

Abstract
Background
Pendred syndrome, an autosomal-recessive disorder characterized by deafness and goiter, is caused by a mutation of SLC26A4, which codes for the anion exchanger pendrin. We investigated the relationship between pendrin expression and deafness using mice that have (Slc26a4+/+ or Slc26a4+/-) or lack (Slc26a4-/-) a complete Slc26a4 gene. Previously, we reported that stria vascularis of adult Slc26a4-/- mice is hyperpigmented and that marginal cells appear disorganized. Here we determine the time course of hyperpigmentation and marginal cell disorganization, and test the hypothesis that inflammation contributes to this tissue degeneration.
Methods
Slc26a4-/- and age-matched control (Slc26a4+/+ or Slc26a4+/-) mice were studied at four postnatal (P) developmental stages: before and after the age that marks the onset of hearing (P10 and P15, respectively), after weaning (P28-41) and adult (P74-170). Degeneration and hyperpigmentation stria vascularis was evaluated by confocal microscopy. Gene expression in stria vascularis was analyzed by microarray and quantitative RT-PCR. In addition, the expression of a select group of genes was quantified in spiral ligament, spleen and liver to evaluate whether expression changes seen in stria vascularis are specific for stria vascularis or systemic in nature.
Results
Degeneration of stria vascularis defined as hyperpigmentation and marginal cells disorganization was not seen at P10 or P15, but occurred after weaning and was associated with staining for CD68, a marker for macrophages. Marginal cells in Slc26a4-/-, however, had a larger apical surface area at P10 and P15. No difference in the expression of Lyzs, C3 and Cd45 was found in stria vascularis of P15 Slc26a4+/- and Slc26a4-/- mice. However, differences in expression were found after weaning and in adult mice. No difference in the expression of markers for acute inflammation, including Il1a, Il6, Il12a, Nos2 and Nos3 were found at P15, after weaning or in adults. The expression of macrophage markers including Ptprc (= Cd45), Cd68, Cd83, Lyzs, Lgals3 (= Mac2 antigen), Msr2, Cathepsins B, S, and K (Ctsb, Ctss, Ctsk) and complement components C1r, C3 and C4 was significantly increased in stria vascularis of adult Slc26a4-/- mice compared to Slc26a4+/+ mice. Expression of macrophage markers Cd45 and Cd84 and complement components C1r and C3 was increased in stria vascularis but not in spiral ligament, liver or spleen of Slc26a4-/- compared to Slc26a4+/- mice. The expression of Lyzs was increased in stria vascularis and spiral ligament but not in liver or spleen.
Conclusion
The data demonstrate that hyperpigmentation of stria vascularis and marginal cell reorganization in Slc26a4-/- mice occur after weaning, coinciding with an invasion of macrophages. The data suggest that macrophage invasion contributes to tissue degeneration in stria vascularis, and that macrophage invasion is restricted to stria vascularis and is not systemic in nature. The delayed onset of degeneration of stria vascularis suggests that a window of opportunity exists to restore/preserve hearing in mice and therefore possibly in humans suffering from Pendred syndrome.
Background
Pendred syndrome is an autosomal recessive disorder that is characterized by profound sensorineural deafness, abnormal iodide transport across the thyroid follicular epithelium and an enlarged vestibular aqueduct [1,2]. It is an important condition as it accounts for 1–10% of all cases of hereditary deafness [3]. Pendred syndrome is caused by mutations of the gene SLC26A4, which codes for the protein pendrin [4]. Hearing loss in Pendred syndrome develops in most cases prelingually, which implies that pendrin is not essential for hearing but that a defective pendrin protein causes hearing loss via a secondary mechanism [3,5]. Most Pendred syndrome patients are euthyroid, although the abnormal iodide transport in the thyroid affects the incorporation of iodide into thyroglobulin [6,7]. It is conceivable that the observed thyroid hyperplasia (goiter), which generally develops around puberty, ensures normal levels of thyroid hormone [2].
Pendrin is a Na+-independent exchanger for anions such as Cl-, I-, HCO3 - and formate [8-10]. Pendrin is expressed in the inner ear, thyroid, kidney, mammary gland, uterus, testes, and placenta [6,11-17]. In the thyroid, pendrin is expressed on the apical side of the thyrocytes and mediates Cl-/I- exchange. In the kidney, pendrin is expressed on the apical side of the non-A, non-B intercalated cells, cytoplasmic regions of the type B intercalated cells of cortical collecting tubules, distal convoluted tubules and connecting tubules and mediates Cl-/HCO3 - exchange [14,18]. Loss of pendrin does not effect the arterial pH but results in a lower urinary pH [19]. In the inner ear, pendrin is localized in the outer sulcus epithelial cells, root cells, apical membranes of spiral prominence surface epithelial cells and in apical membranes of spindle-shaped cells of stria vascularis [11,17].
A model for Pendred syndrome, consisting of mice lacking functional expression of pendrin, has recently been developed [20]. Similar to patients suffering from Pendred syndrome, Slc26a4-/- mice are deaf, have an enlarged vestibular aqueduct and appear to be euthyroid. Mice, in contrast to human patients, do not exhibit goiter. Adult Slc26a4-/- mice do not generate an endocochlear potential, which is generated by stria vascularis and is necessary for normal hearing [15,17].
We have shown that adult Slc26a4-/- mice show signs of degeneration of stria vascularis, including hyperpigmentation and marginal disorganization [15,17]. It remains unclear, however, whether hyperpigmentation and marginal cell disorganization occurred before or after the normal onset of hearing (P10 or P15, respectively). Further, it remains unclear whether the disorganized surface epithelial cells were all marginal cells or whether different cells rose to the epithelial surface of stria vascularis, giving rise to the disorganized appearance. In the present study, we determined the time course of hyperpigmentation and marginal disorganization. Further, we tested the hypothesis that hyperpigmentation and marginal cell disorganization is a consequence of tissue inflammation including an invasion of inflammatory cells.
Methods
Animals
Breeding pairs of Slc26a4-/- and Slc26a4+/+ mice were obtained from the colony of Dr Susan Wall (Emory University, Atlanta, GA, USA) to establish a new colony at KSU. Mice used for this study were anaesthetized either with 4% tribromoethanol (0.014 ml/g body weight i.p.) or pentobarbitol (0.1 mg/g body weight, i.p.) and sacrificed by decapitation or by transcardial perfusion. Transcardial perfusion consisted of Cl-free solution (6 ml, 1 min) followed by Cl- free solution containing 4% paraformaldehyde (24 ml, 4 min). Cl- free solution contained mM 150 mM Na-gluconate, 1.6 mM K2HPO4, 0.4 mM KH2PO4, 4 mM Ca-gluconate2, 1 mM MgSO4 and 5 mM glucose, pH 7.4.
Mice that either express (Slc26a4+/+ or Slc26a4+/-) or lack (Slc26a4-/-) a functional pendrin gene were studied at four developmental stages, before and after the age that marks the onset of hearing at postnatal day 10 (P10) and P15, respectively, after weaning (P30-41) and adult (P74-170). Genotypes were determined by PCR as described previously [20]. All procedures involving animals were approved by the Institutional Animal Care and Use Committee of Kansas State University.
Confocal microscopy of cryosections
Temporal bones from age matched Slc26a4-/- and Slc26a4+/- mice were rendered blood free and fixed by transcardial and perilymphatic perfusion with Cl- free solution containing 4% paraformaldehyde. Temporal bones were decalcified in EDTA, processed through a sucrose gradient and infiltrated with polyethylene glycol. Mid-modiolar cryosections (12 μm, CM3050S, Leica, Nussloch, Germany) were blocked in PBS-TX (137 mM NaCl, 10.1 mM Na2HPO4, 1.8 mM KH2PO4, 2.7 mM KCl, pH 7.4 with 0.2% Triton X 100) and 5% bovine serum albumin. Slides were incubated overnight at 4°C with Alexa-488 conjugated rat anti-mouse CD68 antibody (1:25, Serotec, Raleigh, NC, USA) in PBS-TX with 1–3% BSA. After incubation, slides were washed with PBS-TX, mounted with FluorSave (Calbiochem, La Jolla, CA, USA), and viewed by confocal and laser scanning brightfield microscopy (LSM 510 Meta, Carl Zeiss, Göttingen, Germany).
