

Abstract
X inactivation is the mechanism by which mammals adjust the genetic imbalance that arises from the different numbers of gene-rich X-chromosomes between the sexes. The dosage difference between XX females and XY males is functionally equalized by silencing one of the two X chromosomes in females. This dosage-compensation mechanism seems to have arisen concurrently with early mammalian evolution and is based on the long functional Xist RNA, which is unique to placental mammals. It is likely that previously existing mechanisms for other cellular functions have been recruited and adapted for the evolution of X inactivation. Here, we critically review our understanding of dosage compensation in placental mammals and place these findings in the context of other cellular processes that intersect with mammalian dosage compensation.
Keywords: dosage compensation, Polycomb, X inactivation, Xist
Introduction
Sex determination in mammals is based on the heteromorphic sex chromosomes X and Y. It is believed that the X and Y chromosomes evolved from a pair of homologous autosomes. The evolutionary decay of genes on the Y chromosome results in a different number of gene-rich X-chromosomes being found in males and females, and, therefore, a large-scale genetic imbalance between the sexes (Graves, 2006). To equalize this dosage difference, an epigenetic compensatory system has evolved. Similar XY sex chromosome systems have arisen elsewhere in the animal kingdom; however, the mechanisms by which dosage compensation is achieved in flies and worms, for example (reviewed by Lucchesi et al, 2005), vary from the mammalian mode, which is the focus of this review.
In mammals, dosage compensation is achieved by inactivating one of the two X chromosomes in females. Haplo-insufficiency, resulting from the shut down of an entire chromosome, poses a potential problem; therefore, the presence of a single active X chromosome (Xa) must have conferred an evolutionary advantage and, hence, enabled its conservation in the process of natural selection. There is now evidence to support a second form of dosage compensation in mammals, which involves the upregulation of the Xa to balance the differential dosage between the X chromosome and the autosomes (Gupta et al, 2006; Nguyen & Disteche, 2006). So, what advantage did X inactivation confer and how did it arise in mammals?
Mammalian X inactivation seems to come in two forms: imprinted and random X inactivation. Imprinted X activation occurs in early mammals, such as marsupials, which diverged from placental mammals approximately 180 million years ago. This form of dosage compensation is achieved by inactivating the paternally inherited X chromosome. Imprinted X inactivation can also be found in the extra-embryonic tissues of a subset of placental mammals. The cells forming the embryo in placental mammals undergo random X inactivation, through which either the paternal or the maternal X chromosome is inactivated. This novel mechanism is based on a noncoding RNA called Xist, which is unique to placental mammals and has not been found in marsupials or other vertebrate genomes (Duret et al, 2006). The evolutionary force behind random X inactivation and the emergence of the regulating Xist RNA is unclear; however, it could have been a combination of environmental circumstances and the selection pressure for placental mammals.
It has been suggested that the expansion of placental mammals was a consequence of the change in oxygen levels over the past 200 million years (Falkowski et al, 2005). The placental system requires a high ambient oxygen concentration owing to its inefficient transport from the maternal circulation to the developing embryo. This system became more efficient with the rise in global oxygen levels and resulted in a greater reproductive demand on mammalian females. We speculate that as a consequence of this increased demand on females, the selection pressure for the evolution of advanced mammalian traits became limited to males. The erosion of the Y chromosome, and the consequent emergence of a single X in males, provided a haploid region of the genome that is subject to efficient and stringent selection. Consistent with this, genes that are expressed in brain, muscle and germ cells are enriched on the human X chromosome (Graves, 2006).
Hemizygosity of X-linked genes makes males more prone to the adverse effects of X-linked mutations (Franco & Ballabio, 2006). This situation also occurs in female marsupials, in which imprinted inactivation of the paternal X chromosome unmasks mutations on the maternal Xa. By contrast, random X inactivation in female placental mammals generates a mosaic of cells with either a paternal or maternal Xa, thereby masking the effect of deleterious mutations and increasing reproductive fitness (Franco & Ballabio, 2006). We therefore postulate that the benefit obtained in protecting the female against potentially deleterious mutations puts random X inactivation at a selective advantage and, hence, has become the mechanism of choice in placental mammals.
Random X-inactivation consists of an ordered series of processes. Each cell ensures, in a random manner, that only one X chromosome remains active and that the other X chromosome is inactivated. The differential treatment of the two X chromosomes results in an Xa and an inactive X (Xi), both of which are present in the same female nucleus. The Xist RNA, which is expressed exclusively from, and coats, the Xi (Fig 1), is used to establish the two functionally distinct forms of the X chromosomes. Although RNA components have been commonly used throughout evolution in the regulation of gene expression, Xist is unique in its ability to spread over and encompass the Xi. The proposed mechanisms that underlie Xist function are presented in this review. We also discuss how the Xi can be identified from its epigenetic marks, which represent a chromatin decoration feature that is commonly found in heterochromatic regions in the nucleus. Furthermore, we discuss the role of Polycomb group (PcG) proteins—notorious for maintaining transcriptional repression of developmental control genes in species ranging from flies to mammals—in the faithful maintenance of the Xi throughout cellular divisions.
