H]BP-7,8-diol 9,10-epoxide standard [30]. At least 3 independent sets of the postlabeling reactions were carried out for each sample treated, in order to determine the total PAH-DNA adduct levels.
2.7 Western blot analysis
Microsomal proteins (50 μg) were denatured by boiling for 3 min and separated by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) on 7.5% acrylamide gels (Sigma). After electrophoresis, the proteins were transferred onto a PVDF membrane (Bio Rad, Hercules, CA) in a Mini Blotter apparatus (Bio Rad). The membrane was blocked for 1 h in phosphate buffered saline with 0.05% tween (PBS-T) plus 8% powdered milk and incubated with either of the following primary antibodies for another hour: anti-CYP1A1 (1:1000), anti-CYP1B1 (1:5000) or monoclonal anti-actin A 4700 (1:1500) (Sigma). Human CYP1A1 protein was detected by rabbit polyclonal anti-CYP1A1 prepared against purified recombinant human CYP1A1 protein and kindly provided by F.P. Guengerich (Vanderbilt University School of Medicine, Nashville, TN). Polyclonal rabbit anti-CYP1B1 against human CYP1B1 was provided by C. Marcus (University of New Mexico, Albuquerque, NM). Peroxidase-conjugated anti-rabbit IgG (1:20,000) (Sigma) was applied as a secondary antibody for 30 min. As loading control, βactin was detected by horseradish peroxidase (HRP)-conjugated anti-actin from mouse (A 2028, Sigma) (1: 20,000). After several washes in PBS-T, the immunoreactive proteins were observed by the enhanced chemiluminescence (ECL) detection method, as described by the manufacturer (Amersham Life Science, Arlington Heights, IL). Microsomal proteins (10 μg) from Chinese hamster ovary V79 cells expressing either human CYP1A1 or CYP1B1 were used as positive controls.
3. Results
3.1. Cytotoxicity assay
MCF-7 cells were exposed to varying doses of DBC for 24 h and the cell viability was measured by MTT assay. The results obtained are illustrated in Fig. 3. Cells treated with DBC (0.75–4.5 μM) indicated cell viability similar to that of the controls (DMSO vehicle control or cell free media control). Thus cells remained viable at all DBC doses tested. By contrast, the percentage of viable cells was decreased to about 50-60% after exposure to BP or DBP, confirming the expected toxicity of these compounds at a concentration of 6 and 5 μM, respectively.
Fig. 3.
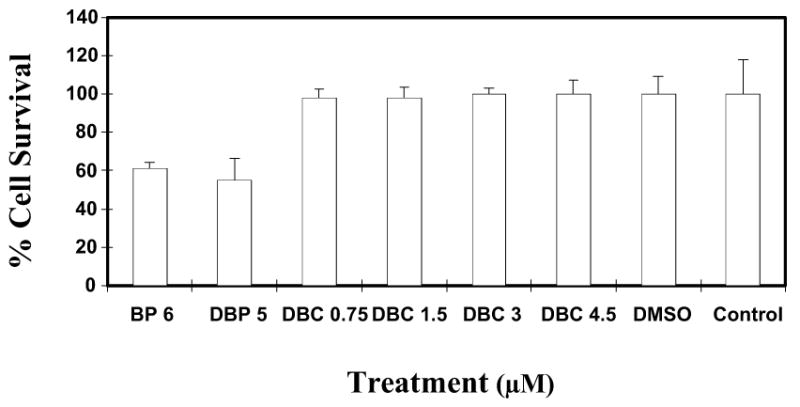
Cytotoxicity assay using MCF-7 cells in culture. MCF-7 cells were exposed to varying doses (0.75-4.5 μM) of DBC. Cells were also treated with BP and DBP at known cytotoxic doses of 6 and 5 μM, respectively, for comparison. Each data point represents the mean of four wells (mean ± SD). The percent cell survival is given in comparison to untreated cells.
3.2. CYP expression
The EROD assay was performed utilizing microsomes from MCF-7 cells exposed to BP, DBP, and DBC, as described in Section 2. Treatment with BP resulted in an increased EROD activity indicative of the induction of CYP enzymes that belong to the family CYP1 (e.g., CYP1A1, CYP1B1). Although the activity measured was found well above background levels, exposure to either of the two hexacyclic hydrocarbons (DBP or DBC) only led to a minor increase in the levels of EROD activity (Fig. 4). This result was confirmed through Western-blot analysis of the expression of CYP1A1 and CYP1B1 (Fig. 5). In comparison to BP, the level of CYP1A1 expression was only low in cells upon treatment with DBP or DBC, while no differences in CYP1B1 expression levels were observed. This may be due to the constitutive expression of this enzyme form in MCF-7 cells [31].
Fig. 4.

EROD assay testing for the induction of enzymes of the CYP1 family (e.g., CYP1A1, CYP1B1) in MCF-7 cells upon treatment with BP, DBP, or DBC. After 48 h of incubation, microsomes were isolated and utilized in the assay as described in Section 2.
Fig. 5.
