

Abstract
We define here a new mechanism through which Mdm2 (mouse double minute 2) regulates p53 activity, by targeting the p53 transcription cofactor JMY. DNA damage causes an increase in JMY protein, and, in a similar manner, small molecule inhibitors of Mdm2 activity induce JMY in unperturbed cells. At a mechanistic level, Mdm2 regulation of JMY requires the Mdm2 RING (really interesting new gene) finger, which promotes the ubiquitin-dependent degradation of JMY. However, regulation of JMY occurs independently of the p53-binding domain in Mdm2 and p53 activity. These results define a new functional relationship between the p53 cofactor JMY and Mdm2, and indicate that transcription cofactors that facilitate p53 activity are important targets for Mdm2 in suppressing the p53 response.
Introduction
The p53 tumour suppressor protein is a transcription factor that has a pivotal role in the prevention of malignant diseases. The ability of p53 to suppress tumorigenesis is mediated through its response to cellular stress, which culminates in cell-cycle arrest, apoptosis or DNA repair (Levine, 1997). In normal cells, p53 is maintained at low levels in a latent form; however, cellular stress results in the rapid stabilization of p53, which then mediates the p53 response through activation of a wide group of target genes (Vousden & Lu, 2002). The importance of the tumour suppressor function of p53 is highlighted by the fact that mutation of the p53 gene is one of the most common aberrations observed in human tumour cells (Hollstein et al, 1994).
In addition to genes involved in cell-cycle arrest and apoptosis, p53 activates genes that are required for its own regulation, the most important of these being mdm2 (mouse double minute 2; Barak et al, 1993). The Mdm2 protein is a key regulator of p53, which targets p53 at multiple levels, including transcriptional activation, stability and subcellular localization (Freedman et al, 1999). Mdm2 promotes the ubiquitination of p53 and then targets it for degradation by the 26S proteasome (Haupt et al, 1997; Kubbutat et al, 1997; Fang et al, 2000), and phosphorylation of certain amino-acid residues in p53 has been correlated with increased stability of p53 and attenuated interaction with Mdm2 (Alarcon-Vargas & Ronai, 2002). Furthermore, p53 ubiquitination has been linked to regulation of its localization; the RING (really interesting new gene) finger domain of Mdm2, which harbours its ubiquitin ligase activity, is essential to relocalize p53 to the cytoplasm (Boyd et al, 2000; Geyer et al, 2000). Thus, ubiquitination-deficient p53 mutants are resistant to Mdm2-mediated nuclear export (Lohrum et al, 2001), suggesting that ubiquitination of p53 can affect its cellular localization and enhance its export.
Another important regulator of p53 activity is the p300/CBP (CREB-binding protein) family of proteins, which act as coactivators for a diverse number of transcription factors including p53 (Chan & La Thangue, 2001). In regulating p53 transcription, p300/CBP proteins form multi-component complexes with both positive and negative regulators of p53 activity, including Mdm2 (Grossman et al, 1998). Mdm2 binds to p300, and Mdm2 mutants that are defective in p300 binding lose their ability to degrade p53 (Zhu et al, 2001), suggesting that p300 is also important in mediating the negative effects of Mdm2. It is noteworthy that in addition to the ubiquitin ligase activity in Mdm2, p300 has a ubiquitin ligase that targets p53 (Grossman et al, 2003).
p300 binds to various cofactors that enhance the p53 response (Coutts & La Thangue, 2005). Two such cofactors, JMY and Strap, are required for p53 activity (Shikama et al, 1999; Demonacos et al, 2001, 2004). JMY and p300 cooperate in augmenting the p53 response and both are recruited to p53 in a protein complex in response to DNA damage (Shikama et al, 1999).
In this study, we considered the possibility that, in addition to p53, Mdm2 regulates cofactors that contribute to p53 activity. In this regard, we find that DNA damage causes JMY to accumulate, and further small molecule inhibitors of Mdm2 induce JMY protein. Furthermore, we show that Mdm2 downregulates JMY activity through a ubiquitin-dependent pathway. This requires the integrity of the Mdm2 RING finger domain, but occurs independently of the p53-binding domain and p53 activity. These results indicate that JMY is regulated by Mdm2 and, more generally, that transcription cofactors that facilitate p53 activity are likely to be important targets for Mdm2 in overcoming the p53 response.
Results And Discussion
JMY accumulates in DNA-damaged cells
We assessed whether JMY was regulated in cells treated with DNA-damaging agents. Under conditions of DNA damage that activated p53, we observed a concomitant increase in JMY levels (Fig 1A,B). We assessed a variety of damaging agents, including ultraviolet light, etoposide and actinomycin D, all of which caused the induction of JMY (Fig 1A–D). The results indicate that JMY is a DNA-damage responsive protein.
