

Abstract
The developmental and oncogenic roles of MYC proteins are well established, but the transcriptional targets mediating their functions remain elusive. Using small interfering RNA-mediated knockdown in breast and cervix carcinoma cell lines, which overexpress c-MYC, we show that c-MYC independently controls metabolism and cell proliferation, and can, depending on the cells, promote or inhibit migration. We identified new c-MYC target genes in these cell lines, and show that selective regulation of some targets correlates with the phenotypic responses of these different cell lines to c-MYC depletion. Notably, we show that a positive regulation of the WNT signalling pathway contributes to c-MYC pro-mitogenic effects in breast and cervix carcinoma cells. We also show that repression of CCL5/RANTES accounts for c-MYC anti-migratory effects in specific breast cancer cells. Our combined genomic and phenotypic analysis indicates that c-MYC functions are cellular-context-dependent and that selectively regulated genes are responsible for its differential properties.
Keywords: c-MYC, cancer, transcription, target genes, proliferation, migration
Introduction
MYC proto-oncogenes (myelocytomatosis viral oncogene homologs) are aberrantly expressed in many cancer types, in part due to genomic alterations, and altered MYC gene expression is highly correlated with advanced disease stage and adverse prognosis (Pelengaris et al, 2002; Vogelstein & Kinzler, 2004). MYC genes encode transcription factors that regulate gene expression in two main ways: activation of E-box- driven genes as heterodimers with MAX (MYC associated factor X); and repression, in particular, of genes driven by Initiator (Inr) elements through inhibitory interactions with MIZ1 (MYC-interacting Zinc finger protein 1; Pelengaris et al, 2002; Patel et al, 2004). MYC proteins have been conserved during evolution but control different cellular functions, such as growth in Drosophila and division in the mouse (Johnston et al, 1999; Trumpp et al, 2001). They are essential during mammalian embryogenesis (Moens et al, 1992; Charron et al, 1992; Stanton et al, 1992; Davis et al, 1993), notably through the control of stem cell and progenitor cell renewal and differentiation (Waikel et al, 2001; Knoepfler et al, 2002; Wilson et al, 2004), and also have roles in angiogenesis, cell migration and metastasis (Biro et al, 1993; Goodman et al, 1997; Wakamatsu et al, 1997; Baudino et al, 2002). Depending on the cellular context, MYC proteins induce either cell proliferation or apoptosis, and they require cooperation with other oncoproteins and inhibition of apoptotic pathways to transform cells (Pelengaris et al, 2002). This implies that they regulate different targets in these contexts. Regulatory cofactors and many transcriptional targets of MYC proteins have been identified (Coller et al, 2000; O'Hagan et al, 2000; Boon et al, 2001; Pelengaris et al, 2002; Fernandez et al, 2003; Patel et al, 2004). However, only a few MYC targets, identified primarily through ectopic expression approaches, have been shown to contribute to MYC functions. It is often not clear whether they are regulated by MYC in natural situations (Patel et al, 2004). Chromatin immunoprecipitation (ChIP) approaches have allowed the identification of genomic MYC-binding sites, confirming previously identified targets and revealing new ones. A significant number of genes are bound by MYC only in specific cells, and only a fraction of these are regulated by MYC (Fernandez et al, 2003; Patel et al, 2004, and references therein). Therefore, promoter binding and gene regulation by MYC proteins should be analysed in natural physiological and pathological conditions to identify the transcriptional targets that mediate their complex functions.
Results And Discussion
Abnormal activation of c-MYC is known to contribute to breast and cervix carcinogenesis (Escot et al, 1986; Ocadiz et al, 1987; Sinn et al, 1987). With the aim of identifying transcriptional targets that mediate the functions of MYC in cells originating from these common cancers, we have used a loss-of-function approach combined with phenotypic and global transcription profiling studies.
In the cell lines examined, all derived from breast carcinomas, except HeLa (cervix carcinoma), only c-MYC, amplified in two cases, is strongly expressed. N-MYC and L-MYC are weakly expressed and show no genomic alterations (supplementary Fig 1 online).