Confocal microscopy of whole-mounts
Temporal bones from age matched Slc26a4-/- and Slc26a4+/- mice were rendered blood free by transcardial perfusion with Cl- free solution. Stria vascularis was obtained by microdissection and fixed for 2 hrs at 4°C in Cl- free solution containing 4% paraformaldehyde, washed twice in Cl- free solution and once in PBS-TX and then blocked with 5% BSA in PBS- TX for 45 min at RT and then washed three times in PBS-TX. Stria vascularis was then incubated overnight at 4°C either with Alexa-488 conjugated rat anti-mouse CD68 antibody (1:25, see above) or goat anti-Kcnq1 primary antibody (1:200, C20, Santa Cruz Biotechnology, Santa Cruz, CA, USA) in PBS-TX with 1–3% BSA. Tissues incubated with goat anti-Kcnq1 primary antibody were washed with PBS-TX and incubated for 1 h at 25°C with chicken anti-goat Alexa 594 secondary antibody, 1:1,000 (Molecular Probes, Eugene, OR, USA) in PBS-TX with 1–3% BSA.
After antibody incubation, stria vascularis was washed in PBS-TX (0.2% Triton-X) and stained with phalloidin conjugated to Alexa 594 (1:40; Invitrogen, Carlsbad, CA, USA), washed three times with PBS-TX, mounted with FluorSave (Calbiochem), and viewed by confocal and laser scanning brightfield microscopy (LSM 510 Meta, Carl Zeiss).
RNA isolation
Temporal bones were removed and stria vascularis and spiral ligament were obtained by microdissection. Microdissection solutions were changed twice and isolated tissues were washed to minimize contamination between tissue fractions. In addition, liver and spleen were collected and rapidly frozen in liquid nitrogen. Total RNA was isolated and residual DNA contamination was removed by DNase treatment (RNeasy micro, Qiagen, Valencia, CA, USA). Frozen samples of liver and spleen were pulverized and homogenized. Total RNA was isolated and freed from DNA contamination (RNeasy mini, Qiagen). Isolated RNA was either used immediately or diluted and stored at -80°C for later analysis (RNA storage solution, Ambion, Austin, TX, USA).
Gene array
Total RNA was isolated from stria vascularis of adult Slc26a4+/+ and Slc26a4-/- mice (P148 ± 3 and P153 ± 1, respectively). The minimal amount of blood present within capillaries was deemed insignificant; no cardiac perfusion was performed. Isolated RNA was concentrated and frozen for shipment to the Biotechnology Support Facility at University of Kansas Medical Center. A total of six chips were run; each chip was used to analyze RNA pooled from two animals. Three chips each were used to analyze RNA expression in stria vascularis of Slc26a4+/+ and Slc26a4-/- mice. RNA was amplified by two rounds of amplification, and cRNA was hybridized to high-density oligonucleotide gene chips (Small sample protocol, version II; mouse 430 2.0 gene chip, Affymetrix, Santa Clara, CA, USA). Gene array data were analyzed using commercial software (GCOS, Affymetrix; Genespring, Silicon Genetics, Redwood City, CA, USA) as well as custom-written macros (Excel, Microsoft, Redmond, WA, USA). Quality metrics conformed with MIAME standards (Table 1). Present/absent calls and averaged signal intensities (average of data obtained from three chips) were used to determine expression and changes in expression levels, respectively.
Table 1.
Quality metrics of gene arrays. Gene array data were deposited at GEO (GSE4749). The required MIAME quality metrics are given as average ± SD.
| Chips | Scale Factor | Average call (All) | Present calls (%) | Background intensity | Noise | Raw Q |
| 6 | 3.05 ± 1.07 | 682.9 ± 23.6 | 42.2 ± 3.1 | 100.3 ± 18.9 | 5.4 ± 1.1 | 3.1 ± 0.5 |
In the tabulated data summaries, 'Intensity' for Slc26a4+/+ and for Slc26a4-/- samples represents averages of data from one or more probes. For example, the gene Slc12a2 is represented on the chip by four probes. Present calls (P) were summarized for all three chips, e.g. 12/12 indicates that this gene was called present by all 12 probes (4 × 3 = 12); 9/15 indicates that the gene is represented by 5 probes on the 3 chips (5 × 3 = 15) and that the gene was called present by 9 of the 15 probes.
Ratios of intensity values (Slc26a4-/- to Slc26a4+/+) were calculated for each probe and averaged. Average ratios > 1.000 were reported as Fold with the Direction 'up'. Average ratios < 1.000 were inverted (1/average ratio) and reported as Fold with the Direction 'down'. Fold values are given in the tables only when the gene was called Present (P) in Slc26a4+/- or Slc26a4-/- samples in at least half of the available probes. The direction of the fold change is only given when it exceeded 1.30. Fold changes lower than 1.30 were not considered significant.
Quantitative RT-PCR
Age and sex matched Slc26a4-/- and Slc26a4+/- mice were rendered blood free by transcardiac perfusion with Cl- free solution. Total RNA was isolated from microdissected stria vascularis and spiral ligaments, as well as from liver and spleen. In each 96-well plate, the expression of seven different genes as well as 18S rRNA was analyzed. Total RNA from stria vascularis and spiral ligaments or from spleen and liver of one Slc26a4-/- and one matched Slc26a4+/- mouse was analyzed in duplicate reactions in parallel to allow paired comparisons (paired t-test). qRT-PCR was performed in the presence of 0.5× SYBR green I on total RNA isolated from individual animals using gene specific primers (One step RT-PCR kit, Qiagen; iCycler, BioRad, Hercules, CA, USA; SYBR green I, Molecular Probes; Table 2). RT was performed for 30 min at 50°C and 15 min at 95°C. PCR consisted of 40 cycles of 1 min at 60°C, 1 min at 72°C, 20 s hot measurement, and 1 min at 94°C. Specificity of primers was verified by sequencing. The generation of a single product of the appropriate size was verified by agarose gel electrophoresis.
Table 2.
Sequences of gene specific primers
| Gene | Primer sequence | Product length (bp) |
| 18S | gag gtt cga aga cga tca ga (sense) | |
| tcg ctc cac caa cta aga ac (antisense) | 317 | |
| C1r | aac aag atg ctg ctg acc (sense) | |
| tat tca agg ctg gag aca tag (antisense) | 302 | |
| C3 | atc cga tac tac acc tacc (sense) | |
| cct ggt ttc ctt caa tcc (antisense) | 307 | |
| Ptprc (Cd45) | tgt tga cag agt tag tga atg g (sense) | |
| gtg gac gag gat gga tgc (antisense) | 323 | |
| Lyzs | cct gct ttc tgt cac tgc tc (sense) | |
| ggt gta atg atg gca aaa cc (antisense) | 235 | |
| Il1a | aca cta tct cag cac cac ttg g (sense) | |
| gca ccc gac ttt gtt ctt tgg (antisense) | 311 | |
| Il6 | ctg caa gag act tcc atc c (sense) | |
| tat atc cag ttt ggt agc atc c (antisense) | 300 | |
| Il12a | tca atc acg cta cct cct c (sense) | |
| ctg ttg tgg aag aag tct ctc (antisense) | 302 | |
| Nos2 | gtt tga cca gag gac cca ga (sense) | |
| acc tga tgt tgc cat tgt tg (antisense) | 354 | |
| Nos3 | cct aca gag cag caa atc ca (sense) | |
| gcc ttt ctc cag ttg ttc ca (antisense) | 202 | |
| Otos | atg cag ccc tgt ctg ctg tgg tgg (sense) | |
| gtc ctc ctg gta ggg aac atg gaa (antisense) | 267 | |
| Tyr | gcc tgt gcc tcc tct aag (sense) | |
| ttc taa tca aga ctc gct tct c (antisense) | 295 |
Template molecules were quantified according to T = 10^log (PCt)/(Eavg^Ct), where PCt is product molecules at Ct, Eavg is the average efficiency and Ct is cycle at which the fluorescence of the product molecules reached a set threshold. Efficiencies for individual reactions was obtained from the slope of the log-linear phase of the growth curve using an Excel-based program (LinRegPCR) [21].