Figure 1.
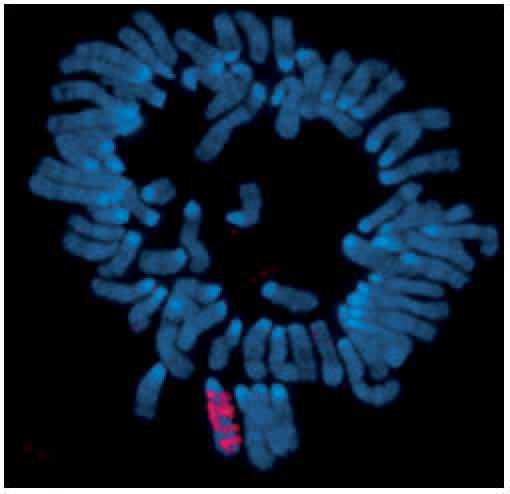
Xist RNA encompasses the X from which it is transcribed. RNA-fluorescence in situ hybridization detecting Xist RNA (red) localized on the inactive X in a preparation of condensed chromosomes from differentiated mouse cells. DNA is counterstained (blue).
From meiotic pairing to counting and choosing the X
Random inactivation poses an additional problem compared with other dosage-compensatory mechanisms because the two X chromosomes are present in different states—active and inactive—within the female nucleus. In imprinted X inactivation, the X chromosomes are distinguished by their parental origin. The inactive state of the paternal X chromosome has been proposed to be a carry-over effect from meiotic sex-chromosome inactivation in the male germline. Asynapsed chromosome segments, resulting from the non-homologous XY chromosome pair, are subject to transcriptional silencing in order for meiosis to proceed (Namekawa et al, 2006; Turner et al, 2006). In mice—in which random X inactivation occurs—a pre-inactivated paternal X arrives at the zygote, which is consistent with the idea of imprinted X inactivation being the ancestral form of random X inactivation (Huynh & Lee, 2003). This paternal Xi is subsequently reactivated in the embryo, followed by random X inactivation in a Xist-dependent manner (Mak et al, 2004; Okamoto et al, 2004).
At the onset of random X inactivation, the number of X chromosomes relative to the number of autosomes is counted, allowing one X to remain active per diploid chromosome set (reviewed by Heard & Disteche, 2006). Counting and choice are controlled by a single locus on the X, known as the X-inactivation centre (Xic), which contains multiple regulatory elements including the Xist gene (Fig 2A). Ectopic Xic transgenes can trigger the initiation of X inactivation in male cells (Chow et al, 2005; Herzing et al, 1997; Lee et al, 1996). The function of Xic has been studied extensively by using gene-targeting techniques in mice. A deletion spanning 65-kbp at the 3′ end of Xist leads to ectopic X inactivation in cells that have only one X chromosome (Clerc & Avner, 1998). Another choice regulator, Tsix RNA, overlaps with the Xist gene and is transcribed in the antisense orientation (Lee et al, 1999). Tsix is initially expressed on both X chromosomes and is downregulated on the Xi before inactivation; conversely, Tsix expression persists longer on the Xa. Heterozygous deletion of Tsix in female cells causes the deletion-bearing chromosome to be inactivated, suggesting that Tsix has a role in choice (Lee & Lu, 1999). Tsix is developmentally regulated by enhancers contained in the Xite and DXPas34 elements. Differential methylation of Xite and the CCCTC-binding factor (CTCF)-binding sites on DXPas34 correlate with X chromosome choice in mice (Boumil et al, 2006). The CTCF protein is associated with chromatin boundaries and has been proposed as a candidate factor involved in choice, although functional evidence is lacking (Chao et al, 2002). Consistent with this, deletion of DXPas34 results in ectopic X-inactivation (Vigneau et al, 2006).
Figure 2.