Figure 1.
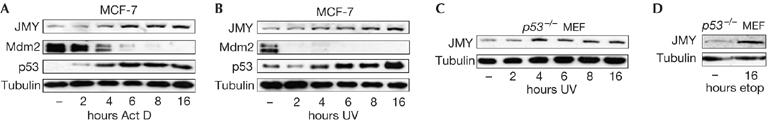
JMY is a DNA-damage responsive protein. (A–D) Michigan Cancer Foundation-7 (MCF-7) or p53−/− mouse embryonic fibroblasts (MEFs) were treated with actinomycin D (Act D; 20 nM), ultraviolet (UV) light (50 J/m2), or etoposide (etop; 10 μM). Cell extracts were collected at the indicated time points and whole-cell extracts were run on 10% SDS–polyacrylamide gel electrophoresis. Endogenous JMY was detected using goat JMY antibody L-16.
Mdm2 regulates the stability of p53 during the DNA damage response (Freedman et al, 1999; Ashcroft et al, 2000); therefore, we reasoned that it might have a similar role in controlling JMY. We assessed the effect of two small molecule inhibitors of Mdm2 activity on JMY levels (Lai et al, 2002; Vassilev et al, 2004). In cells treated with either nutlin-3 or an inhibitor of Mdm2 E3 ligase, there was a specific and significant increase in JMY levels (three- and twofold, respectively; Fig 2A,B), suggesting that Mdm2 negatively regulates JMY activity in unperturbed cells. Nutlin-3 increased JMY levels in a time-dependent manner, with the most noticeable increases observed after 3 h of treatment (Fig 2C). This effect was not due to increases in p53 observed after nutlin-3 treatment (Fig 2C), as similar results were seen in p53−/− mouse embryonic fibroblasts (MEFs; Fig 2D; supplementary Fig 1A online). Furthermore, in p53−/−/Mdm2−/− MEFs, JMY levels were not increased after treatment with either nutlin-3 or the Mdm2 E3 ligase inhibitor (Fig 2A), suggesting that this effect is related to the presence of Mdm2. Consequently, we found that Mdm2 and JMY occurred in the same immunoprecipitate when either Mdm2 or JMY antibodies were used (Fig 2E). These results indicate that Mdm2 negatively regulates and interacts with JMY under physiological conditions.
Figure 2.
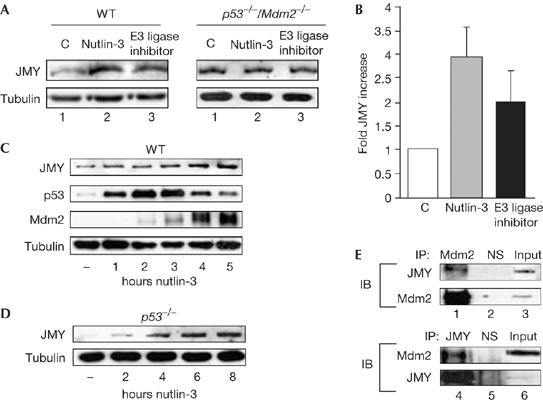
JMY is regulated by mouse double minute 2. (A) Wild-type (WT) and p53−/−/Mdm2−/− mouse embryonic fibroblasts (MEFs) were treated as described with either nutlin-3 (10 μM) or Mdm2 E3 ligase inhibitor (10 μM) for 24 h and cell extracts were immunoblotted with JMY (upper panel) or tubulin (lower panel) antibodies. (B) The relative induction of JMY in WT MEFs compared with untreated cells (n=3, Mdm2 E3 ligase inhibitor; or n=4, nutlin-3; independent experiments). (C) WT MEFs were treated with 10 μM nutlin-3 for the times indicated. Endogenous JMY was detected using goat JMY antibody L-16. (D) p53−/− MEFs were treated with 10 μM nutlin-3 for the times indicated. Endogenous JMY was detected using goat JMY antibody L-16 and tubulin was used as a loading control. (E) Upper panels: endogenous Mdm2 was immunoprecipitated from whole-cell extracts prepared from monocyte chemoattractant factor-7 (MCF-7) cells by using mouse Mdm2 antibody SMP14 (lane 1). Mouse IgG was used as a nonspecific (NS) control (lane 2) and 2% of the extract was loaded in lane 3. Endogenous JMY was detected using goat JMY antibody L-16. Lower panels: endogenous JMY was immunoprecipitated from whole-cell extracts prepared from MCF-7 cells by using goat JMY antibody L-16 (lane 4). Goat IgG was used as an NS control (lane 5) and 2% of the extract was loaded in lane 6. IB, immunoblotting; IP, immunoprecipitation; Mdm2, mouse double minute 2.