The number of product molecules at Ct (PCt) was calculated by amplifying known numbers of 18S rRNA (T18S) molecules according to PCt = T18S × E18S^Ct, where E18S is the average efficiency of all the 18S rRNA reactions. The mass of 18S rRNA in 1 μg of total RNA was estimated to calculate the number of 18S rRNA molecules in a given amount of RNA sample. Calculations were based on three assumptions: firstly, total RNA consists to 100% of 18S and 28S rRNA. Secondly, 18S and 28S rRNA occur in a 1:1 ratio. Thirdly, murine 18S and 28S rRNA contains 1869 and 4712 nucleotides [Genbank: X00686] and [Genbank: X00525], respectively. The mass of 18S rRNA per 1 μg of total RNA was estimated to be 0.284 μg, equivalent to 4.7 × 10-13 mol or 2.8 × 1011 molecules of 18S rRNA (molecular weight of 18S rRNA estimated to be 598,080).
Western blotting
Temporal bones from age matched Slc26a4-/- and Slc26a4+/- mice were rendered blood free by transcardial perfusion with Cl- free solution. Stria vascularis and spiral ligament were isolated by microdissection. Proteins in stria vascularis and spiral ligament from one animal were isolated by heating (10 min, 95°C) in 20 μl of a diluent (Compound B, NanoOrange, Invitrogen). After cooling (20 min, RT), the isolated protein was quantified (NanoOrange, Invitrogen) and either used immediately in Western blots or stored at -80°C.
Proteins (15 μg per well) were denatured in the presence of Laemmli sample buffer (62.5 mM Tris-HCl, pH 6.8, 25% glycerol, 2% SDS, 0.01% bromophenol blue, 5% 2-mercaptoethanol) at 80°C for 5 min and resolved by electrophoresis (150 V for 45 min) in 4–15% polyacrymalide Tris-HCl gradient gels (10 well mini-gel, BioRad laboratories, Inc., Hercules, CA, USA). Molecular weight standards were used to estimate molecular weights (Precision Markers, BioRad). Proteins were transferred in transblot buffer (25 mM Tris, 192 mM glycine, 20% v/v methanol, pH 8.3). (25 V, 1 h; XCell II blot module, Invitrogen) onto a nitrocellulose membrane (0.2 μm, BioRad).
Nitrocellulose membranes were first evaluated for lysozyme expression, then stripped (Restore Western Blot Stripping Buffer, Pierce, Rockford, IL, USA) and evaluated for actin expression. Membranes were blocked in TBS-Tween (20 mM Tris-Cl, pH 7.6 with 0.1% Tween) containing 5% non-fat dry milk powder (BioRad) and incubated (1 hr RT) with rabbit anti-human lysozyme primary antibody (1:500; Dakocytomation, Carpinteria, CA) or rabbit anti-Actin primary antibody (1:1,000; Sigma) in TBS-Tween containing 5% non-fat dry milk. Membranes were washed three times with TBS-Tween and then incubated with horseradish peroxidase conjugated donkey anti-rabbit antibody (1:25,000, Amersham Biosciences) in TBS-Tween containing 5% non-fat dry milk. The membranes were washed again three times with TBS-Tween and then treated with chemiluminescent detection agents (SuperSignal West Femto Maximum Sensitivity Substrate; Pierce), exposed to film (CL-XPosure Film, Pierce), which was developed immediately (XOmat 2000A, Kodak, Rochester, NY, USA).
Statistics
Numeric data are presented at average ± sem, unless specified otherwise. The number (n) of animals, blots or cells is given. Differences were determined by paired t-tests. Significance was assumed at p < 0.05.
Results and Discussion
Cellular reorganization of stria vascularis
Previously, we reported that stria vascularis of adult Slc26a4-/- mice is hyperpigmented and that marginal cells appear disorganized [15,17]. Stria vascularis was obtained from age matched Slc26a4+/- and Slc26a4-/- mice and hyperpigmentation was evaluated by laser-scanning light microscopy. Hyperpigmentation was absent at P10 and P15 but observed after weaning (P33-41) and in adult (P84-96) Slc26a4-/- mice (Fig. 1).
Figure 1.

Hyperpigmentation in stria vascularis. The time course of hyperpigmention during development was determined by laser scanning brightfield microscopy of stria vascularis whole-mounts. Top: images from Slc26a4+/- mice of various ages (P, postnatal day). Bottom: images from Slc26a4-/- mice. Note that hyperpigmentation (arrows) was observed in Slc26a4-/- mice at P33 and P84 but not at P10 or P15. No hyperpigmentation was observed in Slc26a4+/- mice. The scale bar shown in the bottom right image represents 10 μm and pertains to all images in this figure.
Marginal cells can be identified by there expression of the K+ channel Kcnq1 in their apical membrane [17]. To gain information on the identity of surface epithelial cells, expression of Kcnq1 was visualized by immunocytochemistry and F-actin, a marker for tight junctions, was visualized by phalloidin staining. Marginal cells at all ages of Slc26a4+/- mice and at P10 and P15 of Slc26a4-/- mice expressed the K+ channel Kcnq1 evenly. Little variation was observed in cell surface areas at all ages of Slc26a4+/- mice and at P10 and P15 of Slc26a4-/- mice, and variations appeared normally distributed (Figs 2 and 3). The average cell surface area of marginal cells in Slc26a4+/- mice was 104 ± 2 μm2 (n = 120). No significant differences were detected in Slc26a4+/- mice of different ages. In contrast, the average cell surface areas of marginal cells in P10 and P15 Slc26a4-/- mice were significantly larger, 236 ± 12 μm2 (n = 30) and 232 ± 12 μm2 (n = 30), respectively. This enlargement of the apical surface area of marginal cells is consistent with the understanding that Slc26a4+/- and Slc26a4-/- mice do not differ in the total number of marginal cells, and that cell surface areas are enlarged to cover the larger area that results from the enlarged cochlear diameter found in Slc26a4-/- mice.
Figure 2.

Loss of Kcnq1 expression in marginal cells. The time course of marginal cell reorganization during development was determined by confocal immunohistochemistry of Kcnq1 (red) and phalloidin-staining of α-actin (green) of stria vascularis in whole-mounts. Top, images from Slc26a4+/- mice of various ages. Bottom, images from Slc26a4-/- mice. Note that marginal cells of Slc26a4+/- mice expressed Kcnq1 homogeneously in their apical membrane and that apical membrane surface areas displayed little variation in size. Marginal cells of Slc26a4-/- mice at P10 and P15 expressed Kcnq1 homogeneously, too, and apical membrane surface areas showed little variation in size. However, surface areas were larger compared to Slc26a4+/- mice. At P37, expression of Kcnq1 in Slc26a4-/- mice was reduced in some marginal cells and maintained in others. Loss of Kcnq1 expression correlated with an enlargement of the apical membrane surface area and maintenance of Kcnq1 expression correlated with a reduction in the apical membrane surface area. This segregation of marginal cells was more drastic at P96. The scale bar shown in the bottom right image represents 10 μm and pertains to all images in this figure.
Figure 3.

Reorganization of marginal cells. Apical surface areas of marginal cells were measured using images of whole-mounts of stria vascularis as shown in Fig. 2. Frequency histograms (bin size 25 μm2) of surface area measurements were constructed. The number (n) of cells contributing to each histogram is given. Note that surface areas in Slc26a4+/- mice of all ages were normally distributed and narrowly centered at ~ 100 μm2. The distribution of surface areas in Slc26a4-/- mice progressed during development from a distribution centered at ~ 240 μm2 to a wide multimodal distribution with peaks centered at ~ 100 and ~ 240 μm2.
After weaning, marginal cells of Slc26a4-/- mice began to reorganize, leading to a multimodal distribution of cell surface areas (Figs 2 and 3). Most interestingly, marginal cells that reduced their surface areas from ~ 240 to ~ 100 μm2 retained dense expression of Kcnq1, whereas marginal cells that increased their surface area gradually lost Kcnq1 expression. The degree of Kcnq1 staining in stria vascularis of P37 Slc26a4-/- mice correlated inversely with the size of the cell surface area. Stria vascularis of adult Slc26a4-/- mice consisted of a mosaic of Kcnq1-expressing and degenerated marginal cells. Kcnq1-expressing marginal cells are likely to remain functional, given that Kcnq1 is essential for K+ secretion and the endolymphatic K+ concentration was found to be normal (~ 140 mM) in adult Slc26a4-/- mice [17,22,23].