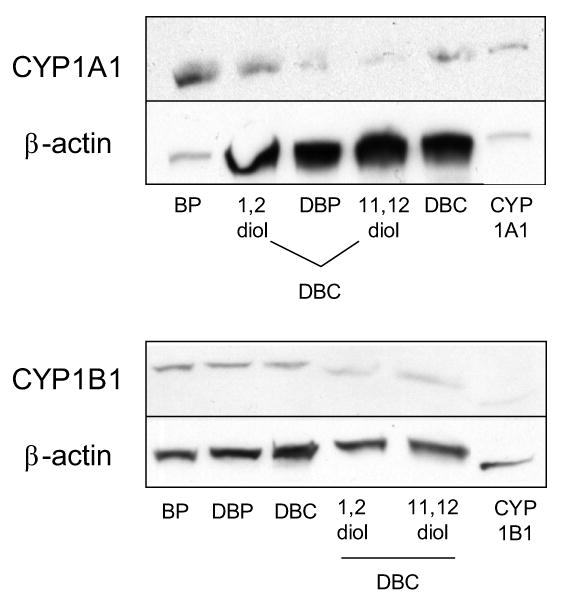
Western-blot analysis of CYP1A1 and CYP1B1 expression in MCF-7 cells treated for 48 h with BP, DBP, DBC, DBC-1,2-diol or DBC-11,12-diol. The assay was performed as described in Section 2.
3.3. DNA adduct formation in MCF-7 cells in culture
Based on the results obtained in the cytotoxicity assay, MCF-7 cells were treated with BP (2 μM), DBP (0.02 μM) and DBC (4.5 μM) as described in Section 2, and the PAH-DNA adducts were 33P-postlabeled and analyzed by HPLC. The HPLC elution profiles of the DNA adducts after 24 or 48 h of exposure are shown in Fig. 6. No detectable adducts were found in cells treated with the solvent (DMSO) alone. While treatment with BP produced only one major DNA adduct eluting at a retention time of approximately 25 minutes (Fig. 6, BP), several adducts were detected after exposure to DBP (DNA adduct peaks eluting between 50 and 80 minutes; Fig. 6, DBP). In contrast, DBC produced only minor and unreproducible amounts of polar adducts in MCF-7 cells that eluted between 25 and 50 minutes under the conditions used (Fig. 6, DBC).
Fig. 6.
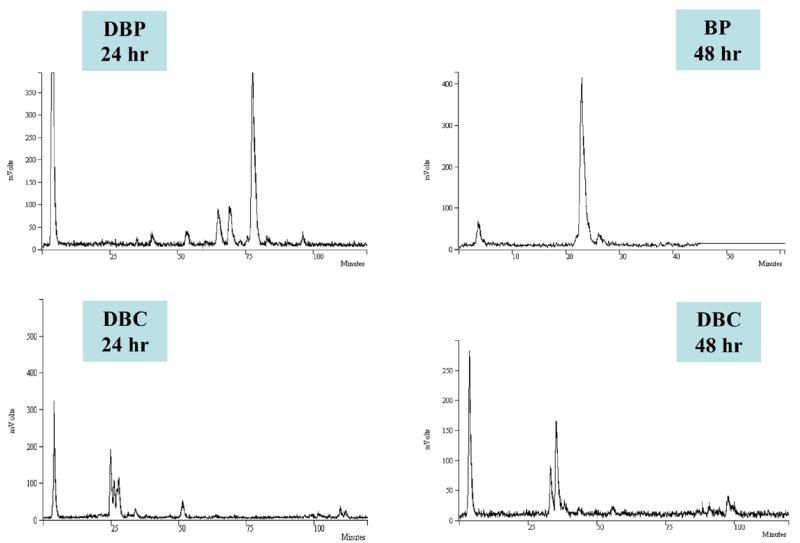
Representative HPLC elution profiles of 33P-postlabeled PAH-DNA adducts formed in MCF-7 cells. Cells were treated for 24 or 48 h with BP (2 μM), DBP (0.2 μM), and DBC (4.5 μM), respectively. DNA was isolated, digested, postlabeled and adducted nucleotides were HPLC separated as described in Section 2. No adducts or elution peaks were obtained when cells were exposed to the solvent (DMSO) only.
Treatment with DBC-1,2-diol (1 μg/ml, 2.8 μM) revealed a major peak at a retention time of about 25 min, with a smaller peak at about 110 min under the elution conditions used (Fig. 7, DBC-1,2-diol). A similar DNA adduct profile was obtained from cells after treatment with anti-DBC-1,2-diol-3,4-epoxide (1 μg/ml, 2.6 μM). The HPLC profiles revealed one major peak eluting at a retention time of 25 min, and several smaller peaks eluting in the range between 75 and 110 min (Fig. 7, anti-DBC-1,2-diol-3,4-epoxide). In contrast, cells treated with DBC-11,12-diol (1 μg/ml, 2.8 μM) for 2 or 48 h did not exhibit any detectable PAH-DNA adducts (Fig. 8, DBC-11,12-diol). On the other hand, treatment with anti-DBC-11,12-diol-13,14-epoxide (1μg/ml, 2.6 μM) again revealed a series of DNA adduct peaks eluting at retention times between 80 and 110 min (Fig. 8, anti-DBC-11,12-diol-13,14-epoxide).
Fig. 7.
