

Abstract
Defects in protein folding and the proteasomal pathway have been linked with many neurodegenerative diseases. PLIC-1 (protein linking IAP to the cytoskeleton) is a ubiquitin-like protein that binds to the ubiquitin-interacting motif (UIM) of the proteasomal subunit S5a. Here, we show that PLIC-1 also binds to the UIM proteins ataxin 3—a deubiquitinating enzyme—HSJ1a—a co-chaperone—and EPS15 (epidermal growth factor substrate 15)—an endocytic protein. Using a polyglutamine (polyQ) disease model, we found that both endogenous PLIC-1 and EPS15 localize to perinuclear aggresomes, and that polyQ enhances their in vivo interaction. We show that knockdown of PLIC-1 and EPS15 by RNA interference reduces aggresome formation. In addition, PLIC-1ΔUBL functions as a dominant-negative mutant, blocking both polyQ transport to aggresomes and the association of EPS15 with dispersed aggregates. We also show that PLIC-1 is upregulated by arsenite-induced protein misfolding. These results indicate a role for PLIC-1 in the protein aggregation-stress pathway, and we propose a novel function for the ubiquitin-like (UBL) domain—by means of UBL–UIM interactions—in transport to aggresomes.
Keywords: PLIC-1, EPS15, aggresomes
Introduction
Ubiquitin modification of proteins is both rapid and reversible, and a regulatory role for ubiquitination is akin to phosphorylation. Ubiquitination can signal proteins for degradation (Pickart, 2001), and regulate protein interactions and intracellular localization (Di Fiore et al, 2003). A domain homologous to ubiquitin is also found in proteins of the type II ubiquitin-like (UBL) family (Hartmann-Petersen & Gordon, 2004). Sequence and structural homology indicates that UBL domains mimic aspects of ubiquitin function, in particular its interaction with the proteasome (Walters et al, 2002). Included in this UBL family is PLIC-1 (protein linking IAP to the cytoskeleton (Wu et al, 1999; Bedford et al, 2001), also known as ubiquilin-1 (Mah et al, 2000). Structurally, PLIC-1 encodes an amino-terminal UBL domain that binds to the second ubiquitin-interacting (UIM-2) motif of S5a, a proteasomal 19S cap subunit (Walters et al, 2002). PLIC-1 also encodes a region rich in asparagine–proline repeats and a carboxy ubiquitin-associated (UBA) domain, which binds to polyubiquitinated proteins (Chen et al, 2001). The ability of PLIC-1 to bind to both ubiquitinated proteins and the proteasome has suggested a role for PLIC-1 as a proteasomal shuttling factor.
Several observations indicate that the UBL domain is crucial for the function of PLIC-1. The yeast homologue of PLIC-1—Dsk2p—was identified through a function-disrupting mutation in the UBL domain (Biggins et al, 1996). The UBL domain is also highly conserved between PLIC-1 and its homologues in sequence (Kleijnen et al, 2000), structure and proteasomal binding (Walters et al, 2002). In addition, an interesting observation is that the UIM domain of S5a recognizes both the UBL domain of PLIC (Walters et al, 2002) and ubiquitin (Fisher et al, 2003) through a conserved surface.
UIM domains have been identified in various proteins and their ubiquitin-binding abilities confirmed (Hofmann & Falquet, 2001). These include the epidermal growth factor substrate 15 (EPS15; Polo et al, 2002), the deubiquitinating enzyme ataxin 3 (AT3; Burnett & Pittman, 2005) and the co-chaperone human neuron-specific DNAJ-like protein 1a (HSJ1a; Westhoff et al, 2005). The identification of UIMs in proteins other than S5a raises the possibility that there might be many UIM-containing proteins that bind to the UBL domain of PLIC-1.
Here, we report that the UBL domain of PLIC-1 binds to the UIM domains of EPS15, AT3 and HSJ1a. Using a polyglutamine (polyQ) disease-model protein, we examined the role of PLIC-1 and the endocytic UIM protein EPS15 in aggresome formation, and identified a novel function for the UBL domain of PLIC-1 in aggregate transport.
Results
The UBL domain binds to multiple UIM proteins
PLIC-1 has been shown to bind to the UIM domains of S5a (Walters et al, 2002). To test whether UIM protein binding is a general property of the UBL domain, we carried out glutathione S-transferase (GST) pulldowns using the UIM proteins AT3, HSJ1a and EPS15. GST fused to the UBL domain of PLIC-1 (GST–PLIC-1UBL) pulled down Myc–AT3 and Myc–HSJ1a, and also green fluorescent protein (GFP)–EPS15 (Fig 1A). Deletion of the UIM domains of either HSJ1a or EPS15 resulted in the loss of binding of these proteins to GST–PLIC-1UBL. In addition, GST–EPS15UIM pulled down Myc–PLIC-1 and GFP–PLIC-1UBL but not a Myc–PLIC-1ΔUBL mutant (Fig 1B). As a control, we used GST–S5a, which, as predicted (Kleijnen et al, 2003), bound to Myc–PLIC-1 and to Myc–PLIC-1ΔUBL, although with lower affinity (Fig 1C). These results indicate that binding of UIM proteins is a common feature of the UBL domain of PLIC-1.