By using small interfering RNA (siRNA), c-MYC was knocked down in BT-474, MCF-7, MDA-MB-231 and HeLa cells (Fig 1A,B). In BT-474, but not other cells, the observed consequence was a reduction of global RNA and protein content and also of cell size (supplementary Fig 2 online). By contrast, c-MYC depletion strongly impaired the proliferation of MCF-7, MDA-MB-231 and HeLa cells, but did not affect that of BT-474 cells, even after 2 weeks (Fig 1C; data not shown). No significant changes in cell-cycle distribution or evidence of cell death were detected (supplementary Fig 3 online), suggesting that the reduced proliferation is due to an overall slower transit through the cell cycle. Distinct c-MYC siRNAs caused similar proliferation defects, which could be rescued by an siRNA-resistant allele of c-MYC (supplementary Fig 4 online), proving their specificity. Finally, c-MYC depletion differentially altered the migration of studied cells, inhibiting that of MDA-MB-231 cells, consistent with the pro-migratory role of c-MYC and N-MYC in other cell types (Biro et al, 1993; Wakamatsu et al, 1997), and enhancing that of MCF-7 cells, implying an anti-migratory role in this cell line (Fig 1D). c-MYC inhibition had no significant effect on the migration of HeLa cells (Fig 1D), and BT-474 cells did not migrate under the conditions tested (data not shown).
Figure 1.
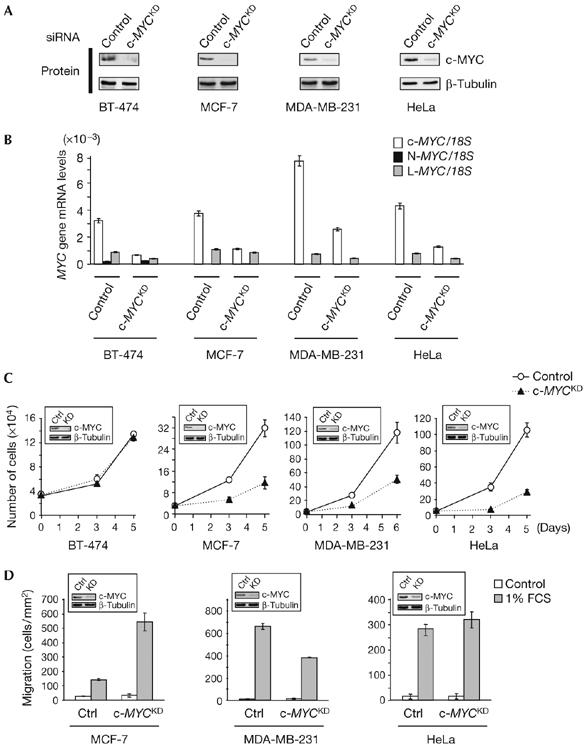
c-MYC depletion has versatile consequences on carcinoma cell proliferation and migration. (A) c-MYC western blot analysis, performed 3 days after transfection, in control and c-MYCKD (knockdown) small interfering RNA (siRNA)-transfected human carcinoma cells. β-Tubulin analysis of the same extracts was used as a control for protein loading. (B) Matching RNA samples were analysed by quantitative radioactive reverse transcription–PCR for expression of the MYC genes. 18S rRNA was used for normalization. Control and c-MYCKD cells were assayed for proliferation at given time points after siRNA transfection (C) and for their ability to migrate in response to fetal calf serum (FCS; D). Plotted values are the means±s.e.m. of three replicates. c-MYC downregulation by siRNA is shown in the insets. Ctrl, control. MYC, myelocytomatosis viral oncogene homologs.
Lack of N-MYC or L-MYC induction (Figs 1B, 2A) and altered MYC-dependent transcription on c-MYC depletion (supplementary Fig 5 online; data not shown) indicated that other MYC proteins do not compensate for c-MYC depletion in the cells studied, in particular in BT-474 cells, which proliferate independently of c-MYC. Therefore, in the cells examined, c-MYC independently controls metabolism, size and proliferation and can exert, depending on the cells, opposite effects on migration.
Figure 2.