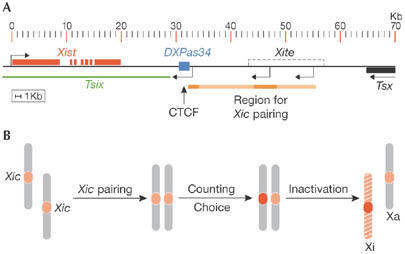
The X-inactivation centre regulates Xist expression to ensure that one X chromosome remains active. (A) Map of the regulatory elements implicated in counting and choice in the mouse Xic locus. The Xist gene, the antisense Tsix RNA, Xite, CCCTC-binding factor (CTCF)-binding sites at DXPas34 and the region implicated for Xic pairing are indicated. (B) Proposed scheme of counting and choice that involves pairing of the Xic loci at the initiation of X-inactivation. Homologous X-chromosomes within one nucleus are shown and the Xic is highlighted in red. Pairing of the Xic loci could activate Xist transcription on one chromosome, therefore enforcing the commitment to become silenced, while the other chromosome remains active. Xa, active X; Xi, inactive X; Xic, X-inactivation centre.
In male cells, Tsix is not required for silencing of the Xist promoter. The existence of a ‘competence factor', which is produced in cells that have more than one X chromosome and predisposes females to initiate X-inactivation by favouring Xist expression, has therefore been proposed (Lee & Lu, 1999). Consistent with this, an ectopic promoter inserted upstream of Xist has been shown to predispose the chromosome to be chosen for inactivation (Nesterova et al, 2003). Conversely, deletions within Xist also affect choice and cause inactivation of the other X chromosome (Marahrens et al, 1998). This indicates that Xist is regulated by positive signals and negative elements, which possibly form an epigenetic switch within Xic (Lee, 2005; Vigneau et al, 2006).
Recently, a model involving physical pairing of the Xic loci to mediate inter-chromosomal communication has received experimental support from an analysis of the nuclear position of the Xic loci in cells undergoing X inactivation (Marahrens, 1999; Bacher et al, 2006; Xu et al, 2006). The Xic loci come within close proximity of each other just at the time when X inactivation is initiated (Fig 2B). This implies that pairing of the Xic loci might provide the necessary communication needed to establish the differential treatment of the homologous X chromosomes. Remarkably, ectopic insertions of Xite and Tsix transgenes can initiate de novo pairing, suggesting a role for these regulatory elements in trans (Xu et al, 2006). Whether the mechanism behind Xic pairing is related to the meiotic pairing of homologous chromosomes is unknown. It is also interesting to note that imprinted X inactivation in the extra-embryonic lineages of some placental mammals depends on Xist (Okamoto et al, 2005). It is open to speculation whether Xist-dependent imprinted X inactivation could have arisen after the evolution of random X inactivation in placental mammals or is an evolutionarily older form.
Using the Xist RNA to shut down an X chromosome
Once the choice is made, expression of the long functional Xist RNA is upregulated on the X chromosome to be inactivated (Chow et al, 2005). Xist RNA molecules then accumulate over the chromosome and initiate silencing. Gene targeting in mice has shown that Xist is required for both imprinted and random X inactivation (Marahrens et al, 1997; Penny et al, 1996), and that ectopic Xist expression in the absence of other Xic sequences can initiate chromosome-wide silencing (Wutz & Jaenisch, 2000). However, initiation of silencing by Xist is restricted to the early stages of differentiation, implying that it is developmentally regulated. Initially, gene silencing is reversible and dependent on Xist; however, at a later stage in differentiation, X-inactivation becomes independent of Xist and irreversible (Csankovszki et al, 1999). Hence, Xist is crucial for initiating silencing, but has a minor role in maintaining the Xi.
Xist localization does not require X chromosome-specific sequences, and ectopic Xist expressed from autosomal transgenes can localize to autosomes and cause silencing, albeit to differing extents (Herzing et al, 1997; Lee et al, 1996; Wutz & Jaenisch, 2000). The observation that Xist spreading and the maintenance of silencing is less efficient outside the X chromosome instigated the idea of ‘way stations' or ‘boosters' at intervals along its length (Fig 3A). Owing to the high density of long interspersed elements (LINEs) on the X chromosome, these regions have been proposed as candidate boosters (Lyon, 2003). A recent study using cells with a t(X;4)37H X;autosome translocation showed that Xist spreading stops close to the X-autosomal translocation point (Popova et al, 2006) and correlates with a severe drop in LINE density on chromosome 4. However, the mechanism underlying the function of distinct booster elements needs to be clarified.
Figure 3.
