


The ESF–Wellcome Trust Conference meeting on Signalling to Chromatin Epigenetics took place between 5 and 9 June 2006 in Hinxton, UK, and was organized by P. Varga-Weisz, M. Turner, N. Divecha and A. Rao.
Introduction
In June 2006, about 90 scientists came together at the Wellcome Trust Conference Centre in Hinxton (near Cambridge, UK), to attend the first European Science Foundation (ESF)–Wellcome Trust Conference on Signalling to Chromatin Epigenetics. This meeting addressed how cellular processes both signal and respond to chromatin. Chromatin is composed of nucleosomes that each comprise 147 base pairs of DNA wrapped around an octamer of two copies of each histone H2A, H2B, H3 and H4. Nucleosomes are folded into higher-order structures that are stabilized by linker histones. Chromatin structure can be altered by enzymes that post-translationally modify histones (for example, through phosphorylation, acetylation, methylation or ubiquitination) or by ATP-driven chromatin-remodelling complexes that alter nucleosome position and/or composition. The role of chromatin in cellular processes, such as transcription, DNA-damage repair and cell differentiation, was discussed at the meeting. In this report, we summarize a few of the highlights from the broad range of topics discussed.
Phosphorylation switching
Histone modifications have an important role in the regulation of gene transcription. L. Mahadevan (Oxford, UK) and co-workers have studied how a particular set of histone modifications controls the rapid induction of the immediate-early genes c-jun and c-fos. Mahadevan showed that on induction by extracellular signals, the extracellular-signal-regulated kinase (ERK) and the mitogen- and stress-activated protein kinase 1/2 (MSK1/2) pathways direct a dynamic turnover of acetylation and phosphorylation marks on histone H3 at these genes (Hazzalin & Mahadevan, 2005). Sequential chromatin immunoprecipitation (ChIP) assays showed that acetylated K9-phosphorylated S10 (H3K9acS10ph) and trimethylated K4 (H3K4me3) forms of histone H3 co-exist on the same nucleosomes at the promoter and at the 5′-coding sequences of these genes when they are transcriptionally active. Intriguingly, H3K9acS10ph is only transiently observed on gene induction, whereas H3K4me3 is present before induction. Mahadevan suggested that H3K4me3 might assist in creating a poised state by favouring rapid H3K9acS10ph on induction. Interestingly, the inhibition of histone deacetylases impairs the induction of c-jun and c-fos. It therefore seems that the rapid turnover of both the acetylation and phosphorylation marks is crucial for proper induction of these transiently expressed genes.
Chromatin undergoes extensive changes throughout B-cell development. During the terminal differentiation of B cells into plasma cells, genome-wide chromatin condensation is observed. P. Sabbatini (London, UK) reported that plasma-cell heterochromatin is enriched in phosphorylated H3S10, which is normally associated with active promoters or mitotic chromosomes. Interestingly, this mark is flanked by H3K9me3, which is a well-known feature of heterochromatin. Previous studies have shown that H3K9me3 allows the association of heterochromatin protein 1 (HP1) with chromatin, but that concomitant H3S10ph abrogates this interaction (reviewed by Hediger & Gasser, 2006). Sabbatini found that aurora B kinase, which is responsible for H3S10ph in mitosis, is expressed during terminal plasma-cell differentiation when HP1 becomes depleted from heterochromatin. Inhibition of aurora B by hesperadin results in the loss of H3S10ph from plasma-cell heterochromatin, and the retention of HP1. These findings suggest that aurora B creates a ‘phospho-switch' that converts chromatin in B cells to a specialized heterochromatin during terminal plasma-cell differentiation. The nature of this specialized heterochromatin is unclear, and it remains to be elucidated whether other heterochromatin factors bind H3K9me3S10ph in terminally differentiated cells.