Mdm2 regulates JMY stability
As Mdm2 targets p53 for degradation (Haupt et al, 1997; Kubbutat et al, 1997; Fang et al, 2000), the interaction between Mdm2 and JMY might alter the levels and stability of JMY. First, we assessed whether Mdm2 altered the subcellular level of JMY in p53−/−/Mdm2−/− MEFs; this allowed us to rule out any contribution from endogenous p53 and Mdm2 to JMY activity. JMY is principally localized to the nucleus (Fig 3A) and when Mdm2 was expressed in p53−/−/Mdm2−/− MEFs we observed the downregulation of JMY (Fig 3B–E). This effect was specific to Mdm2 as the expression of green fluorescent protein (GFP), which also accumulated in nuclei, failed to affect the level of JMY (Fig 3B,C).
Figure 3.
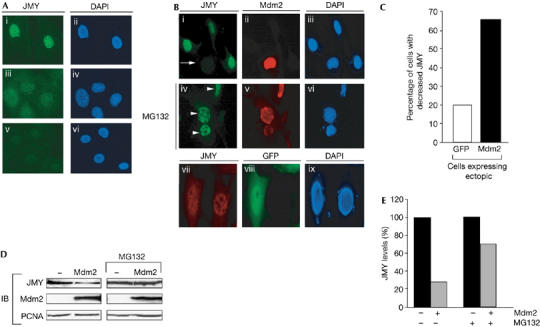
Mouse double minute 2 reduces the level of nuclear JMY. (A) Endogenous JMY was detected in p53−/−/Mdm2−/− mouse embryonic fibroblasts (MEFs) using the JMY peptide antibody 1289 (i). Specific JMY staining was competed using the JMY peptide used to generate rabbit anti-serum (v). 4,6-Diamidino-2-phenylindole (DAPI) staining was used to visualize nuclei (ii, iv, vi). Nonspecific staining was detected using normal rabbit serum (iii). (B) p53−/−/Mdm2−/− MEFs were transfected with pCHDMIA encoding human Mdm2 and processed for immunofluorescence after 24 h. Endogenous JMY was detected using the rabbit anti-JMY antiserum 1289 (i, iv, vii). Exogenous human Mdm2 was detected using the mouse Mdm2 antibody SMP14 (ii, v). DAPI staining was performed to visualize nuclei (iii, vi, ix). The arrow indicates cells that express exogenous Mdm2 and show reduced levels of endogenous JMY. Where indicated (middle row; iv, v, vi), cells were treated with 10 μM MG132 for 8 h before fixation. Arrowheads indicate cells that express exogenous Mdm2 but do not show reduced JMY. As a control treatment, cells were transfected with pEGFP-C1 encoding green fluorescent protein (GFP; vii, viii, ix) and the level of endogenous JMY was monitored (vii). (C) pCHDMIA (expressing Mdm2) or pEGFP-C1 expression vector (500 ng) was transfected into p53−/−/Mdm2−/− MEFs grown on glass coverslips. Cells were stained with rabbit anti-JMY antiserum 1289 and Mdm2 antibody SMP14 or rabbit anti-JMY antiserum only for GFP-expressing cells. Fields of cells were compared for the level of JMY expression in the presence or absence of Mdm2 or GFP. The histogram represents the percentage of cells that had a decrease in JMY staining compared with surrounding cells. Approximately 50 cells were counted for each treatment. (D) p53−/−/Mdm2−/− MEFs were transfected with pCHDMIA encoding Mdm2 or an empty vector together with MG132 (10 μM) as indicated for 8 h before collecting. Endogenous JMY was detected using goat JMY antibody L-16 and Mdm2 was detected using mouse Mdm2 antibody SMP14. Proliferating-cell nuclear antigen (PCNA) was used as a loading control. (E) The percentage change in JMY levels compared with control treatments. IB, immunoblotting; Mdm2, mouse double minute 2.
Second, as the Mdm2 E3 ligase is involved in regulating JMY levels (Fig 2A), and E3 ligase ubiquitinates and promotes degradation by means of proteasomes (Ardley & Robinson, 2005), we investigated whether a similar mechanism was involved in JMY control. In the presence of MG132, Mdm2 failed to cause a reduction in the level of nuclear JMY, in contrast to the effect of wild-type Mdm2 in the absence of MG132 (Fig 3B). Similarly, immunoblotting showed a reduced effect of Mdm2 on JMY in cells treated with MG132 (Fig 3D,E).