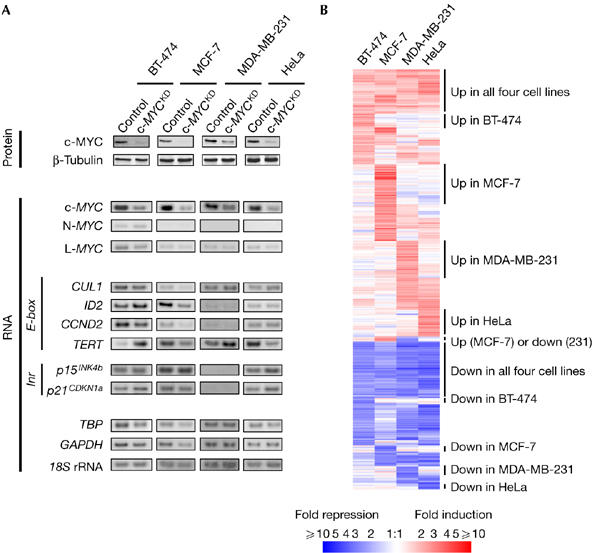
Transcriptional response to c-MYC depletion in human carcinoma cells. (A) c-MYC downregulation in the cells studied does not have a consistent effect on MYC targets expression. Proteins were collected 3 days after control and c-MYCKD (knockdown) short interfering RNA (siRNA) transfection and assayed for c-MYC downregulation. β-Tubulin analysis of the same extracts was used as a control for protein loading. Matching RNA samples were analyzed by quantitative radioactive reverse transcription-PCR for MYC family and MYC target (E-box- and Inr element-driven) genes expression: CUL1 (cullin 1), ID2 (inhibitor of DNA binding 2), CCND2 (cyclin D2), TERT (telomerase reverse transcriptase), P15INK4b (cyclin-dependent kinase 4 inhibitor B), P21CDKN1A (cyclin-dependent kinase inhibitor 1A). TBP (TATA box binding protein), GAPDH (glyceraldehyde-3-phosphate dehydrogenase) and 18S rRNA were used for normalization. (B) Genome-wide patterns of transcriptional response to c-MYC depletion in human carcinoma cells. The cluster diagram depicts microarray data for genes that are regulated by siRNA-mediated c-MYC depletion. Mean fold-change values from independent replicates are given for 741 probe sets showing at least twofold regulation in one or more cell lines. White indicates unchanged expression, and red and blue shades indicate upregulation and downregulation, respectively, relative to control siRNA-transfected cells.
We proposed that the versatile consequences of c-MYC depletion might be due to cell-specific differences in target genes, and therefore searched for differentially regulated genes. In the cells studied, selected previously identified targets were either not altered significantly by c-MYC depletion or their regulation was inconsistent (Fig 2A) and did not correlate with the resulting phenotypes (Fig 1). For example, TERT (telomerase reverse transcriptase), previously shown to be transactivated by c-MYC and to contribute to cell proliferation (Wu et al, 1999; Smith et al, 2003), is either upregulated or downregulated on c-MYC depletion (Fig 2A), irrespective of the proliferative response (Fig 1C). Thus, in the studied tumour cells, these target genes might not contribute to the effects of c-MYC depletion.
To identify targets that might explain the variable effects of c-MYC depletion, we undertook a genome-wide transcriptome analysis using microarrays. We screened for genes with regulation patterns correlating with the c-MYC depletion phenotypes, that is, regulated only when cell proliferation was inhibited or when migration was altered. We found 741 transcripts regulated by twofold or more in at least one of the c-MYC-depleted cell lines. Among these were known MYC targets and many new ones; 65% were upregulated and 35% were downregulated (supplementary Table 1 online). Although 23% were regulated in all four cell lines, 29% were regulated only in two or three out of the four cell lines and 48% in only one of the four cell lines, showing that the cellular context strongly influences MYC transcriptional effects (Fig 2B; supplementary Table 1 online). Interestingly, selective regulation of certain genes correlated with specific c-MYC depletion phenotypes in different cells. We thus examined whether some of these selectively regulated genes could mediate the effects of c-MYC in these different cells and explain their versatility.