Figure 1.
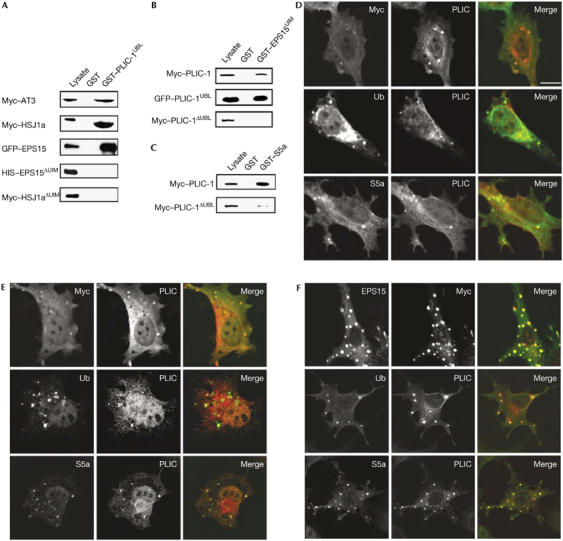
The ubiquitin-like domain of PLIC-1 binds to multiple proteins containing the ubiquitin-interacting motif. Lysates from baby hamster kidney cells transfected with the indicated proteins were pulled down with GST fusion proteins, followed by western blotting with antibodies to the designated epitope tags. (A) In vitro binding of Myc–AT3, Myc–HSJ1a, GFP–EPS15, His–EPS15ΔUIM and Myc–HSJ1aΔUIM to immobilized GST–PLIC-1UBL and GST. (B) In vitro binding of Myc–PLIC-1, GFP–PLIC-1UBL and Myc–PLIC-1ΔUBL to immobilized GST–EPS15UIM and GST. (C) In vitro binding of Myc–PLIC-1 and Myc–PLIC-1ΔUBL to GST–S5a and GST. Lysate is 5% of input for GFP–EPS15 (A) and 10% for all others. PLIC-1–UIM protein structures resemble aggresomes. BHK cells transfected with (D) PLIC-1 and Myc–AT3, (E) PLIC-1 and Myc–HSJ1a and (F) PLIC-1 and His–EPS15 were stained for colocalization, ubiquitinated proteins (Ub) and the proteasomal subunit S5a as designated on the panels. Scale bar, 10 μm. AT3, ataxin 3; BHK cells, baby hamster kidney cells; EPS15, epidermal growth factor substrate 15; GFP, green fluorescent protein; GST, glutathione S-transferase; HIS, 6× histidine; HSJ1a, human neuron-specific DNAJ-like protein 1a; PLIC-1, protein linking IAP to the cytoskeleton; S5a, proteasomal subunit S5a; UBL, ubiquitin-like; UIM, ubiquitin-interacting motif.
PLIC-1–UIM protein structures resemble aggresomes
AT3 and HSJ1a are associated with the clearance of misfolded aggregation-prone proteins (Burnett & Pittman, 2005; Westhoff et al, 2005), and EPS15 is associated with the sorting of monoubiquitinated proteins for endocytosis and lysosomal degradation (Polo et al, 2002). Alone, PLIC-1 showed diffuse expression (supplementary Fig S1a online), whereas coexpression with AT3 or HSJ1a resulted in the formation of small punctate structures containing both proteins (Fig 1D,E). To examine whether the constituents of these punctate structures were associated with the clearance of misfolded aggregation-prone proteins (Johnston et al, 1998), as suggested by AT3 and HSJ1a functions, we co-stained with antibodies to ubiquitinated proteins and the proteasomal subunit S5a, and found that they stained positively for both (Fig 1D,E).
Coexpression of PLIC-1 and EPS15 also resulted in the localization of both proteins in punctate structures (Fig 1F), which were more pronounced than with AT3 and HSJ1a; EPS15 expression alone did not induce such a phenotype (supplementary Fig S1b online). Enlarged early endosomes are commonly associated with the overexpression of endosome-associated proteins. However, these structures did not stain for the early endosomal antigen-1 (supplementary Fig S2 online) but instead for ubiquitin and S5a (Fig 1F), similar to the HSJ1a and AT3 structures.