Mdm2 regulates JMY ubiquitination
To clarify the role of Mdm2, we studied the level of JMY in wild-type MEFs compared with p53−/−/Mdm2−/− MEFs. The constitutive level of JMY was considerably higher in p53−/−/Mdm2−/− MEFs compared with their wild-type counterparts, with p53−/−/Mdm2−/− MEFs showing a 2.8-fold increase (Fig 4A,B). As p53 does not have any direct effect on the level of JMY (Shikama et al, 1999; Fig 1C,D), these results are in accordance with the earlier results that Mdm2 is involved in the downregulation of JMY. Furthermore, when the half-life of JMY was studied in wild-type MEFs and compared with p53−/−/Mdm2−/− cells, the half-life of JMY was considerably shorter in the wild-type MEFs (Fig 4). Moreover, the half-life of JMY in p53−/− MEFs was similar to its half-life in wild-type MEFs (supplementary Fig 1B online).
Figure 4.
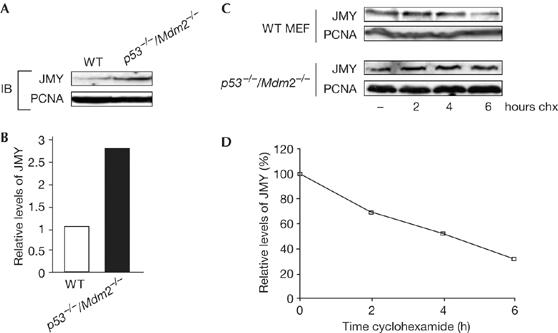
JMY is regulated by a proteasome-dependent pathway. (A) Endogenous JMY was detected in wild-type (WT) and p53−/−/Mdm2−/− mouse embryonic fibroblasts (MEFs) using the JMY L-16 antibody; proliferating-cell nuclear antigen (PCNA) was used as a loading control. (B) The fold increase in JMY level in p53−/−/Mdm2−/− compared with WT MEFs. (C) WT or p53−/−/Mdm2−/− MEFs were treated with 50 μg/ml cyclohexamide (chx) for the times indicated, and cell extracts immunoblotted with the JMY antibody L-16. PCNA was used as a loading control. (D) The relative JMY levels in WT MEFs as a percentage of total JMY in p53−/−/Mdm2−/− MEFs. IB, immunoblotting.
A prediction of the effect of MG132 on JMY protein level is that JMY is directly ubiquitinated. Indeed, JMY was found to be ubiquitinated when coexpressed with His6–ubiquitin (Fig 5A), a result consistent with the effect of MG132 in increasing the level of JMY observed earlier (Fig 3B,D). Coexpression of Mdm2 and JMY increased the polyubiquitination, as shown by the extensive series of slower mobility JMY species in the presence of Mdm2 (Fig 5B). Furthermore, this effect required the integrity of the carboxy-terminal domain, because coexpression of Mdm2 C464A with JMY resulted in lower levels of ubiquitinated derivatives. By contrast, the coexpression of Mdm2 Δ58–59 with JMY resulted in ubiquitinated JMY, with levels that were more similar to those seen in the presence of wild-type Mdm2 (Fig 5B). Similar results were seen in both U2OS cells and p53−/−/Mdm2−/− MEFs (Fig 5A,B; supplementary Fig 1C online). Combined with the earlier results on the effect of the Mdm2 E3 ligase inhibitor (Fig 2A), we have shown that the E3 ligase activity of Mdm2 is involved in regulating JMY ubiquitination.
Figure 5.