Enzymes that modulate post-translational modifications might themselves be post-translationally modified. A. Tarakhovsky (New York, NY, USA) showed that the histone methyltransferase G9a is one such enzyme. G9a recognizes the ARKS motifs that are present around Lys 9 and Lys 27 in the histone H3 tail. Interestingly, G9a itself contains the similar ARKT motif. Tarakhovsky showed that K164 in the ARKT motif is methylated by G9a, and that methylated G9aK164, similar to methylated H3K9, is a binding site for HP1γ in vitro. By analogy with K9/S10 on histone H3, T165 in the G9a ARKT motif might be a target site of the aurora B kinase. Indeed, Tarakhovsky found that aurora B phosphorylates T165 in vitro and that phosphorylation of T165 disrupts HP1 binding to methylated G9a. The auto-methylation of G9a does not affect its histone methyltransferase activity. However, methylation and phosphorylation of G9a are suspected to be important for regulating its targeting to chromatin in vivo.
Histone demethylation
Post-translational histone modifications, such as phosphorylation and acetylation, are reversible. By contrast, lysine methylation of histones was long thought to be irreversible, until Shi and co-workers discovered the lysine-specific demethylase 1 (LSD1) enzyme (Shi et al, 2004). LSD1 is a nuclear amine oxidase homologue that demethylates monomethylated and dimethylated H3K4 through a FAD-dependent oxidation reaction that produces formaldehyde.
The jumonji C (JmjC)-domain-containing histone demethylase 1 (JHDM1) has also been identified recently (Tsukada et al, 2006). JHDM1 preferentially demethylates H3K36me2 through an oxidation reaction that uses Fe(II) and α-ketoglutarate. Hence, this reaction chemically differs from that catalysed by LSD1. Importantly, both LSD1 and JHDM1 were found to act only on monomethylated or dimethylated lysine residues. This raises the question of whether trimethylation is an irreversible chromatin mark or whether an as-yet-unidentified enzyme can reverse trimethylation. Y. Shi (Boston, MA, USA) and P. Cloos (Copenhagen, Denmark) reported the identification of JmjC-domain-containing members of the JMJD2 family that efficiently reverse trimethylation (Cloos et al, 2006; Whetstine et al, 2006). Shi showed that recombinant JMJD2A specifically reversed H3K9me3 or H3K36me3 to dimethyl forms on free histones, whereas Cloos found that JMJD2C/GASC1 (gene amplified in squamous cell carcinoma) demethylates H3K9me3 and H3K9me2 to monomethyl forms on nucleosomes. Further evidence of site and methyl-group specificity by JMJD2 family enzymes was given by Shi, who found that JMJD2A–JMJD2D all act on H3K9me3. By contrast, only JMJD2D acts on H3K9me2, and only JMJD2A and JMJD2C/GASC1 act on H3K36me3. Both groups found that overexpression of either JMJD2A or JMJD2C/GASC1 reduces bulk H3K9me3/H3K36me3 levels in cells. Cloos also observed delocalized HP1 on JMJD2C/GASC1 overexpression, which resulted in the loss of bulk heterochromatin in vivo.