The EPS15 PLIC-1 structures varied in size; therefore, we postulated that they coalesced to form larger structures. To investigate this, we used live imaging of cells transfected with GFP–EPS15 and PLIC-1 (supplementary Fig S3 and Movie 1 online). We observed that small GFP-labelled structures formed and then seemed to be transported, merging into larger structures close to the nucleus. Aggresomes are characterized by the presence of ubiquitinated misfolded proteins and the proteasome, and are formed by the retrograde transport of small aggregates on microtubules to the microtubule-organizing centre (MTOC; Johnston et al, 1998). Nocodazole depolymerization of microtubules, which blocks aggresome formation, inhibited the merging of these structures (supplementary Fig S3 and Movie 2 online); therefore, these EPS15 PLIC-1 structures show several defining characteristics of aggresomes.
Endogenous PLIC-1 and EPS15 localize to aggresomes
AT3 and HSJ1a have been found at aggresomes formed either by an elongated tract of glutamines (polyQ) or by the ΔF508 cystic fibrosis transmembrane conductance regulator mutant and are involved in aggresome biogenesis (Burnett & Pittman, 2005; Westhoff et al, 2005). To investigate whether endogenous PLIC-1 and EPS15 are similarly associated with aggresomes, we used an AU1 epitope tagged expanded polyQ tract protein (AU1-Q78; Toulouse et al, 2005) to induce aggresome formation. AU1-Q78 formed single, large perinuclear aggresomes, which co-stained for both endogenous PLIC-1 and EPS15 (Fig 2A,B). These aggresomes were also characterized by staining with the MTOC marker γ-tubulin (Fig 2C), which is a marker of aggresomes (Johnston et al, 1998). These results show that endogenous PLIC-1 and EPS15 localize to γ-tubulin-positive aggresomes.
Figure 2.
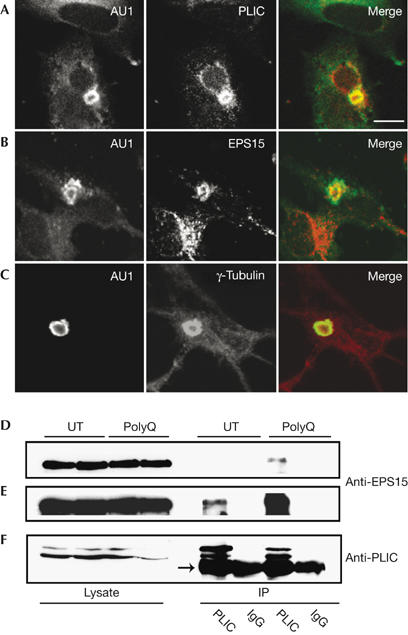
Endogenous PLIC-1 and epidermal growth factor substrate 15 interact and localize to aggresomes. Baby hamster kidney cells were transfected with AU1-Q78 and stained for (A) AU1 and PLIC-1, (B) AU1 and EPS15 or (C) AU1 and γ-tubulin. All proteins localize to aggresomes. Scale bar, 10 μm. Aggregation modulates endogenous PLIC-1 and EPS15 interaction. Lysates from untransfected (UT) or AU1-Q78 (polyQ)-transfected BHK cells were immunoprecipitated (IP) with anti-PLIC-1 or control rabbit IgG and subjected to western blotting with antibodies to EPS15 (D,E) and PLIC-1 (F). (D) Representation of a longer exposure of (E); the arrow indicates heavy-chain IgG. AU1, epitope tag; EPS15, epidermal growth factor substrate 15; PLIC-1, protein linking IAP to the cytoskeleton.
Aggregation modulates PLIC-1 and EPS15 interaction
As PLIC-1 and EPS15 interacted in vitro (Fig 1A), colocalized when overexpressed (Fig 1F), and the endogenous proteins localized to polyQ aggresomes (Fig 2A,B), we examined whether these two proteins interacted endogenously by co-immunoprecipitation and whether aggregation-prone proteins modulate this interaction. We found that endogenous EPS15 co-immunoprecipitated with PLIC-1 and that more EPS15 was bound in AU1-Q78-expressing cells (Fig 2D–F). Neither endogenous EPS15 nor PLIC-1 was detected in control rabbit IgG immunoprecipitates, and AU1-Q78 expression was confirmed by western blot (data not shown). These results indicate that endogenous EPS15 and PLIC-1 interact and that their association is enhanced by the presence of an aggregation-prone protein.