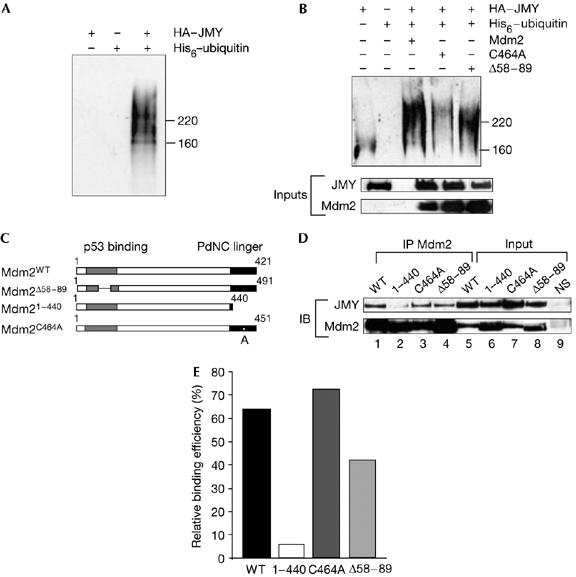
JMY is ubiquitinated. (A) U2OS cells were transfected with 1 μg each of haemagglutinin (HA)–JMY, His6–ubiquitin or HA–JMY and His6–ubiquitin as indicated. Cells were treated with MG132 (10 μM) 6 h before collection and isolation of His-tagged proteins was performed using Ni-NTA agarose. Elutes were run on 7.5% SDS–polyacrylamide gel electrophoresis (SDS-PAGE) and JMY was detected using the goat JMY L-16 antibody. (B) U2OS cells were transfected with 1 μg each of HA–JMY, His6–ubiquitin and Mdm2 (mouse double minute 2) and derivates as denoted. Cells were treated with MG132 (10 μM) 4 h before collection. His-tagged proteins were isolated using Ni-NTA agarose and eluates were run on 7.5% SDS–PAGE. JMY derivatives were detected using the goat JMY L-16 antibody. The lower panel represent 20% of inputs used in His-tagged protein isolation. Whole-cell extracts were run on 10% SDS–PAGE and HA–JMY was detected using HA11 and Mdm2 was detected using SMP14 antibodies. (C) Representation of human Mdm2 and mutant derivatives. (D) U2OS cells were transfected with expression vectors encoding JMY (1 μg) and Mdm2 (2 μg), 1–440 (1 μg), C464A (1 μg) or Δ58–89 (2 μg) to a total of 3.4 μg DNA with vector control, as indicated. Cells were collected after 36 h followed by immunoprecipitation (IP) with the Mdm2 SMP14 antibody (lanes 1–4). JMY was detected with HA antibody HA11 and Mdm2 with SMP14. Because of the increased stability of 1–440 and C464A (Kubbutat et al, 1999), input Mdm2 protein levels were normalized before immunoprecipitation and 10% of each extract is shown (lanes 5–8). Lane 9 shows a control immunoprecipitation performed with a nonspecific (NS) mouse antibody and immunoblotted (IB) with either anti-HA or SMP14. (E) Quantification of the results, shown in (D), for the interaction between JMY and the Mdm2 mutant derivatives. The level of immunoprecipitated JMY was calculated as a relative percentage of the immunoprecipitated Mdm2 derivative, which was given an arbitrary value of 100% throughout to minimize any differences resulting from expression levels.
We investigated the protein domains of Mdm2 involved in the interaction with JMY by studying the binding properties of a series of Mdm2 mutant derivatives (Fig 5C). As expected, full-length Mdm2 and JMY could bind to each other and, similarly, Mdm2 Δ58–89 immunoprecipitated with JMY (with about 30% reduced efficiency). Mdm2 C464A bound to JMY, whereas the C-terminal mutant 1–440, which lacks the RING finger, was much less efficient (Fig 5D,E).
Regulation during the DNA damage response
We considered that Mdm2 might influence apoptosis mediated through the activity of JMY and p53. To assess the importance of JMY in regulating apoptosis, we used short interfering RNA (siRNA) to knock down endogenous JMY (Fig 6A). When JMY siRNA was introduced into DNA-damaged cells and the level of apoptosis was monitored, there was a significant decrease in the proportion of apoptotic cells and a concomitant increase in G1 cells, suggesting that JMY augments apoptosis during the DNA damage response (Fig 6B).
Figure 6.