Signalling from damaged chromatin
It is evident that chromatin has a crucial role in signalling the cellular response to DNA damage. Phosphorylation of the histone variant H2AX occurs rapidly in chromatin that flanks DNA double-strand breaks (DSBs; Fernandez-Capetillo et al, 2004). Ataxia telangiectasia mutated (ATM), ATM and Rad3-related (ATR) and DNA-dependent protein kinase catalytic subunit (DNA-PKcs) are responsible for this phosphorylation, which occurs on the conserved Ser 139 residue at the carboxyl terminus of H2AX. Loss of H2AX results in radiosensitivity, genomic instability and defects in DNA repair. It has long been suspected that phosphorylated H2AX (referred to as γ-H2AX) recruits factors that function in the repair of DNA damage and/or activation of the DNA-damage checkpoint (which delays cell-cycle progression until repair is complete) to sites of DNA damage. S. Jackson (Cambridge, UK) presented results that provide more insight into the function of γ-H2AX in the cellular response to DNA damage. To identify γ-H2AX-interacting proteins, Jackson used a phospho-peptide comprising the phosphorylated H2AX C terminus to pull down mediator of DNA-damage checkpoint 1 (MDC1) and the MRE11, RAD50 and NBS1 (MRN) complex from cellular extracts (Stucki et al, 2005). MDC1 contains a C-terminal BRCT domain, which seems to be essential for interaction with the H2AX phosphopeptide. MDC1 colocalizes with γ-H2AX ionizing-radiation-induced foci (IRIF). Mutation of the MDC1 BRCT domain (S1933Q) or expression of the H2AX phospho-acceptor mutant protein (H2AX–S136/139A) impaired the targeting of MDC1 to IRIF. Furthermore, mutation of the BRCT domain also impaired recruitment of the DNA damage-response factors NBS1, 53BP1 and phosphorylated ATM to IRIF. Jackson proposed a model in which the initial H2AX phosphorylation results from the MRN-mediated recruitment of ATM to DSBs (Fig 1). MDC1 binds to γ-H2AX, recruits more MRN–ATM complex and the new pool of ATM then starts to phosphorylate more H2AX. This cascade of reactions could propagate H2AX phosphorylation over large distances and might be responsible for the maintenance of γ-H2AX IRIF. Consistent with this, downregulation of MDC1 did not abolish γ-H2AX IRIF formation, but impaired its extent and maintenance over time. Finally, Jackson showed that γ-H2AX spreads for 1 Mb on average from critically short telomeres in senescent cells. His group is examining why and how γ-H2AX spreads from telomeres in these cells.
Figure 1.
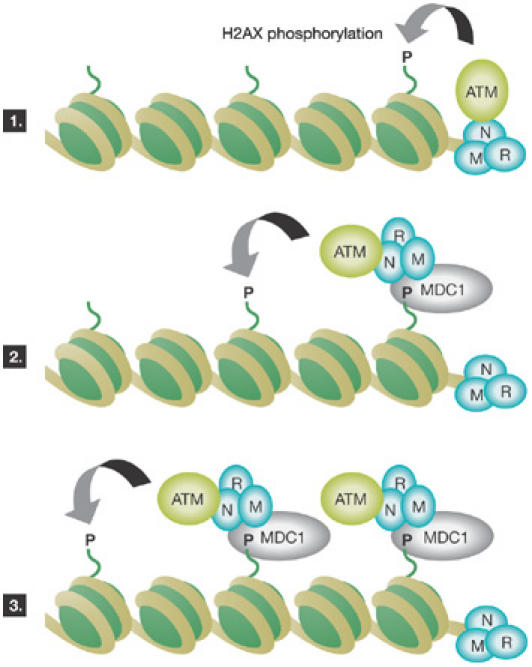
Model of MDC1-regulated phosphorylation of histone H2AX. (1) The MRE11, RAD50 and NBS1 (MRN) complex binds DNA ends at sites of DNA damage and recruits ataxia telangiectasia mutated (ATM), which phosphorylates proximal H2AX. (2,3) Mediator of DNA damage checkpoint 1 (MDC1) binds phosphorylated proximal H2AX and recruits more MRN–ATM. The new pool of ATM phosphorylates more distal H2AX. These events could contribute to the ‘spreading' of H2AX phosphorylation to more distal chromatin regions.
