

Abstract
Mitochondria have crucial roles in the life and death of mammalian cells, and help to orchestrate host antiviral defences. Here, we show that the ubiquitous human pathogen herpes simplex virus (HSV) induces rapid and complete degradation of host mitochondrial DNA during productive infection of cultured mammalian cells. The depletion of mitochondrial DNA requires the viral UL12 gene, which encodes a conserved nuclease with orthologues in all herpesviruses. We show that an amino-terminally truncated UL12 isoform—UL12.5—localizes to mitochondria and triggers mitochondrial DNA depletion in the absence of other HSV gene products. By contrast, full-length UL12, a nuclear protein, has little or no effect on mitochondrial DNA levels. Our data document that HSV inflicts massive genetic damage to a crucial host organelle and show a novel mechanism of virus-induced shutoff of host functions, which is likely to contribute to the cell death and tissue damage caused by this widespread human pathogen.
Keywords: herpes simplex virus, host shutoff, mitochondrial DNA
Introduction
Herpes simplex virus (HSV) is a ubiquitous viral pathogen capable of both productive and latent infections in its human host. During productive infection, HSV rapidly shuts off cellular protein synthesis by depleting most messenger RNAs (mRNAs) encoded by the nuclear genome, thereby dampening host responses to infection and allowing viral mRNAs to dominate the cytoplasmic translational machinery (reviewed by Smiley, 2004). HSV-induced depletion of mRNAs encoded by the nuclear genome is achieved by the concerted action of the viral proteins ICP27 and UL41, which inhibit transcription and processing of cellular nuclear mRNA precursors (Hardwicke & Sandri-Goldin, 1994; Hardy & Sandri-Goldin, 1994; Spencer et al, 1997), and trigger accelerated decay of cytoplasmic mRNAs (Kwong & Frenkel, 1987), respectively. Eukaryotic cells also contain the much smaller mitochondrial genome, which in mammals encodes a subset of the proteins involved in oxidative phosphorylation and the RNA components of the mitochondrial translational machinery (reviewed by Fernandez-Silva et al, 2003). An earlier report indicated that mRNAs encoded by the mitochondrial genome (mt mRNAs) are also depleted during HSV infection (Latchman, 1988); however, the mechanisms involved were not determined. Here, we show that HSV triggers rapid and complete degradation of mt DNA, an effect that is mediated by a viral nuclease with orthologues in all herpesviruses. These data show a novel and irreversible mechanism of viral shutoff of host cell gene expression, which is likely to contribute to HSV pathogenesis. In addition, these data raise the possibility that HSV or other herpesviruses might contribute to somatic mt DNA damage or loss—conditions that have been linked to a increasing list of progressive degenerative diseases in humans (reviewed by Dimauro, 2004; Beal, 2005; Wallace, 2005).
Results And Discussion
We confirmed the earlier report that HSV depletes mRNAs encoded by the mitochondrial genome (Latchman, 1988) by monitoring the levels of the mt mRNAs for cytochrome c oxidase subunit II (COX2) and nicotinamide adenine dinucleotide dehyrogenase subunit 6 (ND6) during productive infection of Vero cells by northern blot hybridization. Both of these mt mRNAs were rapidly lost on infection with the wild-type HSV type 1 (HSV-1) strain KOS (Fig 1A). HSV infection does not markedly reduce the levels of host nuclear DNA (Lomonte & Everett, 1999), although numerous chromosomal aberrations are induced (Fortunato & Spector, 2003). Consistent with these earlier findings, we found that the abundance of sequences hybridizing to an Alu small interspersed repeated element—a prominent component of the nuclear genome—was not altered during infection (supplementary Fig S1C online). In striking contrast, Southern blot analysis showed that the mt DNA sequences detected by the COX2 and ND6 probes were markedly depleted (Fig 1B). Similar results were obtained when the entire mitochondrial genome was used as a probe (supplementary Fig S1B online), indicating that HSV induces global loss of the mitochondrial genome rather than deletion of specific sequences. Mt DNA and mt mRNA were also depleted in HSV-1-infected HeLa cells, human embryonic lung fibroblasts and mouse embryo fibroblasts, and in Vero cells infected with HSV-2 (data not shown). HSV-1-induced depletion of mt DNA was confirmed in live HeLa cells by imaging with PicoGreen (supplementary Fig S2 online), which stains both nuclear and mt DNA (Ashley et al, 2005). Despite the profound loss of mt mRNA and mt DNA, apparently intact mitochondria could be visualized in the infected cells by using MitoTracker Red (supplementary Fig S2D online), which is consistent with the findings of Murata et al (2000) that mitochondria and mitochondrial respiratory function survive largely intact for at least 12 h in HSV-infected cells.
Figure 1.

HSV-1 depletes mt DNA and mt mRNA in infected Vero cells. Mt mRNA (A) and mt DNA (B) were monitored by northern and Southern blot hybridization, respectively, by using probes for COX2 and ND6. Infections with the indicated HSV-1 strains (10 PFU/cell) were allowed to proceed for 4, 8 and 12 h. Mt mRNA (C) and mt DNA (D) loss in Vero cells and 6-5 cells that express UL12 after HSV infection. COX2, cytochrome c oxidase subunit II; HSV-1, herpes simplex virus type 1; mt, mitochondrial; ND6, nicotinamide adenine dinucleotide dehyrogenase subunit 6; PFU, plaque forming unit; WT, wild type.