Expression analysis by gene array
Gene expression in stria vascularis was analyzed by gene array to gain data on the cause of marginal cells reorganization. The quality of the gene array was determined by quality metrics (Table 1), and the quality of the expression analysis was determined by an evaluation of genes that are known to be expressed in stria vascularis (Table 3) and by an evaluation of genes that are known to be expressed in neighboring tissues that could have served as sources of contamination (Table 4). As expected, genes known to be expressed in marginal, intermediate basal and spindle cells were present in Slc26a4+/+ and Slc26a4-/- mice (Table 3). Interestingly, genes known to be expressed in marginal cells were downregulated in Slc26a4-/- mice (Table 3A). In contrast, no change in expression was observed for genes known to be expressed in intermediate and/or basal cells (Table 3B). These data are consistent with a partial degeneration of marginal cells.
Table 3.
Genes that were expected to be expressed in stria vascularis.
| Slc26a4+/+ | Slc26a4-/- | |||||||
| No. | Gene | Description | Intensity | P | Intensity | P | Fold | Direction |
| 1 | Slc12a1 | Na+/2Cl-/K+ cotransporter | 14,664 | 12/12 | 10,656 | 12/12 | 1.33 | down |
| 2 | Atp1a1 | Na+/K+ ATPase, alpha | 7,276 | 9/15 | 5,440 | 9/15 | 2.06 | down |
| 3 | Atp1b1 | Na+/K+ ATPase, beta1 | 10,593 | 12/18 | 8,587 | 12/18 | 1.20 | |
| 4 | Atp1b2 | Na+/K+ ATPase, beta2 | 8,057 | 6/6 | 5,984 | 6/6 | 1.41 | down |
| 5 | Kcnq1 | K+ channel, alpha | 858 | 15/24 | 542 | 11/24 | 2.48 | down |
| 6 | Kcne1 | K+ channel, beta | 9,042 | 6/6 | 3,490 | 6/6 | 2.60 | down |
| 7 | Kcnma1 | K+ channel, BK | 270 | 10/18 | 159 | 9/18 | 1.72 | down |
| 8 | Kcnk1 | K+ channel | 2,667 | 6/6 | 1,670 | 6/6 | 1.92 | down |
| 9 | Slc26a4 | Pendrin | 1,136 | 3/3 | 619 | 3/3 | 1.84 | down |
| 10 | Kcnj10 | K+ channel | 17,169 | 3/3 | 17,005 | 3/3 | 1.01 | |
| 11 | Car2 | Carbonic anhydrase | 9,139 | 3/3 | 9,167 | 3/3 | 1.00 | |
| 12 | Gjb2 | Connexin 26 | 12,785 | 3/3 | 12,629 | 3/3 | 1.01 | |
| 13 | Gjb6 | Connexin 30 | 15,257 | 3/3 | 13,060 | 3/3 | 1.17 | |
| 14 | Sod1 | Superoxide dismutase | 4,149 | 14/15 | 3,538 | 14/15 | 1.21 | |
| 15 | Cat | Catalase | 3,520 | 6/6 | 4,686 | 6/6 | 1.12 | |
| 16 | Gpx1 | Glutathione peroxidase | 2,633 | 3/3 | 4,320 | 3/3 | 1.64 | up |
| 17 | Gpx3 | Glutathione peroxidase | 12,526 | 3/3 | 9,898 | 3/3 | 1.27 | |
| 18 | Gpx4 | Glutathione peroxidase | 11,224 | 6/6 | 10,421 | 6/6 | 1.06 | |
| 19 | Tyr | Tyrosinase | 3,520 | 6/6 | 4,686 | 6/6 | 1.16 | |
Genes that are expected to be expressed in marginal and spindle cells of stria vascularis (1–9) and genes that are expected to be expressed in intermediate and basal cells of stria vascularis (10–19) are listed. For parameters 'P', 'Fold' and 'Direction', see the Gene array section in Methods. Fold changes are given when a gene was called Present (P) in Slc26a4+/- or Slc26a4-/- samples in at least half of the available probes. The direction of the fold change is only given when it exceeded 1.30. Fold changes lower than 1.30 were not considered significant.
Table 4.
Genes that were not expected to be expressed in stria vascularis.
| Slc26a4+/+ | Slc26a4-/- | |||||||
| No. | Gene | Description | Intensity | P | Intensity | P | Fold | Direction |
| 1 | Pres | Prestin | 53 | 0/3 | 8 | 0/3 | ||
| 2 | Kcnq4 | K+ channel | 160 | 0/6 | 160 | 0/6 | ||
| 3 | Atp2b2 | PMCA2 | 35 | 0/9 | 116 | 3/9 | ||
| 4 | Calb1 | Calbinin 28K | 53 | 0/12 | 36 | 0/12 | ||
| 5 | Calb2 | Calretinin | 28 | 0/3 | 27 | 0/3 | ||
| 6 | Ache | Acetylcholinesterase | 57 | 0/3 | 5 | 0/3 | ||
| 7 | Slc1a3 | Glutamate transporter GLAST | 122 | 3/18 | 154 | 8/18 | ||
| 8 | Gria1 | Glutamate receptor, ionotropic | 57 | 0/9 | 55 | 0/9 | ||
| 9 | Atp6v1b1 | H+ ATPase | 58 | 0/3 | 26 | 0/3 | ||
| 10 | Tecta | Tectorin, alpha | 62 | 0/3 | 52 | 0/3 | ||
| 11 | Tectb | Tectorin, beta | 90 | 0/3 | 01 | 0/3 | ||
| 12 | Otog | Otogelin | 230 | 0/3 | 63 | 0/3 | ||
| 13 | Otof | Otoferlin | 231 | 0/3 | 214 | 0/3 | ||
| 14 | Otop1 | Otopetrin 1 | 54 | 0/3 | 62 | 1/3 | ||
| 15 | Otop2 | Otopetrin 2 | 247 | 0/3 | 203 | 0/3 | ||
| 16 | Otop3 | Otopetrin 3 | 60 | 0/3 | 47 | 0/3 | ||
| 17 | Otos | Otospiralin | 636 | 3/3 | 2,368 | 3/3 | 3.73 | up |
| 18 | Slc4a7 | Na+/HCO3 - cotransporter | 398 | 3/3 | 444 | 3/3 | 1.11 | |
| 19 | Kcnj16 | K+ channel | 280 | 2/6 | 239 | 1/6 | ||
Genes that are expected to be expressed in the organ of Corti, the modiolus or the vestibular labyrinth but not in stria vascularis (1–16), and genes that are expected to be expressed in spiral ligament but not in stria vascularis (17–19) are listed. For parameters 'P', 'Fold' and 'Direction', see the Gene array section in Methods. Fold changes are given when a gene was called Present (P) in Slc26a4+/- or Slc26a4-/- samples in at least half of the available probes. The direction of the fold change is only given when it exceeded 1.30. Fold changes lower than 1.30 were not considered significant.
Genes known to be expressed in neighboring tissues were not found in stria vascularis, with the exception of otospiralin (Otos). Otos in known to be expressed in spiral ligament [24], which is the tissue adjacent to stria vascularis (Table 4). Taken together, these data demonstrate that gene array analysis provided reliable data of expression in stria vascularis.
Macrophage invasion of stria vascularis
Gene array analysis revealed that stria vascularis of adult Slc26a4-/- mice expresses markers specific and/or consistent with the presence of macrophages (Table 5). Expression of Mac2 antigen, Itgax, Cd45, Cd83 and Cd68 is limited to leucocytes and hematopoietic cells including macrophages and dendritic cells [25]. Expression of major histocompatibility complex II (MHCII) proteins is limited to antigen-presenting cells including macrophages [25]. The expression of Lysz, a lysosomal enzyme, has been shown to be a marker for monocytes and macrophages [26]. Further, the increased expression of major histocompatibility complex I, of complement components and of cathepsins is consistent with the presence of macrophages although the expression of the gene is not limited to macrophages [27].