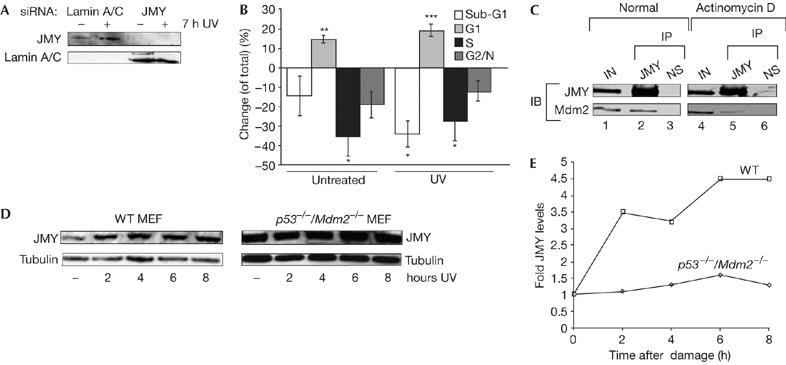
JMY regulation during the DNA damage response. (A) U2OS cells were treated with either JMY or lamin short interfering RNA (siRNA) as indicated. After 72 h, cells were collected followed by immunoblotting with goat JMY antibody L-16 or lamin antibody as indicated. (B) U2OS cells were treated as described with either JMY or lamin siRNA. Seven hours before the collection of cells, they were given either fresh medium or were treated with ultraviolet (UV) light (50 J/m2) and given fresh medium. Cells were analysed by flow cytometry (approximately 20,000 cells). The graph represents the change (expressed as %) in cell-cycle parameters relative to the control lamin siRNA-treated cells. Results represent mean±s.e.m. (n=4 independent experiments). Statistical analysis was performed using the Student's t-test: *P<0.05, **P<0.01, ***P<0.001. (C) U2OS cells were transfected with expression vectors encoding JMY (10 μg) and Mdm2 (10 μg). Cells were left untreated or were treated with actinomycin D (20 nM for 16 h). The cell extracts were immunoprecipitated (IP) with mouse haemagglutinin (HA) antibody (lanes 2, 5) or with mouse Gal4 antibody as a control (lanes 3, 6; NS). Immunoprecipitates were analysed for Mdm2 binding. JMY was detected with a rabbit HA antibody (Y-11) and Mdm2 with the rabbit H-221 antibody. Lanes 1 and 4 show the input (IN; 10% of cell extract). (D) Wild-type (WT) and p53−/−/Mdm2−/− mouse embryonic fibroblasts (MEFs) were treated with UV light (50 J/m2) for the times indicated. Cell extracts were collected and whole-cell extracts were run using 10% SDS–polyacrylamide gel electrophoresis. Endogenous JMY was detected using the goat JMY antibody L-16 and tubulin was used as a loading control. (E) The relative induction of JMY compared with untreated cells. Quantification was performed after normalizing JMY levels compared with loading controls. IB, immunoblotting.
Given the role of JMY in promoting apoptosis, and because the p53/Mdm2 interaction is regulated by the DNA damage response (Alarcon-Vargas & Ronai, 2002), we reasoned that the control of JMY by Mdm2 might be similarly regulated during the DNA damage response. Thus, a JMY–Mdm2 complex was present in cells under unperturbed culture conditions, whereas the amount of Mdm2 bound to JMY was very much reduced in DNA-damaged cells (40% of that present in unperturbed cells; Fig 6C). These results indicate that the interaction between JMY and Mdm2 is regulated under DNA damage conditions, which, furthermore, correlates with the DNA damage induction of JMY (Fig 1). In support of this, JMY levels were induced during damage in wild-type, but not p53−/−/Mdm2−/−, MEFs (Fig 6D,E).
Finally, we studied the effect of Mdm2 on JMY-induced apoptosis. When JMY was introduced into U2OS cells (p53+/+), there was, as expected from previous studies (Fig 6B; Shikama et al, 1999), an increase in the proportion of sub-G1 apoptotic cells (Fig 7A,B). Coexpressing Mdm2 caused a significant reduction in the level of apoptosis, approaching that seen in the absence of JMY (Fig 7A,B). Moreover, the integrity of the C-terminal region, but not the amino-terminal region, was required for Mdm2 to reduce apoptosis because Δ58–89 behaved in a similar manner to wild-type Mdm2, whereas 1–440 and C464A had a minimal effect (Fig 7A,B; supplementary Fig 1D online). These results therefore indicate that there is a functional relationship between JMY and Mdm2 in the control of apoptosis.
Figure 7.
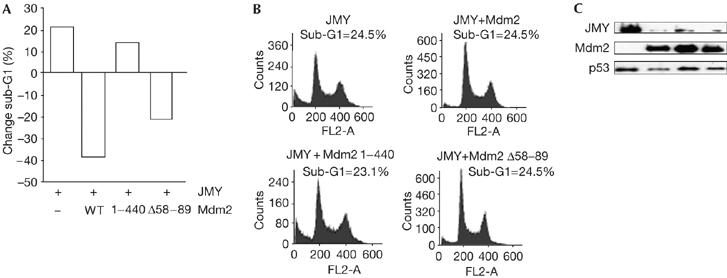
Mouse double minute 2 regulates the functional properties of JMY. (A) U2OS cells were transfected with expression vectors encoding JMY together with expression vectors encoding mouse double minute 2 (Mdm2), Δ58–89 or 1–440, and collected after 48 h. The cell-cycle profile of approximately 10,000 cells was analysed by flow cytometry and the graph represents the relative change (expressed as a %) in sub-G1 cells relative to the control vector treatment. The graphs in (B) show representative fluorescence-activated cell sorting profiles with the total percentage of sub-G1 cells shown for each treatment. The panels in (C) show protein levels for each treatment. Ectopic JMY was detected using HA11, ectopic Mdm2 with SMP14 and endogenous p53 with DO-1. WT, wild type.