PLIC-1 and EPS15 regulate aggresome formation
To investigate whether PLIC-1 and EPS15 are required for aggresome formation, we used RNA interference (RNAi) to knock down their expression, quantified aggresome formation and compared the results with control cells transfected with scrambled RNAi, which were assigned a value of 1. We found that knockdown of PLIC-1 (Fig 3A) significantly inhibited aggresome formation to 0.724±0.070 of control (P<0.05; Fig 3C). Knockdown of EPS15 (Fig 3B) also significantly inhibited aggresome formation to 0.530±0.068 of control (P<0.01; Fig 3C). No significant difference in aggresome formation was found between PLIC-1 and EPS15 knockdown conditions. These reductions were not an effect of cell death, but were due to an increase in the number of cells with diffuse AUI-Q78 expression (Fig 3D). These results indicate that PLIC-1 and EPS15 regulate aggresome formation.
Figure 3.
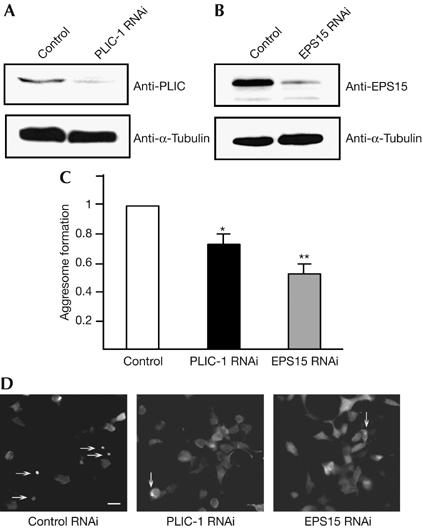
PLIC-1 and epidermal growth factor substrate 15 regulate aggresome formation. Lysates from human embryonic kidney 293 cells transfected with AUI-Q78 and (A) control or PLIC-1 RNAi duplexes or (B) control or EPS15 RNAi duplexes were western blotted with (A) PLIC-1 and (B) EPS15 antibodies. α-Tubulin (A,B) was used as a loading control. (C) Cells transfected as in (A,B) were immunostained with AUI antibodies and 20 random fields of view were quantified for the number of cells containing AUI-Q78 aggresomes. RNAi-transfected cells were normalized to control (arbitrarily assigned a value of 1) for each experiment±s.e.m. (n=3 or more), between-subject analysis of variance was carried out and significant differences from control were found (*P<0.05, **P<0.01) using the Tukey's honestly significant difference post hoc test. (D) Images of cells transfected as in (A,B) and immunostained with AUI antibodies. Fewer cells form aggresomes (arrows) on PLIC-1 and EPS15 knockdown. Also note the increase in cells with diffuse AUI-Q78 expression in these panels. Scale bar, 30 μm. EPS15, epidermal growth factor substrate 15; PLIC-1, protein linking IAP to the cytoskeleton; RNAi, RNA interference.
The UBL domain mediates aggregate transport
To investigate whether the UBL domain—which mediates the EPS15 interaction—is required for aggresome formation, we used a PLIC-1ΔUBL mutant that was compromised in its ability to bind to UIM motifs (Fig 1B), but not to ubiquitinated proteins (supplementary Fig S4 online). When coexpressed with AUI-Q78, PLIC-1ΔUBL severely reduced aggresome formation: γ-tubulin-positive aggresomes formed in less than 1% of co-transfected cells. Instead, we observed numerous AUI-Q78- and Myc–PLIC-1ΔUBL-positive cytoplasmic aggregates (Fig 4A), which did not stain for γ-tubulin (data not shown). When coexpressed with AUI-Q78, Myc–PLIC-1 did not induce this phenotype (Fig 4B), with aggresomes forming in approximately 30% of co-transfected cells. These results indicate that PLIC-1 interacts with protein aggregates before MTOC transport and that the observed Q78-PLIC-1ΔUBL phenotype is a result of aggregate sequestration arising from a block in transport. To confirm that endogenous PLIC-1 recognizes protein aggregates before their MTOC transit, we treated AUI-Q78-transfected cells with nocodazole and then stained for PLIC-1 (Fig 4C). Under these conditions, dispersed AU1-Q78 aggregates formed that co-stained for endogenous PLIC-1.
Figure 4.