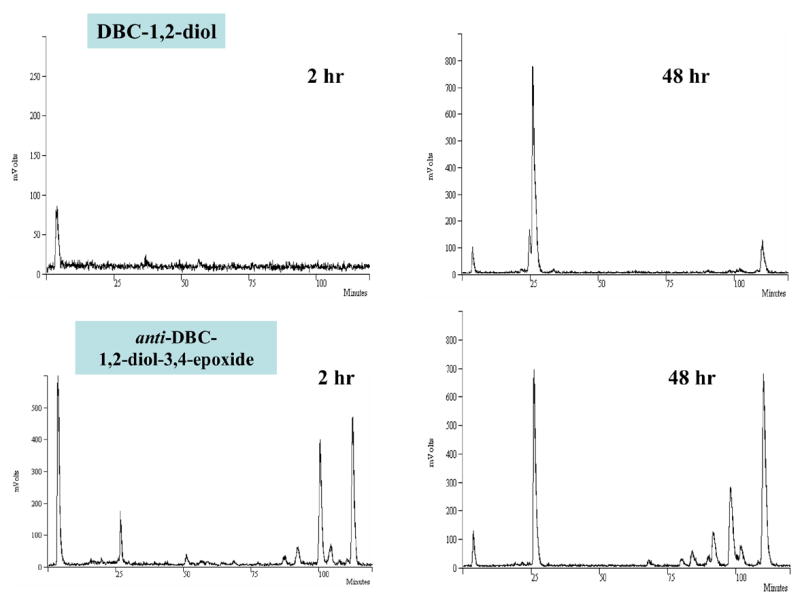
Representative HPLC elution profiles of 33P-postlabeled PAH-DNA adducts formed in MCF-7 cells. Cells were treated for 2 or 48 h with 1 μg/ml (2.8 μM) DBC-1,2-diol or 1 μg/ml (2.6 μM) anti-DBC-1,2-diol-3,4-epoxide. DNA was isolated, digested, postlabeled and adducted nucleotides were HPLC separated as described in Section 2.
Fig. 8.
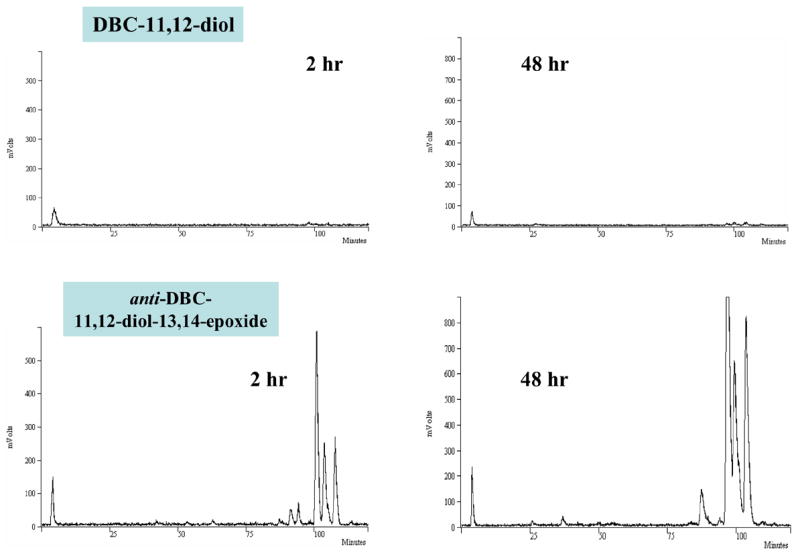
Representative HPLC elution profiles of 33P-postlabeled PAH-DNA adducts formed in MCF-7 cells. Cells were treated for 2 or 48 h with 1 μg/ml (2.8 μM) DBC-11,12-diol or 1 μg/ml (2.6 μM) anti-DBC-11,12-diol-13,14-epoxide. DNA was isolated, digested, postlabeled and adducted nucleotides were HPLC separated as described in Section 2.
Quantitative analyses of the average total DNA binding levels measured are illustrated in Fig. 9. PAH-DNA adduct levels were quantified 48 h after exposure to BP, DBP, and DBC or its diols and anti-diol-epoxides. As mentioned above, DBC bound to DNA to a relatively lower extent (0.6 pmol/mg DNA), as did its 11,12-diol derivative (1.5 pmol/mg DNA). This is contrasted by high binding levels of the 1,2-diol and both anti-diol-epoxides applied in the present study (Fig. 9). The total amount of DNA adducts formed with DBC-1,2-diol was 54 pmol/mg of DNA, while those formed with anti-DBC-1,2-diol-3,4-epoxide and anti-DBC-11,12-diol-13,14-epoxide were 33 and 51 pmol/mg of DNA, respectively. In comparison, BP and DBP gave rise to DNA adduct levels of 10 and 14 pmol/mg of DNA respectively.
Fig. 9.