As noted in the Introduction, HSV depletes host mRNAs encoded by the nuclear genome by the concerted action of the viral proteins ICP27 and UL41. However, analysis of viral mutants showed that neither of these proteins is required for the loss of mt mRNA or mt DNA (Fig 1A,B), indicating that a different mechanism is involved. Depletion, however, required the viral ICP4 protein, a transcriptional activator that is essential for the expression of the viral early and late genes (Preston, 1979), implicating one or more early or late viral gene products. It seemed plausible that a viral nuclease might be involved in depleting mt DNA and/or mRNA. We therefore examined a mutant virus (AN-1; Weller et al, 1990) that is null for the UL12 gene encoding the HSV alkaline deoxyribonuclease—an early protein involved in viral DNA metabolism. Strikingly, the UL12 null mutant failed to deplete either mt mRNA or mt DNA (Fig 1A,B; supplementary Figs S1,S2 online). Moreover, these defects were complemented in the 6-5 cell line, a stably transfected derivative of Vero cells that expresses UL12 gene products on HSV infection (Shao et al, 1993) (Fig 1C,D), confirming that the phenotype of the UL12 mutant originates from the deletion of the UL12 gene rather than from an adventitious mutation located elsewhere in the viral genome. Together, these data raised the possibility that the alkaline nuclease encoded by the UL12 gene mediates the depletion of mt DNA and mt mRNA.
The HSV UL12 gene gives rise to a pair of 3′ co-terminal mRNAs that encode distinct but related proteins: full-length UL12 and an amino-terminally truncated version termed UL12.5, the translation of which is initiated at an internal methionine specified by codon 127 (Martinez et al, 1996a). Both the UL12 and UL12.5 proteins have endonuclease and 5′ → 3′ exonuclease activity on DNA substrates (Hoffmann & Cheng, 1978; Strobel-Fidler & Francke, 1980; Reuven et al, 2004) but only UL12 is required for efficient virus replication in tissue culture (Weller et al, 1990; Martinez et al, 1996b, 2002). UL12 localizes to the nucleus (Reuven et al, 2004) and complexes with the viral single-stranded DNA binding protein ICP8 to form a recombinase (Reuven et al, 2003) that is important in the generation of mature progeny viral DNA (Martinez et al, 1996b; Goldstein & Weller, 1998). UL12.5 is located predominantly in the cytoplasm (Reuven et al, 2004) and cannot fully substitute for UL12 in promoting viral genome maturation (Martinez et al, 1996b); however, its function has yet to be determined. Expanding on these earlier observations, we found that transiently expressed UL12.5 localized to cytoplasmic fibrillar structures that co-stain with an antibody directed against the mitochondrial protein cytochrome c, in addition to fainter nuclear staining in a subset of cells (Fig 2A); by contrast, transiently expressed UL12 was entirely nuclear, as previously described (Reuven et al, 2004). In addition, a C-terminal UL12.5–eGFP (enhanced green fluorescent protein) fusion protein colocalized with the mitochondrial dye MitoTracker Red in live cells, in contrast to the uniform distribution of unmodified eGFP through the cells (Fig 2B). Thus, UL12.5 localizes predominantly to mitochondria, raising the possibility that it has a role in depleting mt DNA.
Figure 2.
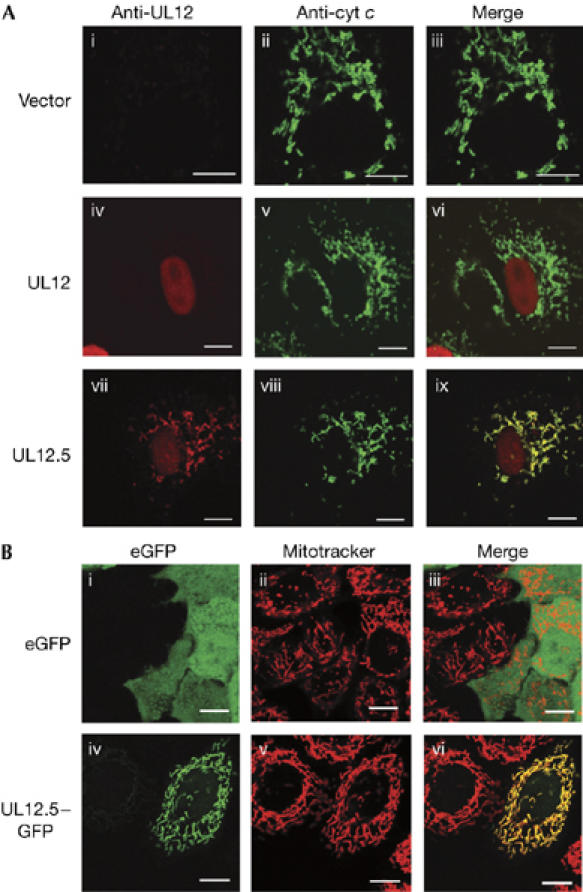
UL12.5 localizes to mitochondria. (A) HeLa cells were transfected with empty vector (i–iii), plasmids expressing UL12 (iv–vi) or UL12.5 (vii–ix) and then analysed by immunofluorescence for UL12/UL12.5 (i,iv,vii; red) and cytochrome c (ii,v,viii; green). The merged images (iii,vi,ix) document the colocalization of UL12.5 with cytochrome c. (B) UL12.5–eGFP fusion protein localizes to mitochondria. Live HeLa cells were transfected with plasmids encoding eGFP (i–iii) or UL12.5–eGFP (iv–vi) and stained with MitoTracker Red. The merged images (iii,vi) document the localization of UL12.5–eGFP to mitochondria. Scale bars, 10 μm. eGFP, enhanced green fluorescent protein.