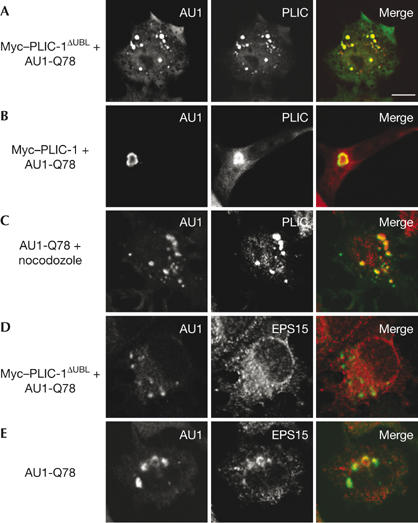
The ubiquitin-like domain of PLIC-1 mediates aggregate transport. (A) Baby hamster kidney cells transfected with Myc–PLIC-1ΔUBL and AUI-Q78 were stained for AU1 and PLIC-1. (B) Myc–PLIC-1- and AUI-Q78-transfected cells were stained for AU1 and PLIC-1. (C) AU1-Q78-transfected cells were treated with nocodazole (1 μM, 2 h) and stained for AU1 and PLIC-1. (D) Myc–PLIC-1ΔUBL- and AUI-Q78-transfected cells were stained for AU1 and EPS15. (E) AU1-Q78-transfected cells were stained for AU1 and EPS15. Scale bar, 10 μm. AU1, epitope tag; EPS15, epidermal growth factor substrate 15; PLIC-1, protein linking IAP to the cytoskeleton; UBL, ubiquitin-like.
To examine whether the Q78-PLIC-1ΔUBL dispersed aggregates resulted from a block in UBL–UIM interactions, we stained transfected cells for EPS15 and found that Q78-PLIC-1ΔUBL aggregates were negative for endogenous EPS15 (Fig 4D), but positive for the proteasome (data not shown). By contrast, we found that EPS15 was normally localized to both AU1-Q78 aggresomes and dispersed aggregates (Fig 4E). These results indicate that PLIC-1ΔUBL blocks both aggregate transport to the MTOC and EPS15 localization to aggregates. Therefore, PLIC-1ΔUBL is a dominant-negative mutant of this process.
PLIC-1 responds to general misfolded protein stress
Our observations have indicated that PLIC-1 recognizes aggregated proteins. This led us to investigate whether PLIC-1 expression is regulated by general cytoplasmic misfolded protein stress, which can be induced by using the heavy-metal poison arsenite (Novoa et al, 2003). To test this, we carried out semiquantitative reverse transcription–PCR (RT–PCR) on RNA isolated from untreated and arsenite-treated cells (Fig 5A); we found that PLIC-1 expression was increased 2.3±0.3-fold by arsenite treatment (Fig 5B). As a positive control, we followed the induction of the C/EBP homologous protein (CHOP), which was potently upregulated by arsenite (Novoa et al, 2003); glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a loading control. These results show that PLIC-1 expression is responsive to the arsenite-induced accumulation of misfolded proteins.
Figure 5.
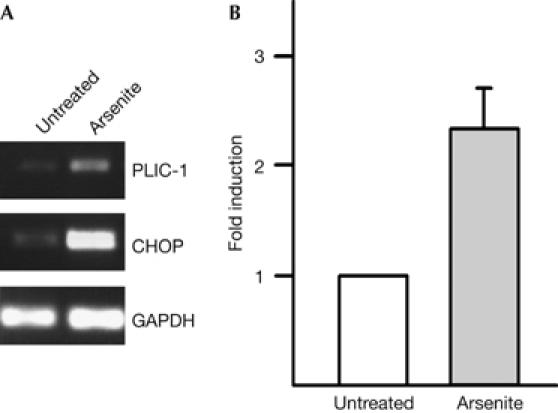
PLIC-1 responds to general misfolded protein stress. (A) Semiquantitative reverse transcription–PCR of RNA from untreated or arsenite-treated (50 μM, 14 h) human hepatoma cells (HepG2) for PLIC-1, CHOP and GAPDH. (B) Fold induction of PLIC-1 (mean±s.e.m., n=3) by arsenite treatment normalized to GAPDH levels. CHOP, C/EBP homologous protein; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; PLIC-1, protein linking IAP to the cytoskeleton.
Discussion
Protein aggregation is a defining characteristic of most neurodegenerative disorders (Ross & Poirier, 2004), and PLIC-1 is genetically associated with one such disease—Alzheimer's disease (Bertram et al, 2005); therefore, our findings are significant to understanding neurodegenerative disease progression. We show that (i) PLIC-1 expression is upregulated in response to an inducer of the general misfolded protein response pathway, (ii) the UBL domain of PLIC-1 binds to UIMs of several proteins and forms complexes in vivo that are sensitive to protein aggregation with the endocytic protein EPS15, (iii) endogenous PLIC-1 and EPS15 are recruited to MTOC-localized polyQ aggresomes, (iv) knockdown of PLIC-1 or EPS15 by RNAi affects aggresome formation, and (v) the loss of the UBL domain of PLIC-1 has a dominant-negative effect on aggregate transport and localization of EPS15 to aggregates. Together, these results indicate that PLIC-1 is a component of the misfolded protein response and that UBL-dependent interactions with EPS15 are important for its function in this pathway.