Mdm2 effect on JMY is ubiquitin-dependent
Although the inhibitory effect of Mdm2 on p53 is well documented (Haupt et al, 1997), our results show that Mdm2 targets proteins involved in the damage response other than p53. Specifically, Mdm2 downregulates the level of JMY protein, which coincides with reduction in JMY-mediated apoptosis. Most importantly, Mdm2 forms a physical complex with JMY and it is likely that the physical interaction between JMY and Mdm2 is involved in Mdm2-dependent control of JMY, although we cannot rule out a role for other, yet to be defined, pathways. The C-terminal region of Mdm2 is necessary for the interaction with JMY; this same domain is required for Mdm2-mediated downregulation of p53 and Mdm2 oncogenic activity (Haupt et al, 1997).
The effect of MG132 on the levels of JMY, the requirement for Mdm2 E3 ligase activity and the increased half-life of JMY in p53−/−/Mdm2−/− cells suggest that Mdm2 targets JMY to a ubiquitin-dependent proteasome degradation pathway. The results highlight many striking similarities in the regulation of JMY and p53 by Mdm2 and suggest, at a more general level, that the RING finger might influence various targets in mediating the effects of Mdm2. In this respect, it will be interesting to explore the role of Mdm2 in regulating other cofactors involved in p53 transcriptional control.
In conclusion, our results define a new functional relationship between JMY and Mdm2, and indicate that JMY provides a significant level of regulation in the p53 response and perhaps an important pathway through which the oncogenic activity of Mdm2 is exerted. More generally, the control of JMY by Mdm2 suggests that diverse points of control, specifically transcription cofactors, are targets for Mdm2 in regulating the p53 response.
Methods
Plasmids and reagents. The following plasmids have previously been described: pcDNA3 HAJMY (Shikama et al, 1999), pCHDMIA, pCHDMΔ58–89, pCHDM C464A, pCHDM1–440 (Chen et al, 1996). pEGFP-C1 was obtained from BD Biosciences (Oxford, UK). His6–ubiquitin was a gift from Dr D. Xirodimas and Dr R. Hay. Nutlin-3 and Hdm2 E3 ligase inhibitor were obtained from Calbiochem (Nottingham, UK).
Antibodies. The following antibodies were used: mouse haemagglutinin (HA)11 anti-HA (Babco, Richmond, CA, USA), rabbit Y-11 anti-HA, mouse SMP14 anti-Mdm2, rabbit H-221 anti-Mdm2, goat L-16 anti-JMY, goat anti-β-tubulin, mouse DO-1 anti-p53, proliferating-cell nuclear antigen, mouse anti-lamin A/C (Santa Cruz, Santa Cruz, CA, USA) and mouse 1C12 anti-p53 (New England Biolabs, Hertfordshire, UK). Rabbit anti-JMY peptide antiserum (antibody 1289) was generated using a peptide corresponding to amino acids 946–968 of mouse JMY (Eurogentec, Seraing, Belgium). For secondary antibodies, alkaline phosphatase-conjugated (Promega, Southampton, UK) or horseradish peroxidase-conjugated (Calbiochem; Dako, Cambridgeshire, UK) anti-immunoglobulin were used. See the supplementary information online for further details.
Supplementary information is available at EMBO reports online (http://www.emboreports.org).
Supplementary Material
supplementary Figure
supplementary Figure Legends and Information
Acknowledgments
We thank M. Caldwell and R. Williams for help in preparation of the manuscript, and M.R.C., C.R.U.K., E.U., L.R.F. and A.I.C.R. for support. H.B. gratefully acknowledges support from the Algerian Government.