To find MYC targets that could contribute to cell proliferation, we investigated those regulated only in MCF-7, MDA-MB-231 and HeLa cells, all three subjected to a proliferation defect on c-MYC depletion. The canonical WNT (wingless-type-family)/β-catenin signalling pathway is important in development and oncogenesis, and contributes to c-MYC transcription (He et al, 1998; Reya & Clevers, 2005). We found that in MDA-MB-231 and HeLa cells, c-MYC depletion led to regulation of several genes encoding components of WNT signalling, with a predicted inhibitory effect on the canonical WNT pathway. Confirming this hypothesis, levels of β-catenin, the accumulation of which reflects active canonical WNT signalling, decreased on c-MYC depletion, particularly in HeLa cells (supplementary Fig 6A online). Among the regulated WNT pathway components, we considered WNT5A as a good candidate mediator of the proliferation defect due to c-MYC depletion. Indeed, we found that WNT5A, known as a tumour suppressor and cell proliferation inhibitor in haematological cancers (Liang et al, 2003), is upregulated in division-deficient c-MYC-depleted MDA-MB231 and HeLa cells (Fig 1C; supplementary Fig 6A online). As c-MYC represses some genes by preventing their transactivation by MIZ-1 (Pelengaris et al, 2002; Patel et al, 2004), we examined the contribution of both factors to the regulation of WNT5A. While causing a moderate increase in WNT5A expression in control MDA-MB-231 and HeLa cells, MIZ-1 knockdown led to reduced WNT5A induction in c-MYC-depleted MDA-MB-231 cells but not HeLa cells (Fig 3A; supplementary Fig 7 online). ChIP analysis showed that c-MYC binds to the WNT5A regulatory region in both MDA-MB-231 and HeLa cells, whereas MIZ-1 binds to this region only in MDA-MB-231 cells (Fig 3B). Therefore, WNT5A repression by c-MYC does not necessarily involve inactivation of MIZ-1, which has an impact on WNT5A expression depending on the cellular context. Ectopic expression and treatment with WNT5A conditioned medium impaired the proliferation of MDA-MB-231 and HeLa cells (Fig 3C,D), and also of other WNT5A-negative breast carcinoma cell lines (data not shown). Finally, WNT5A knockdown partially rescued the proliferation of c-MYC-depleted MDA-MB-231 and HeLa cells (Fig 3E; supplementary Fig 7 online). These results show that, in addition to mediating some of the WNT signalling effects (He et al, 1998; Reya & Clevers, 2005), c-MYC also exerts positive feedback control on this pathway by regulating the expression of some of its components. In particular, direct transcriptional repression of WNT5A contributes to the mitogenic role of c-MYC in some carcinoma cells. In MCF-7 cells, c-MYC depletion was not linked to alterations in WNT signalling (supplementary Fig 6A online), indicating that c-MYC drives their proliferation through a distinct pathway.
Figure 3.
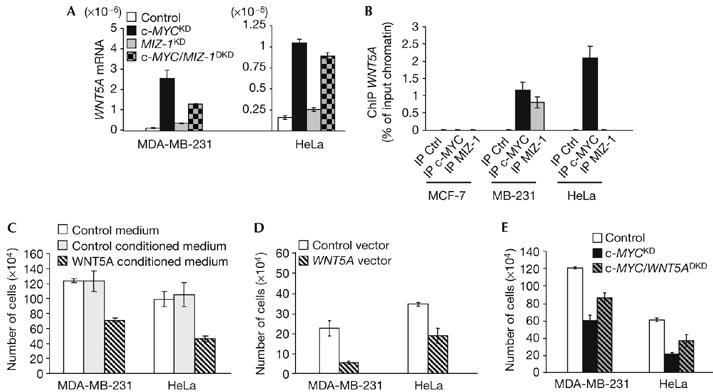
Identification of c-MYC targets involved in carcinoma cell proliferation. (A–E) WNT5A gene repression contributes to the mitogenic function of c-MYC in MDA-MB-231 and HeLa cells. (A) WNT5A gene expression, normalized by 18S ribosomal RNA, was quantified by real-time reverse transcription–PCR analysis in control, c-MYCKD (knockdown), MIZ-1KD or c-MYC/MIZ-1DKD (double knockdown) cells. (B) Chromatin immunoprecipitations (ChIPs) were carried out with antibodies against c-MYC and MIZ-1, or a pre-immune serum (Ctrl). ChIP was evaluated using quantitative real-time PCR analysis, with primers in the WNT5A gene regulatory region. (C–E) MDA-MB-231 and HeLa cells were treated (C) or transfected with the indicated expression vectors (D) or small interfering RNA (E) and counted 6 days later.