Table 5.
Markers for macrophages/dendritic cells.
| Slc26a4+/+ | Slc26a4-/- | |||||||
| No. | Gene | Description | Intensity | P | Intensity | P | Fold | Direction |
| 1 | Ptprc | Cd45 antigen | 226 | 3/3 | 946 | 3/3 | 4.19 | up |
| 2 | Cd83 | Cd83 antigen | 126 | 0/3 | 524 | 3/3 | 4.16 | up |
| 3 | Cd68 | Cd68 antigen | 170 | 3/3 | 1,344 | 3/3 | 7.90 | up |
| 4 | Cd14 | Cd14 antigen | 412 | 1/3 | 619 | 3/3 | 1.50 | up |
| 5 | H2-Aa | MHC class II | 264 | 5/12 | 1,019 | 9/12 | 4.00 | up |
| 6 | H2-Ab | MHC class II | 126 | 0/9 | 279 | 3/9 | 1.86 | up |
| 7 | H2-Eb1 | MHC class II | 429 | 0/3 | 1,010 | 3/3 | 2.35 | up |
| 8 | Lgals3 | Mac2 antigen | 91 | 2/6 | 1,866 | 5/6 | 13.49 | up |
| 9 | Itgax | Integrin alpha X | 125 | 0/3 | 375 | 3/3 | 3.00 | up |
| 10 | Emr1 | Mucin receptor | 750 | 3/3 | 1,547 | 3/3 | 2.06 | up |
| 11 | Lyzs | Lysozyme | 551 | 6/9 | 8257 | 9/9 | 11.78 | up |
| 12 | H2-K1 | MHC class I (D-region) | 1,607 | 13/18 | 4,062 | 15/18 | 2.06 | up |
| 13 | H2-K1 | MHC class I (K-region) | 1,828 | 6/12 | 2,883 | 6/12 | 1.54 | up |
| 14 | H2-K1 | MHC class I (Q-region) | 549 | 1/3 | 890 | 3/3 | 1.62 | up |
| 15 | C1r | Complement | 37 | 2/6 | 266 | 6/6 | 13.87 | up |
| 16 | C1s | Complement | 71 | 0/3 | 200 | 1/3 | ||
| 17 | C1qa | Complement | 355 | 1/3 | 2,905 | 3/3 | 8.19 | up |
| 18 | C1qb | Complement | 552 | 5/6 | 2,001 | 6/6 | 4.43 | up |
| 19 | C2 | Complement | 3,207 | 9/9 | 4,794 | 6/6 | 1.62 | up |
| 20 | C3 | Complement | 11 | 0/3 | 231 | 2/3 | 20.68 | up |
| 21 | C4 | Complement | 1,556 | 3/3 | 4,788 | 3/3 | 3.08 | up |
| 22 | C3ar1 | Complement receptor | 177 | 3/9 | 482 | 6/9 | 2.93 | up |
| 23 | Ctss | Cathepsin S | 2,304 | 3/3 | 7,394 | 3/3 | 3.21 | up |
| 24 | Ctsk | Cathepsin K | 170 | 1/3 | 377 | 3/3 | 2.21 | up |
| 25 | Ctsb | Cathepsin B | 4,172 | 12/15 | 6,576 | 12/15 | 1.90 | up |
| 26 | Msr2 | Macrophage scavenger receptor | 169 | 3/3 | 1140 | 3/3 | 6.75 | up |
| 27 | Mpeg1 | Macrophage expressed gene | 582 | 0/3 | 5,527 | 3/3 | 9.49 | up |
| 28 | Cst3 | Cystatin C | 5,562 | 3/3 | 8,652 | 3/3 | 1.56 | up |
| 29 | Fcgr1 | IgG receptor, high affinity | 98 | 0/6 | 163 | 5/6 | 1.70 | up |
| 30 | Fcgr2b | IgG receptor, low affinity | 112 | 2/15 | 377 | 7/15 | 3.94 | up |
Genes that can serve as markers macrophages/dendritic cells (1–10) and genes that are consistent with the presence of macrophages/dendritic cells (11–27) are listed. For parameters 'P', 'Fold' and 'Direction', see the Gene array section in Methods. Fold changes are given when a gene was called Present (P) in Slc26a4+/- or Slc26a4-/- samples in at least half of the available probes. The direction of the fold change is only given when it exceeded 1.30. Fold changes lower than 1.30 were not considered significant.
Taken together, the data presented in Table 5 suggest that stria vascularis of adult Slc26a4-/- mice is invaded by macrophages. Markers for T-cells, NK-cells, B-cells, neutrophils and for acute inflammation were absent in stria vascularis of Slc26a4+/+ and Slc26a4-/- mice (Tables 6 and 7). The absence of markers of acute inflammation was verified by qRT-PCR of total RNA isolated from stria vascularis and spiral ligament of age and sex matched Slc26a4-/- and Slc26a4+/- mice. No change in expression was found for Il1a, Il6, Il12a, Nos2 and Nos3 at P15 or at P34 (each n = 4–5; data not shown). Thus, no evidence for acute inflammation was obtained. It is conceivable, however, that short periods of acute inflammation escaped detection. Alternatively, it is conceivable that macrophage invasion was not preceded by neutrophil invasion and that macrophage recruitment occurred neutrophil-independently [28]. The source of chemoattractants for monocytes (MCPs) in neutrophil-independent recruitment are endogenously harbored tissue macrophages and mesothelial cells [28]. Such a mechanism could be present in stria vascularis of Slc26a4-/-mice as the cochlea endogenously harbors macrophages in the fibrocytes of the lateral wall [29]. The mechanism of recruitment into stria vascularis, however, remains unclear.
Table 6.
Markers for T, NK and B-cells and Neutrophils.
| Slc26a4+/+ | Slc26a4-/- | |||||||
| No. | Gene | Description | Intensity | P | Intensity | P | Fold | Direction |
| 1 | Tcra | T-cell receptor, alpha | 214 | 0/9 | 190 | 0/9 | ||
| 2 | Tcrb-V13 | T-cell receptor, beta V13 | 136 | 4/36 | 109 | 4/36 | ||
| 3 | Tcrb-J | T-cell receptor, beta V13 | 71 | 2/15 | 50 | 1/15 | ||
| 4 | Tcrg | T-cell receptor, gamma V4 | 54 | 0/15 | 43 | 0/15 | ||
| 5 | Cd3d | Cd3 antigen | 10 | 0/3 | 8 | 0/3 | ||
| 6 | Cd3e | Cd3 antigen | 139 | 0/6 | 74 | 0/6 | ||
| 7 | Cd3g | Cd3 antigen | 43 | 0/3 | 28 | 0/3 | ||
| 8 | Cd3z | Cd3 antigen | 182 | 4/24 | 139 | 3/24 | ||
| 9 | Cd4 | Cd4 antigen | 20 | 0/6 | 149 | 0/6 | ||
| 10 | Cd8a | Cd8 antigen | 144 | 4/15 | 143 | 4/15 | ||
| 11 | Cd8b | Cd8 antigen | 1,313 | 3/6 | 1,343 | 3/6 | ||
| 12 | Ncam1 | Cd56 | 562 | 8/21 | 654 | 12/21 | 1.16 | |
| 13 | Fcgr3 | IgG receptor, low aff, Cd16 | 162 | 2/3 | 401 | 3/3 | 2.48 | up |
| 14 | Sdc1 | Syndecan 1 (Cd138) | 77 | 3/12 | 72 | 3/12 | ||
| 15 | Cd19 | Cd19 antigen | 15 | 0/3 | 13 | 0/3 | ||
| 16 | Mpo | Myeloperoxidase | 159 | 0/3 | 73 | 0/3 | ||
| 17 | Defb1 | Beta-defensin 1 | 66 | 1/6 | 72 | 2/6 | ||
| 18 | Defb2 | Beta-defensin 2 | 23 | 0/3 | 14 | 0/3 | ||
| 19 | Defb3 | Beta-defensin 3 | 31 | 0/3 | 70 | 0/3 | ||
| 20 | Defb4 | Beta-defensin 4 | 85 | 0/3 | 63 | 0/3 | ||
| 21 | Camp | Cathelicidin | 11 | 0/3 | 10 | 0/3 | ||
Genes that can serve as markers for T-cells (1–11), NK-cells (12–13), B-cells (14–15) and neutrophils (16–21) are listed. For parameters 'P', 'Fold' and 'Direction', see the Gene array section in Methods. Fold changes are given when a gene was called Present (P) in Slc26a4+/- or Slc26a4-/- samples in at least half of the available probes. The direction of the fold change is only given when it exceeded 1.30. Fold changes lower than 1.30 were not considered significant.