References
- Alarcon-Vargas D, Ronai Z (2002) p53–Mdm2—the affair that never ends. Carcinogenesis 23: 541–547 [DOI] [PubMed] [Google Scholar]
- Ardley HC, Robinson PA (2005) E3 ubiquitin ligases. Essays Biochem 41: 15–30 [DOI] [PubMed] [Google Scholar]
- Ashcroft M, Taya Y, Vousden KH (2000) Stress signals utilize multiple pathways to stabilize p53. Mol Cell Biol 20: 3224–3233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barak Y, Juven T, Haffner R, Oren M (1993) mdm2 expression is induced by wild type p53 activity. EMBO J 12: 461–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd SD, Tsai KY, Jacks T (2000) An intact HDM2 RING-finger domain is required for nuclear exclusion of p53. Nat Cell Biol 2: 563–568 [DOI] [PubMed] [Google Scholar]
- Chan HM, La Thangue NB (2001) p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J Cell Sci 114: 2363–2373 [DOI] [PubMed] [Google Scholar]
- Chen J, Wu X, Lin J, Levine AJ (1996) mdm-2 inhibits the G1 arrest and apoptosis functions of the p53 tumor suppressor protein. Mol Cell Biol 16: 2445–2452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutts AS, La Thangue NB (2005) The p53 response: emerging levels of co-factor complexity. Biochem Biophys Res Commun 331: 778–785 [DOI] [PubMed] [Google Scholar]
- Demonacos C, Krstic-Demonacos M, La Thangue NB (2001) A TPR motif cofactor contributes to p300 activity in the p53 response. Mol Cell 8: 71–84 [DOI] [PubMed] [Google Scholar]
- Demonacos C, Krstic-Demonacos M, Smith L, Xu D, O'Connor DP, Jansson M, La Thangue NB (2004) A new effector pathway links ATM kinase with the DNA damage response. Nat Cell Biol 6: 968–976 [DOI] [PubMed] [Google Scholar]
- Fang S, Jensen JP, Ludwig RL, Vousden KH, Weissman AM (2000) Mdm2 is a RING finger-dependent ubiquitin protein ligase for itself and p53. J Biol Chem 275: 8945–8951 [DOI] [PubMed] [Google Scholar]
- Freedman DA, Wu L, Levine AJ (1999) Functions of the MDM2 oncoprotein. Cell Mol Life Sci 55: 96–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geyer RK, Yu ZK, Maki CG (2000) The MDM2 RING-finger domain is required to promote p53 nuclear export. Nat Cell Biol 2: 569–573 [DOI] [PubMed] [Google Scholar]
- Grossman SR, Perez M, Kung AL, Joseph M, Mansur C, Xiao ZX, Kumar S, Howley PM, Livingston DM (1998) p300/MDM2 complexes participate in MDM2-mediated p53 degradation. Mol Cell 2: 405–415 [DOI] [PubMed] [Google Scholar]
- Grossman SR, Deato ME, Brignone C, Chan HM, Kung AL, Tagami H, Nakatani Y, Livingston DM (2003) Polyubiquitination of p53 by a ubiquitin ligase activity of p300. Science 300: 342–344 [DOI] [PubMed] [Google Scholar]
- Haupt Y, Maya R, Kazaz A, Oren M (1997) Mdm2 promotes the rapid degradation of p53. Nature 387: 296–299 [DOI] [PubMed] [Google Scholar]
- Hollstein M, Rice K, Greenblatt MS, Soussi T, Fuchs R, Sorlie T, Hovig E, Smith-Sorensen B, Montesano R, Harris CC (1994) Database of p53 gene somatic mutations in human tumors and cell lines. Nucleic Acids Res 22: 3551–3555 [PMC free article] [PubMed] [Google Scholar]
- Kubbutat MH, Jones SN, Vousden KH (1997) Regulation of p53 stability by Mdm2. Nature 387: 299–303 [DOI] [PubMed] [Google Scholar]
- Kubbutat MH, Ludwig RL, Levine AJ, Vousden KH (1999) Analysis of the degradation function of Mdm2. Cell Growth Differ 10: 87–92 [PubMed] [Google Scholar]
- Lai Z et al. (2002) Differentiation of Hdm2-mediated p53 ubiquitination and Hdm2 autoubiquitination activity by small molecular weight inhibitors. Proc Natl Acad Sci USA 99: 14734–14739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine AJ (1997) p53, the cellular gatekeeper for growth and division. Cell 88: 323–331 [DOI] [PubMed] [Google Scholar]
- Lohrum MA, Woods DB, Ludwig RL, Balint E, Vousden KH (2001) C-terminal ubiquitination of p53 contributes to nuclear export. Mol Cell Biol 21: 8521–8532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shikama N, Lee CW, France S, Delavaine L, Lyon J, Krstic-Demonacos M, La Thangue NB (1999) A novel cofactor for p300 that regulates the p53 response. Mol Cell 4: 365–376 [DOI] [PubMed] [Google Scholar]
- Vassilev LT et al. (2004) In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303: 844–848 [DOI] [PubMed] [Google Scholar]
- Vousden KH, Lu X (2002) Live or let die: the cell's response to p53. Nat Rev Cancer 2: 594–604 [DOI] [PubMed] [Google Scholar]
- Zhu Q, Yao J, Wani G, Wani MA, Wani AA (2001) Mdm2 mutant defective in binding p300 promotes ubiquitination but not degradation of p53: evidence for the role of p300 in integrating ubiquitination and proteolysis. J Biol Chem 276: 29695–29701 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
supplementary Figure
supplementary Figure Legends and Information