Identifying this novel function of the UBL domain of PLIC-1 and its binding to UIM proteins other than S5a (Fig 1) suggests that UIM interactions might be a broad property of the UBL domain. Therefore, it will be interesting to examine whether this UBL interacts with other ubiquitin-related domains and also the nature of these interactions. Although an interaction between recombinant EPS15 and PLIC-1 has been reported previously (Regan-Klapisz et al, 2005), here we show that the endogenous proteins interact and that this interaction is enhanced in response to an aggregation-prone protein (Fig 2D–F). The finding that this component of the endocytic machinery can modulate aggresome formation is interesting in the light of a previous report, which demonstrated that the loss of the yeast homologue of EPS15 enhances protein aggregate toxicity (Meriin et al, 2003). Future investigation is needed to examine whether other endocytic components might be required for this role of EPS15.
The observed effects of PLIC-1ΔUBL on aggresome formation (Fig 4) suggest that PLIC-1 sequesters aggregated proteins, when compromised for its UBL-dependent interactions. Simple loss of S5a binding to PLIC-1ΔUBL, leading to impaired proteasomal degradation of AU1-Q78, cannot be reconciled with these results, as pharmacological inhibition of the proteasome promotes aggresome formation (Johnston et al, 1998). Furthermore, the proteasome still binds to PLIC-1ΔUBL (Fig 1C) and is localized to PLIC-1ΔUBL aggregates (data not shown). The link between PLIC-1 and aggregate transport is not unexpected given its ability to recognize ubiquitinated proteins and that misfolded proteins are generally ubiquitinated before degradation (Layfield et al, 2005). However, it remains to be determined precisely how AU1-Q78 aggregates transport to the MTOC and how aggresome formation is blocked.
Altogether, our findings support a model (Fig 6) that proposes a dual function for the UBL domain of PLIC-1 in both proteasomal turnover and transport to the aggresome by competitive UBL–UIM interactions. Under low levels of protein aggregation, PLIC-1 promotes the shuttling of aggregates to the proteasome for their degradation through S5a binding. By contrast, under high levels of protein aggregation, the proteasome becomes overwhelmed and alternative interactions of the UBL domain of PLIC-1, such as with EPS15, are enhanced, promoting the deposition of protein aggregates at the aggresome. Recent data suggest that efficient autophagic removal of the aggresome would follow, reducing cellular toxicity (Iwata et al, 2005).
Figure 6.
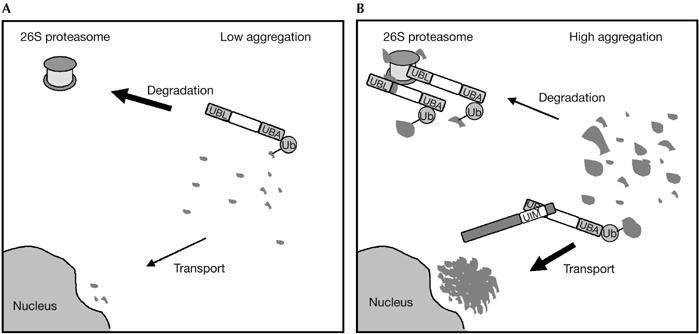
Model for the role of PLIC-1 aggresome formation. (A) Under low levels of protein aggregation, the UBL domain of PLIC-1 shuttles ubiquitinated misfolded proteins to the proteasome through the UIM of S5a. (B) Under high levels, larger ubiquitinated aggregates overwhelm the proteasome, leading to the UBL domain being available for binding to other UIM proteins, which promote aggregate transport to the perinuclear aggresome. PLIC-1, protein linking IAP to the cytoskeleton; Ub, ubiquitin; UBA, ubiquitin-associated domain; UBL, ubiquitin-like; UIM, ubiquitin-interacting motif.
In summary, accumulating evidence (Bertram et al, 2005; Wang et al, 2006), including the findings presented in this report, indicate that PLIC-1 is an important component of the quality control machinery that controls protein aggregation, and prevents cell death and disease.
Methods
Cell culture and treatments. Baby hamster kidney (BHK) cells were cultured in DMEM/F12 (Invitrogen, Carlsbad, CA, USA), 5% CO2 supplemented with 5% FBS, 100 U/ml penicillin/streptomycin and 2 mM L-glutamine, whereas human embryonic kidney 293 (HEK293) cells and human hepatoma (HepG2) cells were cultured in DMEM. Transfections were carried out with Lipofectamine 2000 (Invitrogen). To induce misfolded protein stress, HepG2 cells were treated with 50 μM arsenite or DMSO for 14 h. For microtubule destabilization, cells were incubated with nocodazole (1 μM) for 2 h.