Table 7.
Genes that serve as markers for acute inflammation.
| Slc26a4+/+ | Slc26a4-/- | |||||||
| No. | Gene | Description | Intensity | P | Intensity | P | Fold | Direction |
| 1 | Il1a | Interleukin 1a | 29 | 0/3 | 44 | 0/3 | ||
| 2 | Il1b | Interleukin 1b | 173 | 0/3 | 156 | 0/3 | ||
| 3 | Il6 | Interleukin 6 | 14 | 0/3 | 31 | 0/3 | ||
| 4 | Il12a | Interleukin 12a | 126 | 0/3 | 96 | 0/3 | ||
| 5 | Il12b | Interleukin 12b | 70 | 1/6 | 73 | 1/6 | ||
| 6 | Il10 | Interleukin 10 | 116 | 3/3 | 87 | 3/3 | ||
| 7 | Ifng | Interferon, gamma | 71 | 1/3 | 24 | 0/3 | ||
| 8 | Tnf | Tumor necrosis factor, alpha | 54 | 0/3 | 50 | 0/3 | ||
| 9 | Nos2 | Nitric oxide synthase, inducible | 19 | 0/3 | 27 | 0/3 | ||
Genes that can serve as markers for acute inflammation (1–9) are listed. For parameters 'P', 'Fold' and 'Direction', see the Gene array section in Methods. Fold changes are given when a gene was called Present (P) in Slc26a4+/- or Slc26a4-/- stria vascularis in at least half of the available probes. The direction of the fold change is only given when it exceeded 1.30. Fold changes lower than 1.30 were not considered significant.
Macrophage invasion is restricted to stria vascularis
The observation that stria vascularis of adult mice is invaded by macrophages raises the question whether macrophage invasion is systemic and hence found in other organs, such as spleen and liver, or whether macrophage invasion is restricted to stria vascularis and hence not seen in the adjacent tissue, spiral ligament. This is an important issue, as the inner ear is immunologically responsive to systemic infections [30].
Total RNA was isolated from stria vascularis, spiral ligament, liver and spleen from age and sex matched Slc26a4-/- and Slc26a4+/- mice. The expression of a select group of transcripts was evaluated by qRT-PCR. Possible contamination between stria vascularis and the adjacent spiral ligament was evaluated by quantifying the expression of Otos and tyrosinase (Tyr) under the assumption that Tyr is expressed in stria vascularis and not in spiral ligament, and Otos in spiral ligament and not in stria vascularis. The detected expression level of Tyr in stria vascularis of Slc26a4+/- and Slc26a4-/- mice was 120 ± 33 and 50 ± 13 times higher than in spiral ligament, respectively (n = 6) and the detected expression of Otos in spiral ligament of Slc26a4+/- and Slc26a4-/- mice was 108 ± 32 and 50 ± 28 times higher than in stria vascularis, respectively (n = 3). These data illustrate a small contamination between stria vascularis and spiral ligament. Contamination was larger in Slc26a4-/- mice (2%) than in Slc26a4+/- mice (1%), consistent with the observation during microdissection that stria vascularis from Slc26a4-/- is more fragile.
Expression of complement components and macrophage markers including C1r, C3, Ptprc (= Cd45), Cd83 and Lyzs was increased in stria vascularis of Slc26a4-/- mice compared to Slc26a4+/- mice (Fig. 4). Expression of these genes was not upregulated in spiral ligament with the exception of Lyzs and Cd45. Further, the expression of these genes was not upregulated in liver or spleen (Fig. 4). Taken together, these data suggest that macrophage invasion in Slc26a4-/- mice is restricted to stria vascularis.
Figure 4.

Tissue specificity of macrophage invasion. Transcripts of markers specific for or consistent with the presence of macrophages in stria vascularis, spiral ligament, spleen and liver of Slc26a4+/- and Slc26a4-/- mice at P34 and/or P86 were quantified by qRT-PCR. Significant changes between Slc26a4+/- and Slc26a4-/- mice are marked with an asterisk (*). Numbers between bars represent the number of animal pairs analyzed. Note that significant increases were mainly seen in stria vascularis, to a lesser degree in spiral ligament, but not in spleen or liver. These data suggest that macrophage invasion is specific to stria vascularis.
Time course of macrophage invasion
The finding that macrophage invasion was restricted to stria vascularis raises the question of when in development macrophage invasion occurs. Total RNA was isolated from stria vascularis and spiral ligament from sex and age matched Slc26a4-/- and Slc26a4+/- mice and a select group of transcripts was quantified by qRT-PCR. At P15, no difference in the expression of Lyzs, C3 and Cd45 was found in stria vascularis or in spiral ligament between Slc26a4-/- and Slc26a4+/- mice (Fig. 5). After weaning, and persistent into adulthood, however, the expression of Lyzs, C3 and Cd45 was higher in stria vascularis of Slc26a4-/- mice compared to Slc26a4+/- mice. A similar trend was observed with spiral ligament.
Figure 5.
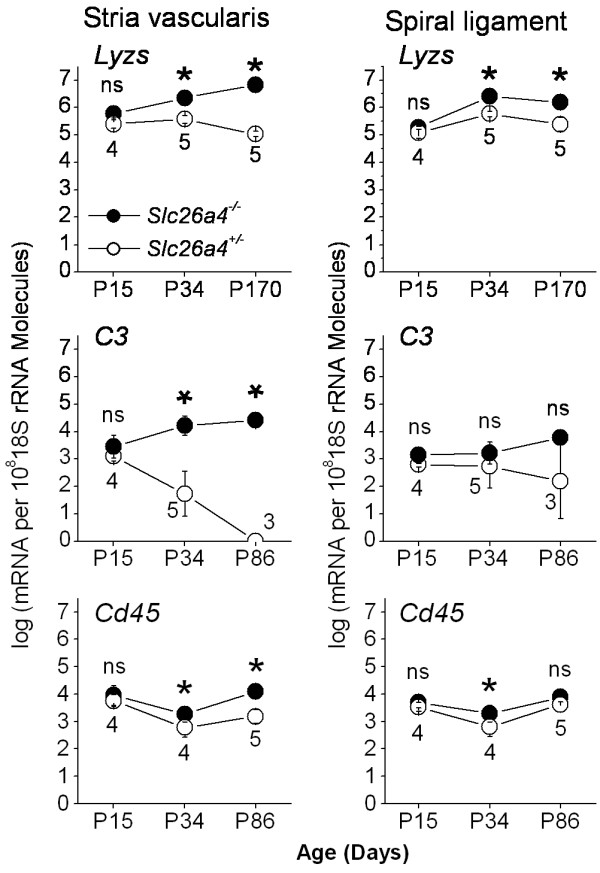
Time course of macrophage invasion. Transcripts of markers specific for or consistent with the presence of macrophages in stria vascularis and spiral ligament of Slc26a4+/- and Slc26a4-/- mice at different ages were quantified by qRT-PCR to determine the time course of macrophage invasion during development. Significant changes between Slc26a4+/- and Slc26a4-/- mice are marked with an asterisk (*). Numbers between symbols represent the number of age-matched animal pairs analyzed. Note that significant differences were not seen before P34, suggesting that macrophage invasion occurred after weaning (P22).
The expression of Lyzs was determined based not only on the transcript level but also on the protein level, which may be less prone to contamination between neighboring tissues. Western blotting revealed that Lyzs protein is upregulated in stria vascularis but not in spiral ligament of post-weaning Slc26a4-/- mice (Fig. 6).
Figure 6.