We also searched for target genes that could explain the migratory responses observed in c-MYC-depleted MCF-7 and MDA-MB-231 cells, thus selectively regulated in these cells. CCL5 (CC chemokine ligand 5) (SCYA5/RANTES), encoding a chemokine that induces immune cell chemotaxis (Schall et al, 1990), was strongly upregulated on c-MYC depletion in MCF-7 cells (Fig 4A, B), which showed, in response, enhanced migration (Fig 1D). On the contrary, in the same cells, MIZ-1 knockdown led to inhibition of basal CCL5 expression and abrogated its induction in response to c-MYC depletion (Fig 4B; supplementary Fig 7 online). Together with the ChIP analysis showing that both c-MYC and MIZ-1 bind to the CCL5 promoter (Fig 4C), these data indicate that in MCF-7 cells c-MYC represses CCL5 by preventing its transactivation by MIZ-1. The fact that these factors also bind to the CCL5 regulatory region in MDA-MB-231 and HeLa cells, in which it is not induced (Fig 4A, C), is consistent with previous studies indicating that MYC binding is not sufficient for regulation (Fernandez et al, 2003; Patel et al, 2004). Recombinant CCL5 promoted serum-triggered (but not basal) migration of MCF-7 cells, whereas blocking of CCL5 function inhibited migration in control MCF-7 cells and partly reverted the increase in migration due to c-MYC depletion (Fig 4D; data not shown). These observations show that CCL5 repression mediates the anti-migratory function of c-MYC in MCF-7 cells, warranting investigation in other cells.
Figure 4.
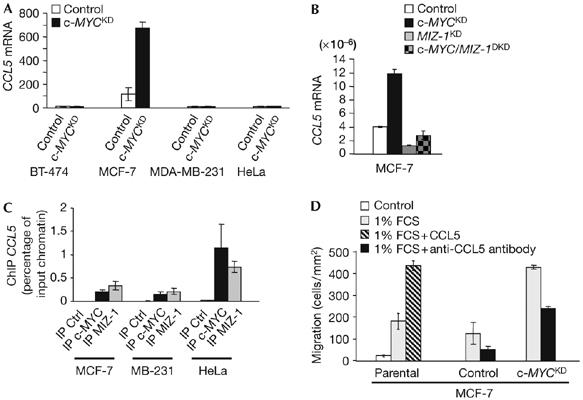
c-MYC represses expression of the CCL5 chemokine, which promotes the migration of MCF-7 cells. (A) Microarray expression profile (mean±s.e.m. from independent replicates) of CCL5 in control and c-MYCKD (knockdown) cells. (B) CCL5 gene expression, normalized by 18S ribosomal RNA, was quantified by real-time reverse transcription–PCR analysis in control, c-MYCKD, MIZ-1KD or c-MYC/MIZ-1DKD (double knockdown) MCF-7 cells. (C) Chromatin immunoprecipitations (ChIPs) were carried out with antibodies against c-MYC and MIZ-1, or a pre-immune serum (Ctrl). ChIP was evaluated using quantitative real-time PCR analysis, with primers in the CCL5 gene regulatory region. (D) Parental control and c-MYCKD MCF-7 cells were treated with fetal calf serum (FCS) and recombinant CCL5 or an anti-CCL5-blocking antibody or left untreated and assayed for migration.
Additional genes, the products of which are known to modulate migration, are induced in c-MYC-depleted MCF-7 or MDA-MB-231 cells, and others, with no established function, are inversely regulated in these cells (supplementary Fig 8 online; supplementary Table 1 online). All are good candidates that might be involved in the opposite migratory responses of these cells to c-MYC depletion (Fig 1D).
In conclusion, our study is the first to analyse, in parallel, the phenotypic and global transcriptional impacts of MYC. We have identified novel mediators of c-MYC functions in cell proliferation and for the first time in cell migration. The contribution of these and further newly identified targets to the physiopathological roles of c-MYC deserves further investigation. Comparison of several cell lines has also shown the importance of the cellular context for the transcriptional and, as a consequence, biological effects of c-MYC. Notably, depending on the cells, c-MYC exerts opposite effects on migration, in correlation with inverse regulation of certain genes, and controls cell proliferation through different pathways. As MYC acts in association with various cofactors (Patel et al, 2004), we propose that the herein reported selective regulation of some target genes in different cell lines is due to cell-dependent variations in the composition of MYC transcriptional complexes. Elucidating this question and also the functions of more c-MYC targets should help to clarify the complex and versatile roles of MYC proteins in physiological and pathological situations.