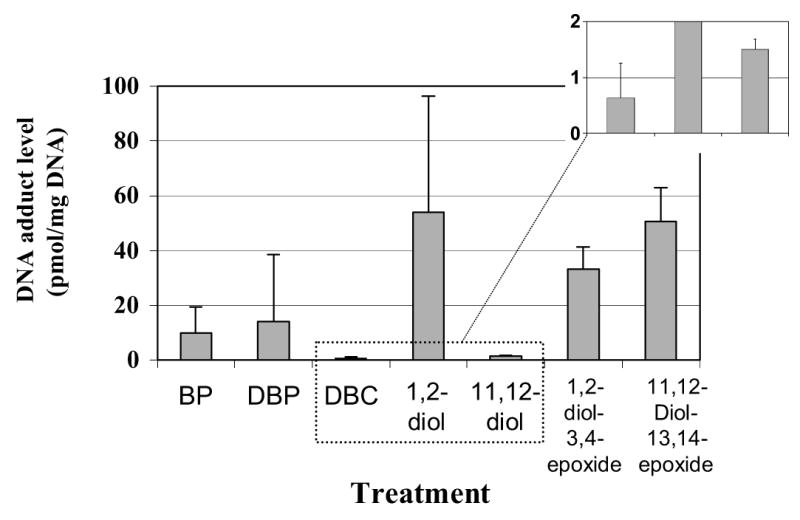
Graphic representation of the average amount of PAH-DNA adducts formed in MCF-7 cells during exposure to BP (2 μM), DBP (2 μM), and 1.5 μg/ml DBC (4.5 μM) or 1 μg/ml of its diol or anti-diol-epoxide derivatives, respectively. Levels are reported in pmol adducts per mg of DNA and mean values ± SD are given for at least 3 independent sets of the postlabeling reaction. The inset depicts adduct levels of DBC and DBC-11,12-diol with an enhanced y-axis.
4. Discussion
For potent carcinogenic PAHs that possess a bay region, i.e. BP or DBA, their carcinogenic power has been causatively linked to the metabolic formation of substantial amounts of bay region anti-diol-epoxides via their metabolic precursors, the trans-dihydrodiols [14,15]. On the other hand, benzo[c]phenanthrene (BPh, Fig. 1), the prototypic PAH harboring a fjord region, is only a weak carcinogen in mouse skin [32,33]. From all trans-dihydrodiols of BPh, the 3,4-diol is the only metabolite that elicits significant biological activities [33,34], yet still about 100-fold lower as compared to its corresponding 3,4-diol 1,2-epoxide [33,35]. It was found that the biological inefficacy of BPh results from a preponderance in metabolic conversion of its K-region, that is position 5-6 [14,36]. Thus stereoisomeric fjord region 3,4-diol 1,2-epoxides of BPh are formed in vivo in rodents only in trace amounts.
In contrast to BPh, the pentacyclic relatives BcC and BgC, both of which harbor a fjord and one or two bay region(s) within the same molecule (Fig. 1), have been reported to be converted into fjord region diol-epoxides in mouse skin to a greater extent [37,38]. Synthetic fjord region diol-epoxides of both benzochrysenes also display exceptionally high intrinsic mutagenic activity in various in vitro test systems [39]. In accordance with this data, BgC has been characterized as being a moderate carcinogen in mouse skin [40], and its fjord region anti-11,12-diol 13,14-epoxide is a potent tumorigen in newborn mice and rat mammary gland [41,42]. Metabolic conversion of BgC toward fjord region diol-epoxides was also confirmed in human MCF-7 cells [43] which express CYP1B1 constitutively [31]. Conversely, studies with V79 cells expressing single CYP enzymes revealed that BcC is activated at other positions in the molecule rather than at its bay (positions 1-4) or fjord region (positions 9-12). Applying human or rodent enzyme forms from the CYP1 family, there was a preponderance of oxidation reactions at positions 3-4 and 7-8 [44]. Despite these in vitro data, BcC is efficiently converted in rodents in vivo into its fjord region anti-9,10-diol 11,12-epoxide [25,38].
The hexacyclic fjord region PAH DBP has been proven to be an exceptionally strong tumorigenic compound [16]. Metabolism and DNA binding studies performed with human cell lines [45,46] or liver preparations from rats pretreated with different inducers of CYP enzymes such as 3-methylcholanthrene [47] or Aroclor 1254 [48] revealed that DBP is effectively activated via its 11,12-diol toward both fjord region syn- and anti-11,12-diol 13,14-epoxide isomers. Both metabolites are highly mutagenic in cell cultures [17] and carcinogenic in newborn mice, rat mammary gland and mouse skin [41,42,49]. In contrast to DBP, the DBC is a unique PAH possessing two fjord regions along with one bay region within the same molecule (Fig. 1). As with all other fjord region PAHs mentioned above, sterical hindrance within its fjord region(s) most likely induces deviation from planarity of the molecule [50]. It has been reported that structural features such as non-planarity may have an important role in determining the potency of a particular PAH as a carcinogen [11,15]. However, activation toward vicinal diol-epoxides and concurrent preponderance for covalent binding at dA sites seem to be the prerequisite for non-planarity vs. planarity to make a difference. So, BgC and DBP are more tumorigenic in rodents than BP because of their very efficient conversion into fjord region diol-epoxides [41,42]. Given the structure of DBC as a merger of BPh, BcC and BgC, one would have expected that this hexacyclic compound would be easily activated toward fjord region diol-epoxides as well. However, and in contrast to BPh [36], BgC [43] and DBP [29,45,46], all of which are activated in human MCF-7 cells toward DNA-binding intermediates to a considerable extent, DBC was found to be only weakly activated in this cell line (Fig. 6 and 9). Accordingly, no cytotoxicity was observed at doses up to 4.5 μM of DBC (Fig. 3).