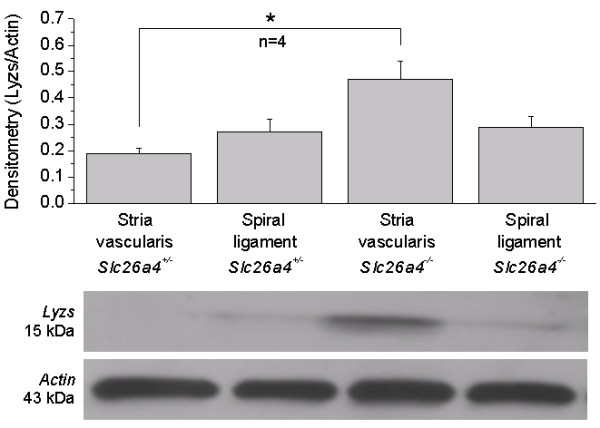
Lysozyme protein expression. Protein expression of lysozyme (Lyzs) in stria vascularis and spiral ligament of post-weaning Slc26a4+/- and Slc26a4-/- mice was determined by Western blotting. Actin expression served as a normalization control. Significant changes between Slc26a4+/- and Slc26a4-/- mice are marked with an asterisk (*). The number of animal pairs (n) is given. Note that protein expression of Lyzs was significantly increased in stria vascularis, but not in spiral ligament.
Macrophage invasion was not only determined by gene expression analysis but also by immunohistochemistry, using CD68 as a marker. The specificity of the anti-CD68 antibody was verified by using bone marrow cells as a positive control and heavily pigmented cells of the vestibular labyrinth as a negative control (Fig. 7). Staining for CD68 was clearly associated with hyperpigmentation in stria vascularis in post-weaning and adult Slc26a4-/- mice (Fig. 7).
Figure 7.
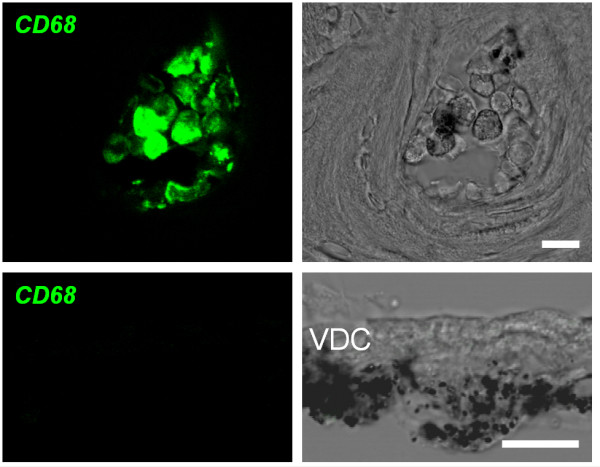
Controls for immunohistochemistry. Controls for CD68 immunohistochemistry were obtained from cryosections of the cochlea. Top: bone marrow cells congregated in cavities of the bony cochlear wall served as a positive control. Bottom: heavily pigmented cells in the connective tissue underneath vestibular dark cells (VDC) served as a negative control. Left: confocal immunohistochemistry of CD68. Right: corresponding bright field images. Scale bars represent 10 μm.
Conclusion
The data demonstrate that hyperpigmentation of stria vascularis and marginal cell reorganization in Slc26a4-/- mice occur after weaning, coinciding with an invasion of macrophages. The data suggest that macrophage invasion contributes to tissue degeneration in stria vascularis, and that macrophage invasion is restricted to stria vascularis and is not systemic in nature. The delayed onset of degeneration of stria vascularis suggests that a window of opportunity exists to restore/preserve hearing in mice and humans suffering from Pendred syndrome.
Competing interests
The author(s) declare that they have no competing interests.
Authors' contributions
SVJ and AO drafted the text and PW finalized the manuscript. SVJ carried out confocal immunocytochemistry and morphometry. SVJ and PW isolated tissue fractions by microdissection. AO designed primers, isolated RNA and performed quantitative RT-PCR. RS prepared gene array data for submission under MIAME standards. RJM carried out confocal immunocytochemistry and Western blotting, SF advised on immunology. PW mined gene array data. SMW, LAE and EDG provided mice prior to the establishment of a colony at KSU. PW conceived the study. All authors have read and approved the final manuscript.
Figure 8.

Macrophage invasion in stria vascularis. Macrophages were visualized by CD68 immunohistochemistry in whole mounts of stria vascularis and cryosections of the cochlear lateral wall. Top row: (a-d), CD68 staining in whole mounts of stria vascularis from P33 Slc26a4+/- and Slc26a4-/- mice. Immunostaining of CD68 (a and d) and corresponding bright field images (b and c) are shown. Second and third row: (e-p), CD68 staining in cryosections of the cochlear lateral wall from P33 and P80 Slc26a4+/- and Slc26a4-/- mice. Immunostaining of CD68 (e, h, k, and n), corresponding bright field images (f, i, l, and o) and merged images (g, j, m, and p) are shown. Note that CD68 expression is restricted to hyperpigmented areas of stria vascularis in Slc26a4-/- mice and that no expression of CD68 was observed in Slc26a4+/- mice. Scale bars represent 10 μm.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Acknowledgments
Acknowledgements
The authors would like to thank Mr. Clark Bloomer, Biotechnology Support Facility at the University of Kansas Medical Center, for expert preparation of targets and hybridization to gene arrays. Laboratory assistance from Nithya Raveendran, Anantha Gollapudi and Theresa Wangemann is gratefully acknowledged. Excel macros in support of gene array analysis were conceived by PW and written by Nithya Raveendran. The creation of the mice was supported in part by the Intramural Program of the National Human Genome Research Institute, National Institutes of Health. Breeding and shipping of Slc26a4+/+ and Slc26a4-/- mice from Emory University was funded by grant NIH-R01-DK52935 to SW. Breeding of the mice at KSU and the work reported in this paper was supported by grant NIH-R01-DC01098 to PW from the National Institutes on Deafness and Other Communication Disorders, National Institutes of Health. The Confocal Core and the Molecular Biology Core at KSU were funded by NIH-P20-RR017686 from the National Institute for Research Resources, National Institutes of Health.
Contributor Information
Sairam V Jabba, Email: sjabba@vet.ksu.edu.
Alisha Oelke, Email: OelkeAlisha@rossmed.edu.dm.
Ruchira Singh, Email: rsingh@vet.ksu.edu.
Rajanikanth J Maganti, Email: rmaganti@vet.ksu.edu.
Sherry Fleming, Email: sdflemin@ksu.edu.
Susan M Wall, Email: smwall@emory.edu.
Lorraine A Everett, Email: wange@vet.ksu.edu.
Eric D Green, Email: egreen@nhgri.nih.gov.
Philine Wangemann, Email: wange@vet.ksu.edu.