Epigenetic memory
Memory of the transcriptional status of genes is crucial for maintaining differentiated states long after differentiation signals have been removed. The Oct3/4 gene encodes a POU domain homeobox transcription factor that is expressed during gametogenesis and in early embryonic cells to maintain totipotency. Oct3/4 is rapidly repressed on implantation, which is a process that can be replicated by the addition of retinoic acid to the growth medium of embryonic stem cells. Y. Bergman (Jerusalem, Israel) described the multistep programme of Oct3/4 inactivation. Initial transcriptional repression is mediated by transiently expressed trans-acting repressors, and is quickly followed by H3K9me of the promoter by the G9a histone methyltransferase and binding of HP1β (Feldman et al, 2006). The promoter subsequently becomes de novo DNA methylated by Dnmt3a/3b. Surprisingly, G9a is required for both H3K9me and DNA methylation of Oct3/4. Bergman found that G9a interacts directly with Dnmt3a/3b to recruit them to the Oct3/4 promoter. This interaction occurs even in histone-methylation-defective SET domain mutants of G9a. Bergman also found that although Oct3/4 is repressed after the retinoic-acid-induced differentiation of G9a-null embryonic stem cells, it quickly reactivates after the removal of the differentiation signal. Interestingly, G9a mutants that are defective in only H3K9 methylation activity or Dnmt3a/3b recruitment are able to prevent Oct3/4 re-expression in the absence of transcriptional repressors. Permanent silencing, however, can only be attained through de novo methylation. Thus, DNA methylation is not only necessary but also sufficient to prevent Oct3/4 reprogramming.
The role of particular histone modifications in the epigenetic maintenance of transcriptional states is a subject of keen debate; however, studies in differentiating primary cells are limited by the large number of cells required for ChIP assays. B. Turner (Birmingham, UK) described a modification of the native ChIP assay that allows analysis of histone modifications at specific target sequences from as few as 50 cells (O'Neill et al, 2006). The method, which is called carrier-ChIP (CChIP), involves mixing a small number of cells of interest with an excess of cells from another species. This allows the preparation of sufficient quantities of chromatin for a standard native ChIP. Species-specific primers are used in subsequent quantitative polymerase chain reactions to assay for the enrichment of a specific target. Turner showed that CChIP can be used to analyse histone modifications at the promoters of Oct3/4, Nanog and Cdx2 in the trophectoderm and inner cell mass dissected from mouse blastocysts.
Turnover of signalling transcription factors
The levels of many transcription factors are carefully controlled in response to the environment of a cell. The recent findings that ubiquitin ligases and proteasome subcomponents are present at many promoters indicate that several transcription factors might be targeted for degradation at the site of their activity. J. Boyes (Leeds, UK) described the complex series of post-translational modifications that regulate the activity and turnover of the GATA-1 transcription factor. The acetylation of GATA-1 potentiates its DNA-binding and transcriptional activity, which is essential for terminal erythroid differentiation. However, acetylation also targets GATA-1 for ubiquitination and degradation. In addition, Boyes found that a further seven sites on GATA-1 must be phosphorylated for ubiquitination to occur. Interestingly, phosphorylation of two mitogen-activated protein kinase target sites only occurs if GATA-1 is able to bind DNA. Boyes suggested that only the active, promoter-bound form of GATA-1 is subject to rapid degradation. Consistent with this, GATA-1 ubiquitination was enhanced in response to stem-cell factor and erythropoietin signalling.
Interchromosomal diplomacy
The question of whether genes that are transcribed by RNA polymerase II (RNAPII) recruit the transcriptional machinery or are themselves recruited to pre-assembled RNAPII complexes has been debated for many years. Recent observations from P. Fraser's group (Cambridge, UK) have done much to support the latter theory. They have found that cis-linked genes often colocalize to common RNAPII-rich ‘transcription factories' when they are expressed (Osborne et al, 2004). The frequency with which genes localize to RNAPII foci is directly proportional to their frequency of expression in a given population of cells. Fraser reported that co-expressed genes in trans colocalize with a frequency higher than expected and are readily detected with the 3C chromosome-interaction assay (Dekker et al, 2002). For example, the c-myc locus on chromosome 8 localizes at the same transcription factory as the IgH locus on chromosome 14 in 25% of B cells. Fraser suggested that this colocalization might favour illegitimate recombination between the IgH and c-myc loci, resulting in the translocations that are common in Burkitt's lymphoma. Fraser also looked at the stability of transcription factories and their interactions with genes after the inhibition of transcription. The RNAPII foci persisted in the absence of transcription, indicating that they might exist by themselves, rather than being established on pre-assembled active genes. The well-characterized interactions between the β-globin locus-control region (LCR) and promoters (dubbed the ‘active chromatin hub'; ACH) in erythrocytes were also found to remain intact despite delocalization from a transcription factory after heat shock. Fraser suggested that ACH formation might not be sufficient to form a transcription factory.