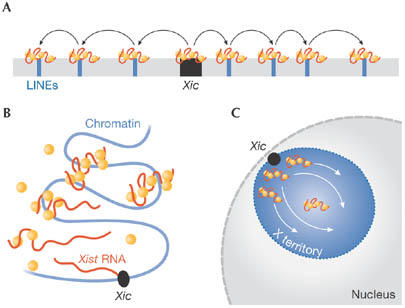
Models for Xist spreading along the chromosome in cis. Xist RNA (red) and protein factors postulated to bind Xist (orange) are shown. (A) Xist spreading along the chromosome by means of ‘way stations' or ‘boosters'. (B) Xist spreading based on a cooperative binding mechanism. Xist spreads in cis from its transcription site, where the high RNA concentration is predicted to nucleate chromatin attachment. (C) Schematic representation of the Xi territory with Xist spreading inward from the Xic to the centre of the chromosome territory. LINEs, long interspersed elements; Xic, X-inactivation centre.
The analysis of functional motifs within the mouse Xist RNA can provide insights into how Xist causes gene silencing. The association of Xist with the chromosome and its ability to trigger silencing are functionally separable (Wutz et al, 2002). Initiation of silencing depends on sequences at the 5′ end of Xist, which contains 7.5 repeats of a motif that is predicted to fold into an RNA structure comprising two stem loops. This structural motif is conserved among all placental mammals and might provide a binding platform for factors that act in gene repression. Xist RNA lacking this motif remains stable and accumulates within the chromosome territory, but no longer induces silencing. Hence, the ability to silence is not a prerequisite for Xist localization. The spreading of Xist RNA along the chromosome is mediated by functionally redundant sequences that are dispersed throughout the remainder of Xist and act synergistically (Wutz et al, 2002).
We have proposed a model based on the idea that these redundant sequences consist of several weak binding sites that facilitate binding to the chromosome and other factors in a cooperative manner. The binding of one factor allows easier binding of the next, resulting in a stable complex. This cooperative interaction drives the spread of Xist from its site of transcription, where a high concentration of Xist molecules is predicted to nucleate chromatin attachment (Fig 3B). Experimental evidence for the cooperative binding of Xist complexes is currently lacking. Nonetheless, this model is consistent with an added layer of complexity in an interphase nucleus. Considering that the Xic is positioned at the periphery of the chromosome (Chaumeil et al, 2006), Xist spreading must be directed towards the centre of the chromosomal territory (Fig 3C). Therefore, a spreading model based on the linear sequence of the X chromosome might be an oversimplification.
Xist is exceptional among cellular RNAs owing to its ability to encompass and silence an entire chromosome. Intriguingly, dosage compensation in flies involves the roX1 and roX2 RNAs, which target the dosage-compensation complex to the single male X and mediate transcriptional upregulation (Lucchesi et al, 2005). RNA has been recently shown to be involved in the activation of Polycomb-repressed genes in the fly (Sanchez-Elsner et al, 2006). It seems possible that such an RNA might have been adapted at the onset of mammalian evolution for the emergence of Xist, although no ancestor has been detected to date (Duret et al, 2006).
Organization of the chromosome territory of the Xi
One way to ensure the recognition of only the elected Xi, and no other chromosome, is to physically reorganize and decorate the chromatin with distinguishable marks. During differentiation, the Xi forms a condensed and highly compacted heterochromatic structure that is characterized by the absence of intronic RNA and RNA polymerase II (Okamoto et al, 2004). Nongenic sequences, such as centromeric and genomic repeats, have been detected within the territory of the Xi, whereas genes are predominantly found at the periphery (Clemson et al, 2006). Recently it has been shown that Xist creates a transcriptionally silent nuclear compartment, which consists of intergenic and repetitive DNA, and that the 5′ end of Xist RNA is required for the organization of repressed genes into the nongenic silent domain (Chaumeil et al, 2006).
Reorganization of the Xi territory is also reflected in the recruitment of factors and chromatin modifications (Table 1; Chow et al, 2005; Lucchesi et al, 2005). Interestingly, scaffold attachment factor-A (SAF-A), which is known to bind satellite DNA and scaffold attachment regions/matrix attachment regions (SARs/MARs), is enriched on the Xi (Fackelmayer, 2005). SARs and MARs have been implicated in higher-order chromatin organization. In the presence of nucleic acids, SAF-A proteins form multimers; this process is a prerequisite for their specific binding to SAR sequences and indicates a cooperative binding mechanism. Xist RNA and SAF-A are retained in nuclear matrix preparations after DNA and chromatin have been extracted (Fackelmayer, 2005). Intriguingly, both LINEs and MAR sequences are (A+T)-rich, and some MARs have been shown to overlap with LINE repeats. Together, this evidence suggests roles for SAF-A and the nuclear scaffold in X-inactivation.
Table 1.