Direct application of the two putative fjord region diol-epoxide precursors of DBC, the 1,2- and 11,12-diols (Fig. 2), revealed that only the 1,2-diol derivative can be further efficiently converted by MCF-7 cells into DNA-binding species (Fig. 7 and 8). While both diol-epoxides bind to DNA to a comparable extent (Fig. 9), only the 1,2-diol is transformed into its corresponding vicinal diol-epoxide. On the contrary, the 11,12-diol is barely accepted as a substrate by MCF-7-expressed CYP1B1. Again, this is in sharp contrast to the MCF-7-mediated activation of the precursor diols of fjord region diol-epoxides of BPh [36], BgC [43] and DBP [29].
Altogether, human mammary carcinoma MCF-7 cells only weakly activate DBC toward DNA-binding metabolites. Although the 1,2-diol intermediate is accepted as a substrate, this derivative was not formed from the parent compound in considerable amounts. Similarly, liver homogenates obtained from rats pretreated with phenobarbital were also shown to be incapable of metabolizing DBC at its 1,2-position [26]. However, these preparations were quite effective in generating the 11,12-diol by conversion of the parent hydrocarbon [26]. Since MCF-7 cells are unable to convert the 11,12-diol further into DNA-binding diol-epoxides, it is not clear whether or not this metabolite is formed in these human cells and whether there are species-specific differences in the activation of DBC. Further studies are warranted in order to clarify the role of individual CYP enzymes and the possible species specificity in the activation of this unique hexacyclic aromatic hydrocarbon.
Acknowledgments
This work was supported by grant CA 28825, DHHS, from the National Cancer Institute. This publication was made possible, in part, by the Cell Culture Core of the Environmental Health Science Center, Oregon State University, funded by grant P30 ES00210 from the National Institute of Environmental Health Sciences. The authors wish to acknowledge Herminio Garcia for his initial assistance in this project. He was supported by Grant ES 007316. We also acknowledge the technical assistance with the HPLC and MTT assay provided by Tamara Musafia.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Luch A. Polycyclic aromatic hydrocarbon-induced carcinogenesis – an integrated view. In: Luch A, editor. The Carcinogenic Effects of Polycyclic Aromatic Hydrocarbons. Imperial College Press; London: 2005. pp. 379–452. [Google Scholar]
- 2.Hoffmann D, Hoffmann I. Tobacco smoke components. Beitr Tabakforsch. 1998;18:49–52. [Google Scholar]
- 3.Mumford JL, Li X, Hu F, Lu XB, Chuang JC. Human exposure and dosimetry of polycyclic aromatic hydrocarbons in urine from Xuan Wei, China with high lung cancer mortality associated with exposure to unvented coal smoke. Carcinogenesis. 1995;16:3031–3036. doi: 10.1093/carcin/16.12.3031. [DOI] [PubMed] [Google Scholar]
- 4.Bartsch H. DNA adducts in human carcinogenesis: etiological relevance and structure-activity relationship. Mutat Res. 1996;340:67–79. doi: 10.1016/s0165-1110(96)90040-8. [DOI] [PubMed] [Google Scholar]
- 5.Phillips DH. Smoking-related DNA and protein adducts in human tissues. Carcinogenesis. 2002;23:1979–2004. doi: 10.1093/carcin/23.12.1979. [DOI] [PubMed] [Google Scholar]
- 6.Perera FP, Estabrook A, Hewer A, Channing K, Rundle A, Mooney LA, Whyatt R, Phillips DH. Carcinogen-DNA adducts in human breast tissue. Cancer Epidemiol Biomarkers Prev. 1995;4:233–238. [PubMed] [Google Scholar]
- 7.Li D, Wang M, Dhingra K, Hittelman WN. Aromatic DNA adducts in adjacent tissues of breast cancer patients: clues to breast cancer etiology. Cancer Res. 1996;56:287–293. [PubMed] [Google Scholar]
- 8.Amin S, El-Bayoumy K. Tumorigenicity of polycyclic aromatic hydrocarbons. In: Luch A, editor. The Carcinogenic Effects of Polycyclic Aromatic Hydrocarbons. Imperial College Press; London: 2005. pp. 315–351. [Google Scholar]