References
- Pendred V. Deaf-mutism and goitre. Lancet. 1896;11:532. doi: 10.1016/S0140-6736(01)74403-0. [DOI] [Google Scholar]
- Reardon W, OMahoney CF, Trembath R, Jan H, Phelps PD. Enlarged vestibular aqueduct: a radiological marker of pendred syndrome, and mutation of the PDS gene. QJM. 2000;93:99–104. doi: 10.1093/qjmed/93.2.99. [DOI] [PubMed] [Google Scholar]
- Fraser GR. Association of congenital deafness with goitre (Pendred's syndrome). A study of 207 families. Ann Hum Genet. 1965;28:201–249. doi: 10.1111/j.1469-1809.1964.tb00479.x. [DOI] [PubMed] [Google Scholar]
- Everett LA, Glaser B, Beck JC, Idol JR, Buchs A, Heyman M, Adawi F, Hazani E, Nassir E, Baxevanis AD, Sheffield VC, Green ED. Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS) Nat Genet. 1997;17:411–422. doi: 10.1038/ng1297-411. [DOI] [PubMed] [Google Scholar]
- Blons H, Feldmann D, Duval V, Messaz O, Denoyelle F, Loundon N, Sergout-Allaoui A, Houang M, Duriez F, Lacombe D, Delobel B, Leman J, Catros H, Journel H, Drouin-Garraud V, Obstoy MF, Toutain A, Oden S, Toublanc JE, Couderc R, Petit C, Garabedian EN, Marlin S. Screening of SLC26A4 (PDS) gene in Pendred's syndrome: a large spectrum of mutations in France and phenotypic heterogeneity. Clin Genet. 2004;66:333–340. doi: 10.1111/j.1399-0004.2004.00296.x. [DOI] [PubMed] [Google Scholar]
- Bidart JM, Mian C, Lazar V, Russo D, Filetti S, Caillou B, Schlumberger M. Expression of pendrin and the Pendred syndrome (PDS) gene in human thyroid tissues. J Clin Endocrinol Metab. 2000;85:2028–2033. doi: 10.1210/jc.85.5.2028. [DOI] [PubMed] [Google Scholar]
- Royaux IE, Suzuki K, Mori A, Katoh R, Everett LA, Kohn LD, Green ED. Pendrin, the protein encoded by the Pendred syndrome gene (PDS), is an apical porter of iodide in the thyroid and is regulated by thyroglobulin in FRTL-5 cells. Endocrinology. 2000;141:839–845. doi: 10.1210/en.141.2.839. [DOI] [PubMed] [Google Scholar]
- Scott DA, Karniski LP. Human pendrin expressed in Xenopus laevis oocytes mediates chloride/formate exchange. Am J Physiol Cell Physiol. 2000;278:C207–C211. doi: 10.1152/ajpcell.2000.278.1.C207. [DOI] [PubMed] [Google Scholar]
- Scott DA, Wang R, Kreman TM, Sheffield VC, Karniski LP. The Pendred syndrome gene encodes a chloride-iodide transport protein. Nat Genet. 1999;21:440–443. doi: 10.1038/7783. [DOI] [PubMed] [Google Scholar]
- Chambard JM, Ashmore JF. Sugar transport by mammalian members of the SLC26 superfamily of anion-bicarbonate exchangers. J Physiol. 2003;550:667–677. doi: 10.1113/jphysiol.2003.039321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett LA, Morsli H, Wu DK, Green ED. Expression pattern of the mouse ortholog of the Pendred's syndrome gene (Pds) suggests a key role for pendrin in the inner ear. Proc Natl Acad Sci USA. 1999;96:9727–9732. doi: 10.1073/pnas.96.17.9727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacroix L, Mian C, Caillou B, Talbot M, Filetti S, Schlumberger M, Bidart JM. Na(+)/I(-) symporter and Pendred syndrome gene and protein expressions in human extra-thyroidal tissues. Eur J Endocrinol. 2001;144:297–302. doi: 10.1530/eje.0.1440297. [DOI] [PubMed] [Google Scholar]
- Rillema JA, Hill MA. Prolactin regulation of the pendrin-iodide transporter in the mammary gland. Am J Physiol Endocrinol Metab. 2003;284:E25–E28. doi: 10.1152/ajpendo.00383.2002. [DOI] [PubMed] [Google Scholar]
- Royaux IE, Wall SM, Karniski LP, Everett LA, Suzuki K, Knepper MA, Green ED. Pendrin, encoded by the Pendred syndrome gene, resides in the apical region of renal intercalated cells and mediates bicarbonate secretion. Proc Natl Acad Sci USA. 2001;98:4221–4226. doi: 10.1073/pnas.071516798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royaux IE, Belyantseva IA, Wu T, Kachar B, Everett LA, Marcus DC, Green ED. Localization and functional studies of pendrin in the mouse inner ear provide insight about the etiology of deafness in pendred syndrome. J Assoc Res Otolaryngol. 2003;4:394–404. doi: 10.1007/s10162-002-3052-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Royaux IE, Everett LA, Mori-Aoki A, Suzuki S, Nakamura K, Sakai T, Katoh R, Toda S, Green ED, Kohn LD. Expression of PDS/Pds, the Pendred syndrome gene, in endometrium. J Clin Endocrinol Metab. 2002;87:938. doi: 10.1210/jc.87.2.938. [DOI] [PubMed] [Google Scholar]
- Wangemann P, Itza EM, Albrecht B, Wu T, Jabba SV, Maganti RJ, Lee JH, Everett LA, Wall SM, Royaux IE, Green ED, Marcus DC. Loss of KCNJ10 protein expression abolishes endocochlear potential and causes deafness in Pendred syndrome mouse model. BMC Med. 2004;2:30. doi: 10.1186/1741-7015-2-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall SM, Hassell KA, Royaux IE, Green ED, Chang JY, Shipley GL, Verlander JW. Localization of pendrin in mouse kidney. Am J Physiol Renal Physiol. 2003;284:F229–F241. doi: 10.1152/ajprenal.00147.2002. [DOI] [PubMed] [Google Scholar]
- Kim YH, Verlander JW, Matthews SW, Kurtz I, Shin W, Weiner ID, Everett LA, Green ED, Nielsen S, Wall SM. Intercalated cell H+/OH- transporter expression is reduced in Slc26a4 null mice. Am J Physiol Renal Physiol. 2005;289:F1262–F1272. doi: 10.1152/ajprenal.00206.2005. [DOI] [PubMed] [Google Scholar]
- Everett LA, Belyantseva IA, Noben-Trauth K, Cantos R, Chen A, Thakkar SI, Hoogstraten-Miller SL, Kachar B, Wu DK, Green ED. Targeted disruption of mouse Pds provides insight about the inner-ear defects encountered in Pendred syndrome. Hum Mol Genet. 2001;10:153–161. doi: 10.1093/hmg/10.2.153. [DOI] [PubMed] [Google Scholar]
- Ramakers C, Ruijter JM, Deprez RH, Moorman AF. Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neurosci Lett. 2003;339:62–66. doi: 10.1016/S0304-3940(02)01423-4. [DOI] [PubMed] [Google Scholar]
- Vetter DE, Mann JR, Wangemann P, Liu Z, McLaughlin KJ, Lesage F, Marcus DC, Lazdunski M, Heinemann SF, Barhanin J. Inner ear defects induced by null mutation of isk gene. Neuron. 1996;17:1251–1264. doi: 10.1016/S0896-6273(00)80255-X. [DOI] [PubMed] [Google Scholar]
- Wangemann P, Liu J, Marcus DC. Ion transport mechanisms responsible for K+ secretion and the transepithelial voltage across marginal cells of stria vascularis in vitro. Hear Res. 1995;84:19–29. doi: 10.1016/0378-5955(95)00009-S. [DOI] [PubMed] [Google Scholar]
- El Amraoui A, Cohen-Salmon M, Petit C, Simmler MC. Spatiotemporal expression of otogelin in the developing and adult mouse inner ear. Hear Res. 2001;158:151–159. doi: 10.1016/S0378-5955(01)00312-4. [DOI] [PubMed] [Google Scholar]
- Fujiwara N, Kobayashi K. Macrophages in inflammation. Curr Drug Targets Inflamm Allergy. 2005;4:281–286. doi: 10.2174/1568010054022024. [DOI] [PubMed] [Google Scholar]
- Solanes N, Rigol M, Ramirez J, Segales J, Roque M, Marimon JM, Perez-Villa F, Roig E, Sanz G, Heras M. Histological basis of the porcine femoral artery for vascular research. Anat Histol Embryol. 2005;34:105–111. doi: 10.1111/j.1439-0264.2004.00580.x. [DOI] [PubMed] [Google Scholar]
- Goldsby R, Kindt T, Osborne B. Immunology. 4. New York: W.H. Freeman & Company; 2000. [Google Scholar]
- Henderson RB, Hobbs JA, Mathies M, Hogg N. Rapid recruitment of inflammatory monocytes is independent of neutrophil migration. Blood. 2003;102:328–335. doi: 10.1182/blood-2002-10-3228. [DOI] [PubMed] [Google Scholar]
- Hirose K, Discolo CM, Keasler JR, Ransohoff R. Mononuclear phagocytes migrate into the murine cochlea after acoustic trauma. J Comp Neurol. 2005;489:180–194. doi: 10.1002/cne.20619. [DOI] [PubMed] [Google Scholar]
- Hashimoto S, Billings P, Harris JP, Firestein GS, Keithley EM. Innate immunity contributes to cochlear adaptive immune responses. Audiol Neurootol. 2005;10:35–43. doi: 10.1159/000082306. [DOI] [PubMed] [Google Scholar]