Features of the inactive X territory
| Feature | Description |
|---|---|
| Xist RNA | Long functional RNA initiates silencing in cis |
| PcG proteins | PRC2 (Eed, Ezh2, Suz12) PRC1 (Bmi-1, Phc1, Phc2, Cbx2, Cbx7, Ring1a, Ring1b) |
| Modifications | Histone H3 Lys27 trimethylation (by PRC2) Histone H2A Lys119 ubiquitination (by PRC1) Histone H4 Lys20 monomethylation Histone H3 Lys9 dimethylation Histone H4 hypoacetylation |
| Histone variants | MacroH2A Histone H1 |
| Other proteins | SAF-A (nuclear matrix-associated) BRCA1 |
| Features | Exclusion of RNA polymerase II Late replication Exclusion of histone H2ABbd |
BRCA1, breast cancer 1; PcG, Polycomb group proteins; PRC, Polycomb-repressive complex; SAF-A, scaffold attachment factor-A.
Finally, the Xi might be not only spatially but also temporally separated from other chromosomes. Replication asynchrony of the Xi relative to other chromosomes is observed with the Xi replicating early in S-phase in preimplantation embryos and replicating late in embryonic cells after implantation (reviewed in Chow et al, 2005). Late replication of the Xi is observed after the onset of embryonic stem cell differentiation, but not in Xist-expressing undifferentiated embryonic stem cells (Wutz & Jaenisch, 2000). Full reorganization of the chromatin of the Xi therefore takes place during cellular differentiation.
Setting up stable silencing with Polycomb
Once the silent state of the Xi is created in early development, the repressed status is maintained throughout subsequent cell divisions. Xist only has a minor role in the maintenance of the Xi, whereas multiple epigenetic marks, including DNA methylation, late replication and hypo-acetylation of histone H4, act synergistically in the maintenance of X-inactivation (Fig 4A; Lucchesi et al, 2005). The mechanism that underlies the transition between the initiation and maintenance phases is poorly understood, although recent findings suggest the involvement of PcG proteins in this process.
Figure 4.
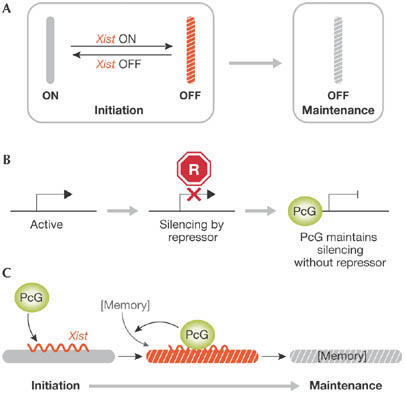
Establishing a stable inactive X for the maintenance of X inactivation. (A) Xist triggers reversible chromosomal silencing in the initiation phase of X-inactivation. In differentiated cells, the silent state no longer depends on Xist and is irreversible. (B) A model of Polycomb group (PcG) protein action to maintain repressed states of developmental control genes. A repressor (red) acts transiently and shuts down gene expression. PcG proteins bind and maintain the repression independent of the repressor. (C) A model depicting the establishment of a chromosomal memory by PcG complexes in the initiation phase of X inactivation. The memory could either be a factor, a chromatin modification or a structural change on the chromosome. Xi, inactive X.
PcG proteins are conserved from flies to mammals and maintain transcriptional repression of developmental control genes (Fig 4B; Bantignies & Cavalli, 2006; Ringrose & Paro, 2004). Two biochemically distinct PcG protein complexes—Polycomb-repressive complex 1 (PRC1) and PRC2—have catalytic activity. PRC1 and PRC2 mediate histone H2A Lys119 ubiquitylation (H2AK119ub1) and histone H3 Lys27 trimethylation (H3K27me3), respectively. As H3K27me3 enhances PRC1 binding to chromatin, PRC2 has been proposed to have a recruitment function (Bantignies & Cavalli, 2006).
PRC1 and PRC2 are recruited to the Xi early in X-inactivation, establishing H3K27me3 and H2AK119ub1 marks along the Xi (de Napoles et al, 2004; Fang et al, 2004; Plath et al, 2003). PcG localization to the Xi and, as well as the persistence of the associated histone marks, is strictly dependent on Xist, irrespective of the developmental state of the cells (Kohlmaier et al, 2004; Schoeftner et al, 2006). Notably, a silencing-deficient Xist RNA can recruit PRC1 and PRC2, showing that their recruitment alone is not sufficient to initiate X-inactivation (Kohlmaier et al, 2004; Plath et al, 2003). These observations are in contrast to the traditional role of PcG proteins in the maintenance of gene repression, but suggest a function for the PcG complexes in the establishment of a chromatin structure on the Xi for the maintenance of X-inactivation (Fig 4C).