Antibodies and plasmids. For details, see the supplementary information online.
Immunofluorescence. BHK cells plated on poly-L-lysine-coated coverslips were processed for immunofluorescence as described by Bedford et al (2001), except for γ-tubulin staining, for which cells were fixed in 1:1 methanol:acetone. Images were acquired on a Zeiss LSM410 confocal microscope.
Immunoprecipitation. Cells were lysed (20 mM Tris–HCl, pH 7.6, 50 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% SDS, plus phosphatase and protease inhibitors) at 4°C for 1 h. Nuclei and insoluble material were removed by centrifugation. Antigen–antibody complexes were immunoprecipitated with 2 μg anti-PLIC or rabbit IgG and Protein-A–Sepharose (Amersham, UK) at 4°C for 2 h. Beads were washed three times in lysis buffer and analysed by western blot as described previously (Bedford et al, 2001).
Glutathione S-transferase affinity chromatography. Transfected cells were lysed (20 mM Hepes, pH 7.4, 50 mM NaCl, 10% glycerol, 0.5% Triton X-100 plus protease inhibitors) at 4°C for 1 h. Nuclei and insoluble material were removed by centrifugation. GST fusion proteins on glutathione–agarose (Sigma, St Louis, MO, USA), prepared as described previously (Bedford et al, 2001), were incubated with the lysates for 1 h, washed three times with buffer and analysed by western blot.
Semiquantitative reverse transcription–PCR. Total RNA was isolated using the TRIZOL reagent (Invitrogen). RT–PCR was carried out using 1 μg of complementary DNA. The PCR cycle number for each primer set was chosen on the basis of a preliminary study determining the linear range of amplification for each molecule. For PCR primers and further details, see the supplementary information online.
RNA interference. At 72 h and 24 h before fixation, HEK293 cells were transfected with 20 nM EPS15 (Huang et al, 2004), PLIC-1 (Massey et al, 2005) or scrambled short interfering RNA duplexes (Dharmacon, Lafayette, CO, USA) using HiPerFect (Qiagen, Hilden, Germany). In addition, at 24 h, cells were also transfected with 2.5 μg pAUI-Q78 using Lipofectamine 2000 (Invitrogen). Cells were then processed for immunofluorescence as described above, and the number of cells with aggresomes was quantified.
Supplementary information is available at EMBO reports online (http://www.emboreports.org).
Supplementary Material
supplementary Information
supplementary Figures
supplementary Movie 1
supplementary Movie 2
Acknowledgments
We thank M. Cheetham, E. Fon, P. McPherson and G. Rouleau for constructs. This work was supported by the Canadian Institute of Research (CIHR). R.H. is a recipient of a CIHR Canada Graduate Scholarship Master's Award, and F.B. is a CIHR-Canada Research Chair—Tier II.
References
- Bedford FK et al. (2001) GABAA receptor cell surface number and subunit stability are regulated by the ubiquitin-like protein PLIC-1. Nat Neurosci 4: 908–916 [DOI] [PubMed] [Google Scholar]
- Bertram L et al. (2005) Family-based association between Alzheimer's disease and variants in UBQLN1. N Engl J Med 352: 884–894 [DOI] [PubMed] [Google Scholar]
- Biggins S, Ivanovska I, Rose MD (1996) Yeast ubiquitin-like genes are involved in duplication of the microtubule organizing center. J Cell Biol 133: 1331–1346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnett BG, Pittman RN (2005) The polyglutamine neurodegenerative protein ataxin 3 regulates aggresome formation. Proc Natl Acad Sci USA 102: 4330–4335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Shinde U, Ortolan TG, Madura K (2001) Ubiquitin-associated (UBA) domains in Rad23 bind ubiquitin and promote inhibition of multi-ubiquitin chain assembly. EMBO Rep 2: 933–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Fiore PP, Polo S, Hofmann K (2003) When ubiquitin meets ubiquitin receptors: a signalling connection. Nat Rev Mol Cell Biol 4: 491–497 [DOI] [PubMed] [Google Scholar]
- Fisher RD, Wang B, Alam SL, Higginson DS, Robinson H, Sundquist WI, Hill CP (2003) Structure and ubiquitin binding of the ubiquitin-interacting motif. J Biol Chem 278: 28976–28984 [DOI] [PubMed] [Google Scholar]