Methods
Constructs. A c-MYC construct was obtained by cloning the c-MYC complementary DNA into the pcDNA3Neo vector (Invitrogen, Carlsbad, CA, USA), and the derived rescue construct by introducing silent mutations in the c-MYC siRNA recognition site by site-directed mutagenesis (sequences of primers are given in supplementary Table 2 online). Control and WNT5A constructs have been described previously (Jonsson & Andersson, 2001). E-box-luciferase reporter constructs were generated by cloning wild-type and mutant E-box oligonucleotides (sequences are given in supplementary Table 2 online) into pTK-Luc, and the INR-luciferase reporter construct was generated by cloning the CDKN1a promoter into pGL3-Luc (Promega, Madison, WI, USA).
Cell culture and transfections. All cell lines were cultured in DMEM with 10% fetal calf serum (FCS), except BT-474 (RPMI1640 with 10% FCS). When specified, 20 ng/ml of RANTES/CCL5 (R&D Systems, Minneapolis, MN, USA) or 0.5 μg/ml rabbit polyclonal anti-human RANTES/CCL5-blocking antibody (Peprotech, Rocky Hill, NJ, USA) was added to the medium. WNT5A-conditioned medium was collected from WNT5A-expressing L cells (ATCC-LGC Promochem, Molsheim, France; CRL-2814). L-cell-conditioned medium (ATCC-LGC Promochem; CRL-2648) was used as a control. The media were filtered through a 0.45 μm filter and applied to cell cultures. Plasmids were transfected using Lipofectamine 2000 (Invitrogen) and siRNA, 21-mer oligoribonucleotide duplexes (Qiagen, Hilden, Germany), using Oligofectamine (Invitrogen). Two different siRNAs were used independently to inhibit c-MYC, one siRNA to inhibit MIZ-1 and WNT5A, and a bacterial LACZ siRNA was used as a control (supplementary Table 2 online).
Proliferation, cell cycle and migration analysis. For cell proliferation assays, cells were collected for counting from triplicates at the indicated time points after transfection or treatment. Cell-cycle analysis was carried out by flow cytometry after propidium iodide staining. For migration assays, cells were collected 3 days after transfection in siRNA experiments, serum-starved and plated in Transwell chambers, as described previously (Marone et al, 2004), with or without 1% FCS, RANTES/CCL5 or anti-RANTES/CCL5-blocking antibody (as described above). Cells were allowed to migrate for 3 h (MDA-MB-231), 6 h (HeLa) or 24 h (MCF-7) and then stained and counted.
Western blotting. Whole-cell protein extracts were analysed by western blotting, as described previously (Marone et al, 2004), using primary antibodies against c-MYC (9E10, Santa Cruz Biotechnology, Santa Cruz, CA, USA), β-catenin (Transduction Laboratories, BD Biosciences, San Jose, CA, USA) and β-tubulin (D10, Santa Cruz Biotechnology).
RNA isolation, reverse transcription–PCR and microarray analysis. RNA purification and quantitative radioactive reverse transcription–PCR and real-time quantitative PCR were carried out as described previously (Cappellen et al, 2002). Primers and PCR conditions are indicated in supplementary Table 2 online. Sequences of EF1α primers were kindly provided by François Radvanyi (CNRS-Institut Curie, Paris, France). For microarray analysis, RNA from each cell line was extracted 3 days after siRNA transfection from two or three independent replicates and profiled on Affymetrix GeneChip® Hu133 arrays, as described previously (Cappellen et al, 2002). Data were analysed using Affymetrix MAS4.0 and GeneSpring 7.0 (Agilent Technologies) softwares. Genes were selected as regulated if their expression deviated twofold or more in c-MYC-depleted cells in comparison with the corresponding control cells. Raw data have been deposited in the NCBI GEO database (http://www.ncbi.nlm.nih.gov/geo/) and are accessible through the accession number GSE5823.
Chromatin immunoprecipitation analyses. ChIP was carried out essentially as described previously (Fernandez et al, 2003), using 2 μg of antibodies against either c-MYC (N-262, Santa Cruz Biotechnology) or MIZ-1 (N-17, Santa Cruz Biotechnology) to precipitate chromatin from 107 cells. Precipitation with a pre-immune serum was used as a control. The precipitated chromatin was purified by phenol–chloroform extraction and ethanol precipitation. DNA fragments interacting with the transcription factors of interest were detected by real-time PCR using appropriate primers (supplementary Table 2 online).