The function of PRC2 in X inactivation has been studied by disrupting the embryonic ectoderm development (Eed) gene, which is an essential component of PRC2. In the absence of PRC2 function, H3K27me3 is no longer enriched on the Xi; PRC2 is not required for the initiation or maintenance of random X inactivation (Kalantry & Magnuson, 2006; Schoeftner et al, 2006). Moreover, PRC2 is required for the maintenance of imprinted X-inactivation in differentiating trophoblast cells, but is dispensable for Xi maintenance in most cells of the extra-embryonic lineages (Kalantry et al, 2006). An interesting observation is that Ring1b, but not Mph1 and Mph2, which are all members of the PRC1 complex, can be recruited by Xist in the absence of PRC2 and mediates H2AK119ub1 (Schoeftner et al, 2006). This implies that components of PRC1 and PRC2 can be recruited in parallel. Given the essential roles of PRC1 and PRC2 during early embryogenesis, as shown by the lethality caused by the disruption of Ring1b and Eed in mice, we propose that both complexes could act redundantly in X-inactivation. Alternatively, PRC1 function could be crucial for X inactivation in embryonic cells, whereas PRC2 might be of less importance. How PcG proteins are recruited is yet to be resolved, and X-inactivation might provide a good model to study the mechanism of Polycomb silencing from a new angle.
During the maintenance phase of X inactivation, Xist alone is not sufficient for the recruitment of PcG proteins; an additional factor, which we have introduced as chromosomal ‘memory', is required (Kohlmaier et al, 2004). The molecular nature of this ‘memory' is unclear. Xist expression early in differentiation establishes memory, and both Xist and memory are required for H3K27me3 and H2AK119ub1 on the Xi later in differentiation (Kohlmaier et al, 2004; Schoeftner et al, 2006). The establishment of memory is observed when the state of gene silencing becomes stable and irreversible. This suggests a role for memory in the transition from the initiation to the maintenance of X inactivation. A function for PcG complexes in the establishment of memory has been proposed, but awaits experimental confirmation.
Conclusion
X inactivation is a model for the developmentally controlled formation of silent chromatin. A number of pathways act in a stepwise manner to establish a stable state of repression. Progress has been made in understanding the molecular details of X inactivation, but the method by which Xist initiates silencing remains unknown. It is also unclear how the process of X-inactivation intertwines with cellular differentiation, and the molecular details of the timing of counting and the restriction of Xist in initiating silencing remain to be defined. Future studies of X-inactivation might therefore also lead to a better understanding of stem cells and of the process of cellular differentiation.

Martin Leeb, Anton Wutz, Karen Ng & Dieter Pullirsch
Acknowledgments
We apologize to all the authors whose work could not be cited. We thank L. Klein for critically reading the manuscript. This work was supported by the Institute of Molecular Pathology (IMP) through Boehringer Ingelheim, and by grants from the Austrian Ministry of Science Genome Research in Austria (GEN-AU) project and the Austrian Science Fund (FWF).
References
- Bacher CP, Guggiari M, Brors B, Augui S, Clerc P, Avner P, Eils R, Heard E (2006) Transient colocalization of X-inactivation centres accompanies the initiation of X inactivation. Nat Cell Biol 8: 293–299 [DOI] [PubMed] [Google Scholar]
- Bantignies F, Cavalli G (2006) Cellular memory and dynamic regulation of polycomb group proteins. Curr Opin Cell Biol 18: 275–283 [DOI] [PubMed] [Google Scholar]
- Boumil RM, Ogawa Y, Sun BK, Huynh KD, Lee JT (2006) Differential methylation of Xite and CTCF sites in Tsix mirrors the pattern of X-inactivation choice in mice. Mol Cell Biol 26: 2109–2117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao W, Huynh KD, Spencer RJ, Davidow LS, Lee JT (2002) CTCF, a candidate trans-acting factor for X-inactivation choice. Science 295: 345–347 [DOI] [PubMed] [Google Scholar]