Further evidence for interchromosomal interactions between the cytokine gene loci in T cells was provided by R. Flavell (New Haven, CT, USA). Naive T cells differentiate into T-helper 1 (Th1) or Th2 effector cells after antigenic stimulation. Previous studies have found that Th1-specific and Th2-specific cytokines are rapidly expressed on antigenic stimulation, indicating that they might be poised for transcription. Using the 3C assay, Flavell reported that regulatory elements from the Th1, Th2 and interferon-γ (Ifnγ) gene loci from mouse chromosomes 17, 11 and 10, respectively, were in contact with one another in naive T cells (Fig 2). The colocalization of all three cytokine loci was observed in 20% of naive T cells by fluorescence in situ hybridization (FISH). Interestingly, these interactions are mono-allelic and are lost on T-cell differentiation (Spilianakis et al, 2005). When the Ifnγ and Th2 cytokine genes are poised for expression in naive T cells, the Ifnγ promoter interacts with the Rad50-hypersensitive site 6 (RHS6) element from the Th2 LCR (Fig 2). Colocalization of the Ifnγ and Rad50 loci was observed in 40% of naive T cells by using FISH (Fig 3). Deletion of RHS7 not only abrogates this interaction, but also delays and reduces Ifnγ expression, indicating that the Ifnγ gene on chromosome 10 uses the Rad50 LCR on chromosome 11. Flavell found that the interchromosomal interactions are not co-dependent or competitive, as the lymphotoxin/tumour necrosis factor-α (Lt/Tnfα) gene cluster on chromosome 17 can be deleted without affecting Th2–Ifnγ interactions. Interchromosomal regulation might allow a single site of control or negotiation, where the cytokine loci respond to stimuli to establish either a Th1 or Th2 response.
Figure 2.
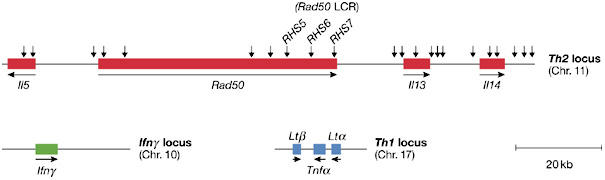
Spatial organization of cytokine loci. Schematic representation of the murine Th2 (Il3/Rad50/Il13/Il4), interferon-γ (Ifnγ) and Th1 (Ltβ/Tnfα/Ltα) loci used in 3C chromosome-interaction analyses by Flavell and co-workers (Spilianakis et al, 2005). Vertical arrows on the top represent the DNaseI-hypersensitive sites so far characterized at the Th2 locus.
Figure 3.
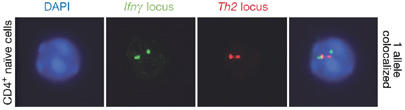
Interchromosomal interaction between the interferon-γ and Th2 loci revealed by fluorescence in situ hybridization. CD4+ naive T cells were hybridized with a SpectrumGreen-dUTP-labelled probe for the interferon-γ (Ifnγ) locus and a rhodamine–dUTP-labelled probe for the Th2 locus. 4′,6-diamidino-2-phenylindole (DAPI) staining of nuclei shows the presence of DNA. Figure courtesy of R. Flavell and co-workers (Spilianakis et al, 2005).