Supplementary information is available at EMBO reports online (http://www.emboreports.org).
Supplementary Material
supplementary Figures
supplementary Table 1
supplementary Table 2
Acknowledgments
We thank E. Oakeley and H. Angliker for help with microarray analysis, I. Boschke and H. Kohler for help with flow cytometry analysis, M. Šuša for discussions and A. Badache for help with migration experiments, discussions and critical reading of the manuscript. D.C. and T.S. were supported by grants from the Swiss Cancer League. The laboratory of N.E.H. was supported by the Novartis Research Foundation, Friedrich Miescher Institute for Biomedical Research (FMI).
References
- Baudino TA, McKay C, Pendeville-Samain H, Nilsson JA, Maclean KH, White EL, Davis AC, Ihle JN, Cleveland JL (2002) c-Myc is essential for vasculogenesis and angiogenesis during development and tumor progression. Genes Dev 16: 2530–2543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biro S, Fu YM, Yu ZX, Epstein SE (1993) Inhibitory effects of antisense oligodeoxynucleotides targeting c-myc mRNA on smooth muscle cell proliferation and migration. Proc Natl Acad Sci USA 90: 654–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boon K et al. (2001) N-myc enhances the expression of a large set of genes functioning in ribosome biogenesis and protein synthesis. EMBO J 20: 1383–1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappellen D, Luong-Nguyen NH, Bongiovanni S, Grenet O, Wanke C, Susa M (2002) Transcriptional program of mouse osteoclast differentiation governed by the macrophage colony-stimulating factor and the ligand for the receptor activator of NF-κB. J Biol Chem 277: 21971–21982 [DOI] [PubMed] [Google Scholar]
- Charron J, Malynn BA, Fisher P, Stewart V, Jeannotte L, Goff SP, Robertson EJ, Alt FW (1992) Embryonic lethality in mice homozygous for a targeted disruption of the N-myc gene. Genes Dev 6: 2248–2257 [DOI] [PubMed] [Google Scholar]
- Coller HA, Grandori C, Tamayo P, Colbert T, Lander ES, Eisenman RN, Golub TR (2000) Expression analysis with oligonucleotide microarrays reveals that MYC regulates genes involved in growth, cell cycle, signaling, and adhesion. Proc Natl Acad Sci USA 97: 3260–3265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis AC, Wims M, Spotts GD, Hann SR, Bradley A (1993) A null c-myc mutation causes lethality before 10.5 days of gestation in homozygotes and reduced fertility in heterozygous female mice. Genes Dev 7: 671–682 [DOI] [PubMed] [Google Scholar]
- Escot C, Theillet C, Lidereau R, Spyratos F, Champeme MH, Gest J, Callahan R (1986) Genetic alteration of the c-myc protooncogene (MYC) in human primary breast carcinomas. Proc Natl Acad Sci USA 83: 4834–4838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez PC, Frank SR, Wang L, Schroeder M, Liu S, Greene J, Cocito A, Amati B (2003) Genomic targets of the human c-Myc protein. Genes Dev 17: 1115–1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman LA, Liu BC, Thiele CJ, Schmidt ML, Cohn SL, Yamashiro JM, Pai DS, Ikegaki N, Wada RK (1997) Modulation of N-myc expression alters the invasiveness of neuroblastoma. Clin Exp Metast 15: 130–139 [DOI] [PubMed] [Google Scholar]
- He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW (1998) Identification of c-MYC as a target of the APC pathway. Science 281: 1509–1512 [DOI] [PubMed] [Google Scholar]
- Johnston LA, Prober DA, Edgar BA, Eisenman RN, Gallant P (1999) Drosophila myc regulates cellular growth during development. Cell 17: 779–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson M, Andersson T (2001) Repression of Wnt-5a impairs DDR1 phosphorylation and modifies adhesion and migration of mammary cells. J Cell Sci 114: 2043–2053 [DOI] [PubMed] [Google Scholar]