- Chaumeil J, Le Baccon P, Wutz A, Heard E (2006) A novel role for Xist RNA in the formation of a repressive nuclear compartment into which genes are recruited when silenced. Genes Dev 20: 2223–2237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow JC, Yen Z, Ziesche SM, Brown CJ (2005) Silencing of the mammalian X chromosome. Annu Rev Genomics Hum Genet 6: 69–92 [DOI] [PubMed] [Google Scholar]
- Clemson CM, Hall LL, Byron M, McNeil J, Lawrence JB (2006) The X chromosome is organized into a gene-rich outer rim and an internal core containing silenced nongenic sequences. Proc Natl Acad Sci USA 103: 7688–7693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clerc P, Avner P (1998) Role of the region 3′ to Xist exon 6 in the counting process of X-chromosome inactivation. Nat Genet 19: 249–253 [DOI] [PubMed] [Google Scholar]
- Csankovszki G, Panning B, Bates B, Pehrson JR, Jaenisch R (1999) Conditional deletion of Xist disrupts histone macroH2A localization but not maintenance of X inactivation. Nat Genet 22: 323–324 [DOI] [PubMed] [Google Scholar]
- de Napoles M et al. (2004) Polycomb group proteins Ring1A/B link ubiquitylation of histone H2A to heritable gene silencing and X inactivation. Dev Cell 7: 663–676 [DOI] [PubMed] [Google Scholar]
- Duret L, Chureau C, Samain S, Weissenbach J, Avner P (2006) The xist RNA gene evolved in eutherians by pseudogenization of a protein-coding gene. Science 312: 1653–1655 [DOI] [PubMed] [Google Scholar]
- Fackelmayer FO (2005) A stable proteinaceous structure in the territory of inactive X chromosomes. J Biol Chem 280: 1720–1723 [DOI] [PubMed] [Google Scholar]
- Falkowski PG, Katz ME, Milligan AJ, Fennel K, Cramer BS, Aubry MP, Berner RA, Novacek MJ, Zapol WM (2005) The rise of oxygen over the past 205 million years and the evolution of large placental mammals. Science 309: 2202–2204 [DOI] [PubMed] [Google Scholar]
- Fang J, Chen T, Chadwick B, Li E, Zhang Y (2004) Ring1b-mediated H2A ubiquitination associates with inactive X chromosomes and is involved in initiation of X inactivation. J Biol Chem 279: 52812–52815 [DOI] [PubMed] [Google Scholar]
- Franco B, Ballabio A (2006) X-inactivation and human disease: X-linked dominant male-lethal disorders. Curr Opin Genet Dev 16: 254–259 [DOI] [PubMed] [Google Scholar]
- Graves JA (2006) Sex chromosome specialization and degeneration in mammals. Cell 124: 901–914 [DOI] [PubMed] [Google Scholar]
- Gupta V, Parisi M, Sturgill D, Nuttall R, Doctolero M, Dudko OK, Malley JD, Eastman PS, Oliver B (2006) Global analysis of X-chromosome dosage compensation. J Biol 5: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heard E, Disteche CM (2006) Dosage compensation in mammals: fine-tuning the expression of the X chromosome. Genes Dev 20: 1848–1867 [DOI] [PubMed] [Google Scholar]
- Herzing LB, Romer JT, Horn JM, Ashworth A (1997) Xist has properties of the X-chromosome inactivation centre. Nature 386: 272–275 [DOI] [PubMed] [Google Scholar]
- Huynh KD, Lee JT (2003) Inheritance of a pre-inactivated paternal X chromosome in early mouse embryos. Nature 426: 857–862 [DOI] [PubMed] [Google Scholar]
- Kalantry S, Magnuson T (2006) The Polycomb group protein EED is dispensable for the initiation of random X-chromosome inactivation. PLoS Genet 2: e66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalantry S, Mills KC, Yee D, Otte AP, Panning B, Magnuson T (2006) The Polycomb group protein Eed protects the inactive X-chromosome from differentiation-induced reactivation. Nat Cell Biol 8: 195–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlmaier A, Savarese F, Lachner M, Martens J, Jenuwein T, Wutz A (2004) A chromosomal memory triggered by Xist regulates histone methylation in X inactivation. PLoS Biol 2: E171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JT (2005) Regulation of X-chromosome counting by Tsix and Xite sequences. Science 309: 768–771 [DOI] [PubMed] [Google Scholar]
- Lee JT, Lu N (1999) Targeted mutagenesis of Tsix leads to nonrandom X inactivation. Cell 99: 47–57 [DOI] [PubMed] [Google Scholar]
- Lee JT, Strauss WM, Dausman JA, Jaenisch R (1996) A 450 kb transgene displays properties of the mammalian X-inactivation center. Cell 86: 83–94 [DOI] [PubMed] [Google Scholar]
- Lee JT, Davidow LS, Warshawsky D (1999) Tsix, a gene antisense to Xist at the X-inactivation centre. Nat Genet 21: 400–404 [DOI] [PubMed] [Google Scholar]