- 9.Gammon MD, Sagiv SK, Eng SM, Shantakumar S, Gaudet MM, Teitelbaum SL, Britton JA, Terry MB, Wang LW, Wang Q, Stellman SD, Beyea J, Hatch M, Kabat GC, Wolff MS, Levin B, Neugut AI, Santella RM. Polycyclic aromatic hydrocarbon-DNA adducts and breast cancer: a pooled analysis. Arch Environ Health. 2004;59:640–649. doi: 10.1080/00039890409602948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arif JM, Dresler C, Clapper ML, Gairola CG, Srinivasan C, Lubet RA, Gupta RC. Lung DNA adducts detected in human smokers are unrelated to typical polyaromatic carcinogens. Res Toxicol. 2006;19:295–299. doi: 10.1021/tx0502443. [DOI] [PubMed] [Google Scholar]
- 11.Dipple A, Peltonen K, Cheng SC, Hilton BD. Chemistry of DNA adduct formation by dihydrodiol epoxides of polycyclic aromatic hydrocarbons. In: Garrigues P, Lamotte M, editors. Polycyclic Aromatic Hydrocarbons: Synthesis, Properties, Analytical Measurements, Occurrence and Biological Effects. Gordon and Breach Publishers; Philadelphia, PA: 1993. pp. 807–816. [Google Scholar]
- 12.Thakker DR, Yagi H, Lu AY, Levin W, Conney AH. Metabolism of benzo[a]pyrene: conversion of (±)-trans-7,8-dihydroxy-7,8-dihydrobenzo[a]pyrene to highly mutagenic 7,8-diol-9,10-epoxides. Proc Natl Acad Sci USA. 1976;73:3381–3385. doi: 10.1073/pnas.73.10.3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mlcoch J, Fuchs J, Oesch F, Platt KL. Characterization of DNA adducts at the bay region of dibenz[a,h]anthracene formed in vitro. Carcinogenesis. 1993;14:469–473. doi: 10.1093/carcin/14.3.469. [DOI] [PubMed] [Google Scholar]
- 14.Thakker DR, Yagi H, Levin W, Wood AW, Conney AH, Jerina DM. Polycyclic aromatic hydrocarbons: metabolic activation to ultimate carcinogens. In: Anders MW, editor. Bioactivation of Foreign Compounds. Academic Press; London: 1985. pp. 177–242. [Google Scholar]
- 15.Luch A, Baird WM. Metabolic activation and detoxification of polycyclic aromatic hydrocarbons. In: Luch A, editor. The Carcinogenic Effects of Polycyclic Aromatic Hydrocarbons. Imperial College Press; London: 2005. pp. 19–96. [Google Scholar]
- 16.Higginbotham S, RamaKrishna NV, Johansson SL, Rogan EG, Cavalieri EL. Tumor-initiating activity and carcinogenicity of dibenzo[a, l]pyrene versus.7,12-dimethylbenz[a]anthracene and benzo[a]pyrene at low doses in mouse skin. Carcinogenesis. 1993;14:875–878. doi: 10.1093/carcin/14.5.875. [DOI] [PubMed] [Google Scholar]
- 17.Luch A, Glatt H, Platt KL, Oesch F, Seidel A. Synthesis and mutagenicity of the diastereomeric fjord-region 11,12-dihydrodiol 13,14-epoxides of dibenzo[a,l]pyrene. Carcinogenesis. 1994;15:2507–2516. doi: 10.1093/carcin/15.11.2507. [DOI] [PubMed] [Google Scholar]
- 18.Mahadevan B, Dashwood WM, Luch A, Pecaj A, Doehmer J, Seidel A, Pereira C, Baird WM. Mutations induced by (−)-anti-11R,12S-dihydrodiol 13S,14R-epoxide of dibenzo[a,l]pyrene in the coding region of the hypoxanthine phosphoribosyltransferase (Hprt) gene in Chinese hamster V79 cells. Environ Mol Mutagen. 2003;41:131–139. doi: 10.1002/em.10136. [DOI] [PubMed] [Google Scholar]
- 19.Phillips DH, Hewer A, Seidel A, Steinbrecher T, Schrode R, Oesch F, Glatt H. Relationship between mutagenicity and DNA adduct formation in mammalian cells for fjord- and bay-region diol-epoxides of polycyclic aromatic hydrocarbons. Chem Biol Interact. 1991;8:177–186. doi: 10.1016/0009-2797(91)90023-z. [DOI] [PubMed] [Google Scholar]
- 20.Naegeli H, Geacintov NE. Mechanisms of repair of polycyclic aromatic hydrocarbon-induced DNA damage. In: Luch A, editor. The Carcinogenic Effects of Polycyclic Aromatic Hydrocarbons. Imperial College Press; London: 2005. pp. 211–258. [Google Scholar]
- 21.Shou M, Korzekwa KR, Crespi CL, Gonzalez FJ, Gelboin HV. The role of 12 cDNA-expressed human, rodent, rabbit cytochromes P450 in the metabolism of benzo[a]pyrene and benzo[a]pyrene trans-7,8-dihydrodiol. Mol Carcinog. 1994;10:159–168. doi: 10.1002/mc.2940100307. [DOI] [PubMed] [Google Scholar]
- 22.Luch A, Coffing SL, Tang YM, Schneider A, Soballa V, Greim H, Jefcoate CR, Seidel A, Greenlee WF, Baird WM, Doehmer J. Stable expression of human cytochrome P450 1B1 in V79 Chinese hamster cells and metabolically catalyzed DNA adduct formation of dibenzo[a,l]pyrene. Chem Res Toxicol. 1998;11:686–695. doi: 10.1021/tx970236p. [DOI] [PubMed] [Google Scholar]