- Knoepfler PS, Cheng PF, Eisenman RN (2002) N-myc is essential during neurogenesis for the rapid expansion of progenitor cell populations and the inhibition of neuronal differentiation. Genes Dev 16: 2699–2712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang H, Chen Q, Coles AH, Anderson SJ, Pihan G, Bradley A, Gerstein R, Jurecic R, Jones SN (2003) Wnt5a inhibits B cell proliferation and functions as a tumor suppressor in hematopoietic tissue. Cancer Cell 4: 349–360 [DOI] [PubMed] [Google Scholar]
- Marone R, Hess D, Dankort D, Muller WJ, Hynes NE, Badache A (2004) Memo mediates ErbB2-driven cell motility. Nat Cell Biol 6: 515–522 [DOI] [PubMed] [Google Scholar]
- Moens CB, Auerbach AB, Conlon RA, Joyner AL, Rossant J (1992) A targeted mutation reveals a role for N-myc in branching morphogenesis in the embryonic mouse lung. Genes Dev 6: 691–704 [DOI] [PubMed] [Google Scholar]
- Ocadiz R, Sauceda R, Cruz M, Graef AM, Gariglio P (1987) High correlation between molecular alterations of the c-myc oncogene and carcinoma of the uterine cervix. Cancer Res 47: 4173–4177 [PubMed] [Google Scholar]
- O'Hagan RC et al. (2000) Gene-target recognition among members of the myc superfamily and implications for oncogenesis. Nat Genet 24: 113–119 [DOI] [PubMed] [Google Scholar]
- Patel JH, Loboda AP, Showe MK, Showe LC, McMahon SB (2004) Analysis of genomic targets reveals complex functions of MYC. Nat Rev Cancer 4: 562–568 [DOI] [PubMed] [Google Scholar]
- Pelengaris S, Khan M, Evan G (2002) c-MYC: more than just a matter of life and death. Nat Rev Cancer 2: 764–776 [DOI] [PubMed] [Google Scholar]
- Reya T, Clevers H (2005) Wnt signaling in stem cells and cancer. Nature 434: 843–850 [DOI] [PubMed] [Google Scholar]
- Schall TJ, Bacon K, Toy KJ, Goeddel DV (1990) Selective attraction of monocytes and T lymphocytes of the memory phenotype by cytokine RANTES. Nature 347: 669–671 [DOI] [PubMed] [Google Scholar]
- Sinn E, Muller W, Pattengale P, Tepler I, Wallace R, Leder P (1987) Coexpression of MMTV/v-Ha-ras and MMTV/c-myc genes in transgenic mice: synergistic action of oncogenes in vivo. Cell 49: 465–475 [DOI] [PubMed] [Google Scholar]
- Smith LL, Coller HA, Roberts JM (2003) Telomerase modulates expression of growth-controlling genes and enhances cell proliferation. Nat Cell Biol 5: 474–479 [DOI] [PubMed] [Google Scholar]
- Stanton BR, Perkins AS, Tessarollo L, Sassoon DA, Parada LF (1992) Loss of N-myc function results in embryonic lethality and failure of the epithelial component of the embryo to develop. Genes Dev 6: 2235–2247 [DOI] [PubMed] [Google Scholar]
- Trumpp A, Refaeli Y, Oskarsson T, Gasser S, Murphy M, Martin GR, Bishop JM (2001) c-Myc regulates mammalian body size by controlling cell number but not cell size. Nature 414: 768–773 [DOI] [PubMed] [Google Scholar]
- Vogelstein B, Kinzler KW (2004) Cancer genes and the pathways they control. Nat Med 10: 789–799 [DOI] [PubMed] [Google Scholar]
- Waikel RL, Kawachi Y, Waikel PA, Wang XJ, Roop DR (2001) Deregulated expression of c-Myc depletes epidermal stem cells. Nat Genet 28: 165–168 [DOI] [PubMed] [Google Scholar]
- Wakamatsu Y, Watanabe Y, Nakamura H, Kondoh H (1997) Regulation of the neural crest cell fate by N-myc: promotion of ventral migration and neuronal differentiation. Development 124: 1953–1956 [DOI] [PubMed] [Google Scholar]
- Wilson A, Murphy MJ, Oskarsson T, Kaloulis K, Bettess MD, Oser GM, Pasche AC, Knabenhans C, Macdonald HR, Trumpp A (2004) c-Myc controls the balance between hematopoietic stem cell self-renewal and differentiation. Genes Dev 18: 2747–2763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu KJ, Grandori C, Amacker M, Simon-Vermot N, Polack A, Lingner J, Dalla-Favera R (1999) Direct activation of TERT transcription by c-MYC. Nat Genet 21: 220–224 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
supplementary Figures
supplementary Table 1
supplementary Table 2