- Hartmann-Petersen R, Gordon C (2004) Integral UBL domain proteins: a family of proteasome interacting proteins. Semin Cell Dev Biol 15: 247–259 [DOI] [PubMed] [Google Scholar]
- Hofmann K, Falquet L (2001) A ubiquitin-interacting motif conserved in components of the proteasomal and lysosomal protein degradation systems. Trends Biochem Sci 26: 347–350 [DOI] [PubMed] [Google Scholar]
- Huang F, Khvorova A, Marshall W, Sorkin A (2004) Analysis of clathrin-mediated endocytosis of epidermal growth factor receptor by RNA interference. J Biol Chem 279: 16657–16661 [DOI] [PubMed] [Google Scholar]
- Iwata A, Riley BE, Johnston JA, Kopito RR (2005) HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J Biol Chem 280: 40282–40292 [DOI] [PubMed] [Google Scholar]
- Johnston JA, Ward CL, Kopito RR (1998) Aggresomes: a cellular response to misfolded proteins. J Cell Biol 143: 1883–1898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleijnen MF, Shih AH, Zhou P, Kumar S, Soccio RE, Kedersha NL, Gill G, Howley PM (2000) The hPLIC proteins may provide a link between the ubiquitination machinery and the proteasome. Mol Cell 6: 409–419 [DOI] [PubMed] [Google Scholar]
- Kleijnen MF, Alarcon RM, Howley PM (2003) The ubiquitin-associated domain of hPLIC-2 interacts with the proteasome. Mol Biol Cell 14: 3868–3875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layfield R, Lowe J, Bedford L (2005) The ubiquitin–proteasome system and neurodegenerative disorders. Essays Biochem 41: 157–171 [DOI] [PubMed] [Google Scholar]
- Mah AL, Perry G, Smith MA, Monteiro MJ (2000) Identification of ubiquilin, a novel presenilin interactor that increases presenilin protein accumulation. J Cell Biol 151: 847–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massey LK, Mah AL, Monteiro MJ (2005) Ubiquilin regulates presenilin endoproteolysis and modulates γ-secretase components, Pen-2 and nicastrin. Biochem J 391: 513–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meriin AB et al. (2003) Aggregation of expanded polyglutamine domain in yeast leads to defects in endocytosis. Mol Cell Biol 23: 7554–7565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novoa I, Zhang Y, Zeng H, Jungreis R, Harding HP, Ron D (2003) Stress-induced gene expression requires programmed recovery from translational repression. EMBO J 22: 1180–1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickart CM (2001) Mechanisms underlying ubiquitination. Annu Rev Biochem 70: 503–533 [DOI] [PubMed] [Google Scholar]
- Polo S, Sigismund S, Faretta M, Guidi M, Capua MR, Bossi G, Chen H, De Camilli P, Di Fiore PP (2002) A single motif responsible for ubiquitin recognition and monoubiquitination in endocytic proteins. Nature 416: 451–455 [DOI] [PubMed] [Google Scholar]
- Regan-Klapisz E et al. (2005) Ubiquilin recruits Eps15 into ubiquitin-rich cytoplasmic aggregates via a UIM–UBL interaction. J Cell Sci 118: 4437–4450 [DOI] [PubMed] [Google Scholar]
- Ross CA, Poirier MA (2004) Protein aggregation and neurodegenerative disease. Nat Med 10: S10–S17 [DOI] [PubMed] [Google Scholar]
- Toulouse A, Au-Yeung F, Gaspar C, Roussel J, Dion P, Rouleau GA (2005) Ribosomal frameshifting on MJD-1 transcripts with long CAG tracts. Hum Mol Genet 14: 2649–2660 [DOI] [PubMed] [Google Scholar]
- Walters KJ, Kleijnen MF, Goh AM, Wagner G, Howley PM (2002) Structural studies of the interaction between ubiquitin family proteins and proteasome subunit S5a. Biochemistry 41: 1767–1777 [DOI] [PubMed] [Google Scholar]
- Wang H, Lim PJ, Yin C, Rieckher M, Vogel BE, Monteiro MJ (2006) Suppression of polyglutamine-induced toxicity in cell and animal models of Huntington's disease by ubiquilin. Hum Mol Genet 15: 1025–1041 [DOI] [PubMed] [Google Scholar]
- Westhoff B, Chapple JP, van der Spuy J, Hohfeld J, Cheetham ME (2005) HSJ1 is a neuronal shuttling factor for the sorting of chaperone clients to the proteasome. Curr Biol 15: 1058–1064 [DOI] [PubMed] [Google Scholar]
- Wu AL, Wang J, Zheleznyak A, Brown EJ (1999) Ubiquitin-related proteins regulate interaction of vimentin intermediate filaments with the plasma membrane. Mol Cell 4: 619–625 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
supplementary Information
supplementary Figures
supplementary Movie 1
supplementary Movie 2