- 23.Luch A, Platt KL, Seidel A. Synthesis of fjord region tetraols and their use in hepatic biotransformation studies of dihydrodiols of benzo[c]chrysene, benzo[g]chrysene and dibenzo[a,l]pyrene. Carcinogenesis. 1998;19:639–648. doi: 10.1093/carcin/19.4.639. [DOI] [PubMed] [Google Scholar]
- 24.Luch A, Seidel A, Glatt H, Platt KL. Metabolic activation of the (+)-S,S- and (−)-R,R-enantiomers of trans-11,12-dihydroxy-11,12-dihydrodibenzo[a,l]pyrene: stereoselectivity, DNA adduct formation, and mutagenicity in Chinese hamster V79 cells. Chem Res Toxicol. 1997;10:1161–1170. doi: 10.1021/tx970005i. [DOI] [PubMed] [Google Scholar]
- 25.Amin S, Lin JM, Krzeminski J, Boyiri T, Desai D, El-Bayoumy K. Metabolism of benzo[c]chrysene and comparative mammary gland tumorigenesis of benzo[c]chrysene bay and fjord region diol epoxides in female CD rats. Chem Res Toxicol. 2003;16:227–231. doi: 10.1021/tx0200921. [DOI] [PubMed] [Google Scholar]
- 26.Sharma AK, Lin JM, Desai D, Amin S. Convenient synthesis of dibenzo[c,p]chrysene and its possible proximate and ultimate carcinogens: in vitro metabolism and DNA adduction studies. J Org Chem. 2005;70:4962–4970. doi: 10.1021/jo040291k. [DOI] [PubMed] [Google Scholar]
- 27.Sauvain JJ, Vu Duc T, Huynh CK. Development of an analytical method for the simultaneous determination of 15 carcinogenic polycyclic aromatic hydrocarbons and polycyclic aromatic nitrogen heterocyclic compounds: application to diesel particulates. Fresenius J Anal Chem. 2001;371:966–974. doi: 10.1007/s002160101047. [DOI] [PubMed] [Google Scholar]
- 28.Otto S, Marcus C, Pidgeon C, Jefcoate C. A novel adrenocorticotropin-inducible cytochrome. P450 from rat adrenal microsomes catalyzes polycyclic aromatic hydrocarbon metabolism. Endocrinology. 1991;129:970–982. doi: 10.1210/endo-129-2-970. [DOI] [PubMed] [Google Scholar]
- 29.Ralston SL, Coffing SL, Seidel A, Luch A, Platt KL, Baird WM. Stereoselective activation of dibenzo[a,l]pyrene and its trans-11,12-dihydrodiol to fjord region 11,12-diol 13,14-epoxides in a human mammary carcinoma MCF-7 cell-mediated V79 cell mutation assay. Chem Res Toxicol. 1997;10:687–693. doi: 10.1021/tx9700275. [DOI] [PubMed] [Google Scholar]
- 30.Lau HH, Baird WM. Detection and identification of benzo[a]pyrene-DNA adducts by [35S]phosphorothioate labeling and HPLC. Carcinogenesis. 1991;12:885–893. doi: 10.1093/carcin/12.5.885. [DOI] [PubMed] [Google Scholar]
- 31.Christou M, Savas Ü, Spink DC, Gierthy JF, Jefcoate CR. Co-expression of human CYP1A1 and human analogue of cytochrome P450-EF in response to 2,3,7,8-tetrachloro-dibenzo-p-dioxin in the human mammary carcinoma-derived MCF-7 cells. Carcinogenesis. 1994;15:725–732. doi: 10.1093/carcin/15.4.725. [DOI] [PubMed] [Google Scholar]
- 32.Levin W, Chang RL, Wood A, Thakker DR, Yagi H, Jerina DM. Tumorigenicity of optical isomers of the diastereomeric bay-region 3,4-diol-1,2-epoxides of benzo[c]phenanthrene in murine tumor models. Cancer Res. 1986;46:2257–2261. [PubMed] [Google Scholar]
- 33.Levin W, Wood AW, Chang RL, Ittah Y, Croisy-Delcey M, Yagi H, Conney AH, Jerina DM. Exceptionally high tumor-initiating activity of benzo[c]phenanthrene bay-region diol-epoxides on mouse skin. Cancer Res. 1980;40:3910–3914. [PubMed] [Google Scholar]
- 34.Wood AW, Chang RL, Levin W, Ryan DE, Thomas PE, Croisy-Delcey M, Ittah Y, Yagi H, Jerina DM, Conney AH. Mutagenicity of the dihydrodiols and bay-region diol-epoxides of benzo[c]phenanthrene in bacterial and mammalian cells. Cancer Res. 1980;40:2876–2883. [PubMed] [Google Scholar]
- 35.Jerina DM, Sayer JM, Yagi H, Croisy-Delcey M, Ittah Y, Thakker DR, Wood AW, Chang RL, Levin W, Conney AH. Highly tumorigenic bay-region diol epoxides from the weak carcinogen benzo[c]phenanthrene. In: Snyder R, Parke DV, Kocsis JJ, Jollow DJ, Gibson CG, Witmer CM, editors. Advances in Experimental Medical Biology: Biological Reactive Intermediates IIA. Plenum Publishing Co; New York: 1982. pp. 501–523. [DOI] [PubMed] [Google Scholar]