Concluding remarks
The first ESF–Wellcome Trust Conference on Signalling to Chromatin Epigenetics brought together researchers from several fields to discuss how cells signal to and from chromatin. A key finding presented at the meeting was that the rapid turnover of histone modifications, such as phosphorylation and acetylation, can be important for transcription and cell differentiation. The identification of a growing family of histone demethylases indicates that histone methylation, which is often considered to be a stable mark, can also be dynamic and subject to turnover. These findings highlight a technical challenge that we all face: to accurately decipher the histone code, not only must we look in the right ‘place', we must also look at the right time. Another key finding was that interchromosomal interactions might be a common feature of co-regulated genes, particularly those with expression levels on different chromosomes that must be coordinated or reciprocated in response to common signals. If such interchromosomal sharing of regulatory elements occurs frequently in the genome, we need to consider the consequences for illegitimate crosstalk. This meeting highlighted how common epigenetic mechanisms can emerge from the comparison of the many diverse processes that signal to and from chromatin.

Adam G. West

Haico van Attikum
Acknowledgments
H.v.A. is supported by fellowships from the European Molecular Biology Organization (EMBO) and the Human Frontier Science Program (HFSP). Research in the A.G.W. group is supported by the UK Biotechnology and Biological Sciences Research Council (BBSRC), the European Commission Marie Curie Actions, the John Robertson Bequest, the UK Medical Research Council (MRC) and the Association for International Cancer Research (AICR). We thank the speakers for allowing us to discuss their presentations and apologize to those whose work could not be included due to space limitations.
References
- Cloos PA, Christensen J, Agger K, Maiolica A, Rappsilber J, Antal T, Hansen KH, Helin K (2006) The putative oncogene GASC1 demethylates tri- and dimethylated lysine 9 on histone H3. Nature 442: 307–311 [DOI] [PubMed] [Google Scholar]
- Dekker J, Rippe K, Dekker M, Kleckner N (2002) Capturing chromosome conformation. Science 295: 1306–1311 [DOI] [PubMed] [Google Scholar]
- Feldman N, Gerson A, Fang J, Li E, Zhang Y, Shinkai Y, Cedar H, Bergman Y (2006) G9a-mediated irreversible epigenetic inactivation of Oct-3/4 during early embryogenesis. Nat Cell Biol 8: 188–194 [DOI] [PubMed] [Google Scholar]
- Fernandez-Capetillo O, Lee A, Nussenzweig M, Nussenzweig A (2004) H2AX: the histone guardian of the genome. DNA Repair (Amst) 3: 959–967 [DOI] [PubMed] [Google Scholar]
- Hazzalin CA, Mahadevan LC (2005) Dynamic acetylation of all lysine 4-methylated histone H3 in the mouse nucleus: analysis at c-fos and c-jun. PLoS Biol 3: e393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hediger F, Gasser SM (2006) Heterochromatin protein 1: don't judge the book by its cover! Curr Opin Genet Dev 16: 143–150 [DOI] [PubMed] [Google Scholar]
- O'Neill LP, Vermilyea MD, Turner BM (2006) Epigenetic characterization of the early embryo with a chromatin immunoprecipitation protocol applicable to small cell populations. Nat Genet 38: 835–841 [DOI] [PubMed] [Google Scholar]
- Osborne CS et al. (2004) Active genes dynamically colocalize to shared sites of ongoing transcription. Nat Genet 36: 1065–1071 [DOI] [PubMed] [Google Scholar]
- Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y (2004) Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119: 941–953 [DOI] [PubMed] [Google Scholar]
- Spilianakis CG, Lalioti MD, Town T, Lee GR, Flavell RA (2005) Interchromosomal associations between alternatively expressed loci. Nature 435: 637–645 [DOI] [PubMed] [Google Scholar]
- Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP (2005) MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 123: 1213–1226 [DOI] [PubMed] [Google Scholar]
- Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, Zhang Y (2006) Histone demethylation by a family of JmjC domain-containing proteins. Nature 439: 811–816 [DOI] [PubMed] [Google Scholar]
- Whetstine JR et al. (2006) Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell 125: 467–481 [DOI] [PubMed] [Google Scholar]