- Lucchesi JC, Kelly WG, Panning B (2005) Chromatin remodeling in dosage compensation. Annu Rev Genet 39: 615–651 [DOI] [PubMed] [Google Scholar]
- Lyon MF (2003) The Lyon and the LINE hypothesis. Semin Cell Dev Biol 14: 313–318 [DOI] [PubMed] [Google Scholar]
- Mak W, Nesterova TB, de Napoles M, Appanah R, Yamanaka S, Otte AP, Brockdorff N (2004) Reactivation of the paternal X chromosome in early mouse embryos. Science 303: 666–669 [DOI] [PubMed] [Google Scholar]
- Marahrens Y (1999) X-inactivation by chromosomal pairing events. Genes Dev 13: 2624–2632 [DOI] [PubMed] [Google Scholar]
- Marahrens Y, Panning B, Dausman J, Strauss W, Jaenisch R (1997) Xist-deficient mice are defective in dosage compensation but not spermatogenesis. Genes Dev 11: 156–166 [DOI] [PubMed] [Google Scholar]
- Marahrens Y, Loring J, Jaenisch R (1998) Role of the Xist gene in X chromosome choosing. Cell 92: 657–664 [DOI] [PubMed] [Google Scholar]
- Namekawa SH, Park PJ, Zhang LF, Shima JE, McCarrey JR, Griswold MD, Lee JT (2006) Postmeiotic sex chromatin in the male germline of mice. Curr Biol 16: 660–667 [DOI] [PubMed] [Google Scholar]
- Nesterova TB, Johnston CM, Appanah R, Newall AE, Godwin J, Alexiou M, Brockdorff N (2003) Skewing X chromosome choice by modulating sense transcription across the Xist locus. Genes Dev 17: 2177–2190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen DK, Disteche CM (2006) Dosage compensation of the active X chromosome in mammals. Nat Genet 38: 47–53 [DOI] [PubMed] [Google Scholar]
- Okamoto I, Otte AP, Allis CD, Reinberg D, Heard E (2004) Epigenetic dynamics of imprinted X inactivation during early mouse development. Science 303: 644–649 [DOI] [PubMed] [Google Scholar]
- Okamoto I, Arnaud D, Le Baccon P, Otte AP, Disteche CM, Avner P, Heard E (2005) Evidence for de novo imprinted X-chromosome inactivation independent of meiotic inactivation in mice. Nature 438: 369–373 [DOI] [PubMed] [Google Scholar]
- Penny GD, Kay GF, Sheardown SA, Rastan S, Brockdorff N (1996) Requirement for Xist in X chromosome inactivation. Nature 379: 131–137 [DOI] [PubMed] [Google Scholar]
- Plath K, Fang J, Mlynarczyk-Evans SK, Cao R, Worringer KA, Wang H, de la Cruz CC, Otte AP, Panning B, Zhang Y (2003) Role of histone H3 lysine 27 methylation in X inactivation. Science 300: 131–135 [DOI] [PubMed] [Google Scholar]
- Popova BC, Tada T, Takagi N, Brockdorff N, Nesterova TB (2006) Attenuated spread of X-inactivation in an X;autosome translocation. Proc Natl Acad Sci USA 103: 7706–7711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringrose L, Paro R (2004) Epigenetic regulation of cellular memory by the Polycomb and Trithorax group proteins. Annu Rev Genet 38: 413–443 [DOI] [PubMed] [Google Scholar]
- Sanchez-Elsner T, Gou D, Kremmer E, Sauer F (2006) Noncoding RNAs of trithorax response elements recruit Drosophila Ash1 to Ultrabithorax. Science 311: 1118–1123 [DOI] [PubMed] [Google Scholar]
- Schoeftner S, Sengupta AK, Kubicek S, Mechtler K, Spahn L, Koseki H, Jenuwein T, Wutz A (2006) Recruitment of PRC1 function at the initiation of X inactivation independent of PRC2 and silencing. EMBO J 25: 3110–3122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner JM, Mahadevaiah SK, Ellis PJ, Mitchell MJ, Burgoyne PS (2006) Pachytene asynapsis drives meiotic sex chromosome inactivation and leads to substantial postmeiotic repression in spermatids. Dev Cell 10: 521–529 [DOI] [PubMed] [Google Scholar]
- Vigneau S, Augui S, Navarro P, Avner P, Clerc P (2006) An essential role for the DXPas34 tandem repeat and Tsix transcription in the counting process of X chromosome inactivation. Proc Natl Acad Sci USA 103: 7390–7395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wutz A, Jaenisch R (2000) A shift from reversible to irreversible X inactivation is triggered during ES cell differentiation. Mol Cell 5: 695–705 [DOI] [PubMed] [Google Scholar]
- Wutz A, Rasmussen TP, Jaenisch R (2002) Chromosomal silencing and localization are mediated by different domains of Xist RNA. Nat Genet 30: 167–174 [DOI] [PubMed] [Google Scholar]
- Xu N, Tsai CL, Lee JT (2006) Transient homologous chromosome pairing marks the onset of X inactivation. Science 311: 1149–1152 [DOI] [PubMed] [Google Scholar]