- 36.Einolf HJ, Amin S, Yagi H, Jerina DM, Baird WM. Benzo[c]phenanthrene is activated to DNA-binding diol epoxides in the human mammary carcinoma cell line MCF-7 but only limited activation occurs in mouse skin. Carcinogenesis. 1996;17:2237–2244. doi: 10.1093/carcin/17.10.2237. [DOI] [PubMed] [Google Scholar]
- 37.Giles AS, Seidel A, Phillips DH. Covalent DNA adducts formed in mouse epidermis by benzo[g]chrysene. Carcinogenesis. 1996;17:1331–1336. doi: 10.1093/carcin/17.6.1331. [DOI] [PubMed] [Google Scholar]
- 38.Giles AS, Seidel A, Phillips DH. Covalent DNA adducts formed by benzo[c]chrysene in mouse epidermis and by benzo[c]chrysene fjord-region diol-epoxides reacted with DNA and polynucleotides. Chem Res Toxicol. 1997;10:1275–1284. doi: 10.1021/tx970114x. [DOI] [PubMed] [Google Scholar]
- 39.Glatt HR, Piée A, Pauly K, Steinbrecher T, Schrode R, Oesch F, Seidel A. Fjord- and bay-region diol-epoxides investigated for stability, SOS induction in Escherichia coli, and mutagenicity in Salmonella typhimurium and mammalian cells. Cancer Res. 1991;51:1659–1667. [PubMed] [Google Scholar]
- 40.Hartwell JL. Survey of compounds which have been tested for carcinogenic activity, Federal Security Agency, Public Health Service Publication No. 149. National Cancer Institute, US Government Printing Office; Washington D.C: 1951. [Google Scholar]
- 41.Amin S, Krzeminski J, Rivenson A, Kurtzke C, Hecht SS, El-Bayoumy K. Mammary carcinogenicity in female CD rats of fjord region diol epoxides of benzo[c]phenanthrene, benzo[g]chrysene and dibenzo[a,l]pyrene. Carcinogenesis. 1995;16:1971–1974. doi: 10.1093/carcin/16.8.1971. [DOI] [PubMed] [Google Scholar]
- 42.Amin S, Desai D, Dai W, Harvey RG, Hecht SS. Tumorigenicity in newborn mice of fjord region and other sterically hindered diol epoxides of benzo[g]chrysene, dibenzo[a,l]pyrene (dibenzo[def,p]chrysene), 4H-cyclopenta[def]chrysene and fluoranthene. Carcinogenesis. 1995;16:2813–2817. doi: 10.1093/carcin/16.11.2813. [DOI] [PubMed] [Google Scholar]
- 43.Agarwal R, Coffing SL, Baird WM, Kiselyov AS, Harvey RG, Dipple A. Metabolic activation of benzo[g]chrysene in the human mammary carcinoma cell line MCF-7. Cancer Res. 1997;57:415–419. [PubMed] [Google Scholar]
- 44.Jacob J, Raab G, Schober W, Frank H, Luch A, Doehmer J, Seidel A. Species-dependent metabolism of benzo[c]chrysene mediated by c-DNA-expressed human, rodent and fish cytochrome P450 enzymes. Polycycl Aromat Compd. 2000;21:109–121. [Google Scholar]
- 45.Ralston SL, Lau HHS, Seidel A, Luch A, Platt KL, Baird WM. The potent carcinogen dibenzo[a,l]pyrene is metabolically activated to fjord-region 11,12-diol-13,14-epoxides in human mammary carcinoma MCF-7 cell cultures. Cancer Res. 1994;54:887–890. [PubMed] [Google Scholar]
- 46.Ralston SL, Seidel A, Luch A, Platt KL, Baird WM. Stereoselective activation of dibenzo[a,l]pyrene to (−)-anti-(11R,12S,13S,14R)- and (+)-syn-(11S,12R,13S,14R)-11,12-diol-13,14-epoxides which bind extensively to deoxyadenosine residues of DNA in the human mammary carcinoma cell line MCF-7. Carcinogenesis. 1995;16:2899–2907. doi: 10.1093/carcin/16.12.2899. [DOI] [PubMed] [Google Scholar]
- 47.Devanesan PD, Cremonesi P, Nunnally JE, Rogan EG, Cavalieri EL. Metabolism and mutagenicity of dibenzo[a,e]pyrene and the very potent environmental carcinogen dibenzo[a,l]pyrene, Chem. Res Toxicol. 1990;3:580–586. doi: 10.1021/tx00018a014. [DOI] [PubMed] [Google Scholar]
- 48.Arif JM, Gupta RC. Microsome-mediated bioactivation of dibenzo[a,l]pyrene and identification of DNA adducts by 32P-postlabeling. Carcinogenesis. 1997;18:1999–2007. doi: 10.1093/carcin/18.10.1999. [DOI] [PubMed] [Google Scholar]
- 49.Gill HS, Kole PL, Wiley JC, Li KM, Higginbotham S, Rogan EG, Cavalieri EL. Synthesis and tumor-initiating activity in mouse skin of dibenzo[a,l]pyrene syn- and anti-fjord-region diolepoxides. Carcinogenesis. 1994;15:2455–2460. doi: 10.1093/carcin/15.11.2455. [DOI] [PubMed] [Google Scholar]
- 50.Herndon WC, Nowak PC, Connor DA, Lin P. Empirical model calculations for thermodynamic and structural properties of condensed polycyclic aromatic hydrocarbons. J Am Chem Soc. 1992;114:41–47. [Google Scholar]