We investigated whether UL12 and/or UL12.5 are able to provoke mt DNA loss in the absence of other HSV gene products. 143B osteosarcoma cells were co-transfected with UL12 or UL12.5 expression plasmids and a vector encoding an orange variant of monomeric red fluorescent protein derived from Discosoma sp (mOrange (Shaner et al, 2004), included to allow identification of transfected cells). Transfected cells transiently expressing mOrange were isolated 24 h later by using a fluorescence activated cell sorter, plated at low cell density and then incubated for 2 days to allow several rounds of cell division. The resulting small colonies were then evaluated for mt DNA by staining with PicoGreen (Fig 3). Control cells transfected with empty vector invariably showed numerous cytoplasmic PicoGreen staining foci (Fig 3A,Bi,iv), which have been shown to correspond to mt DNA nucleoids (Ashley et al, 2005). As expected, these foci were associated with mitochondria identified by using MitoTracker Far Red (Fig 3Ai,ii). Strikingly, approximately 70% of the colonies arising from cells transfected with the UL12.5 expression vector were uniformly devoid of PicoGreen staining mitochondrial foci (Fig 3Biii,vi). A further 9% of the colonies showed a mosaic phenotype, being composed of a mixture of positive and negative cells (data not shown). By contrast, more than 99% of the colonies arising from the UL12 transfection showed a PicoGreen staining pattern that was indistinguishable from that obtained with empty vector (Fig 3Bii,v). These data indicate that UL12.5 is sufficient to deplete mt DNA in the absence of other HSV proteins, whereas full-length UL12 has little, if any activity in this transient transfection assay. However, we note that in HSV-infected cells, the full-length UL12 open reading gives rise to significant levels of truncated protein products with electrophoretic mobilities similar to those of UL12.5 (Martinez et al, 2002), probably by proteolysis, and it is possible that these truncated products share with UL12.5 the ability to localize to mitochondria and deplete mt DNA. Further studies are required to test this possibility.
Figure 3.

UL12.5 is sufficient for mt DNA depletion. (A) Cytoplasmic PicoGreen foci localize to mitochondria. 143B cells co-transfected with empty vector and pcDNA-mOrange plasmid were stained with PicoGreen (green) and MitoTracker Far Red (red). The mOrange signal is not shown. Panel ii shows a higher magnification of the boxed region in panel i, documenting the association of PicoGreen-stained mt DNA nucleoids with the MitoTracker signal. (B) 143B cells were co-transfected with pcDNA-mOrange and empty vector (i,iv), plasmids expressing UL12 (ii,v) or UL12.5 (iii,vi). Colonies arising from individual cells expressing mOrange were examined by staining with PicoGreen and Mitotracker Far Red two days after plating. The mOrange signal is not shown. Panels iv, v and vi show a higher magnification of the boxed regions in panels i, ii and iii, respectively. Scale bars, 10 μm. mt, mitochondrial.
Transient expression of UL12.5 did not trigger the release of cytochrome c from mitochondria (Fig 2) or induce clear morphological signs of apoptosis; moreover, the transfected cells were able to undergo at least several rounds of cell division (Fig 3). These results indicate that transient expression of UL12.5 does not immediately induce lethal cellular damage, despite the profound depletion of mt DNA.
Mammalian cells that have been completely depleted of mt DNA (rho0 cells) through long-term exposure to ethidium bromide, or other agents that prevent mt DNA replication, eventually lose their capacity for oxidative phosphorylation and hence require supplementary uridine and pyruvate to proliferate (King & Attardi, 1989). To determine if the rho0 cells that are rapidly generated on transient expression of UL12.5 show similar properties, individual 143B cells transiently expressing UL12.5 were sorted into the wells of a 96-well plate one day after transfection and incubated in medium supplemented with pyruvate and uridine for several weeks. The resulting clonal cell lines were then isolated, expanded and propagated in the same medium. Five of seven such cell lines examined were devoid of mt DNA, as judged by staining with PicoGreen (Fig 4A; additional data not shown). These rho0 derivatives were unable to divide in normal tissue culture medium (Fig 4B), confirming their respiratory defect. These data show that transient expression of UL12.5 provides a rapid and efficient method of generating viable rho0 mammalian cells. The method does not rely on inhibiting mt DNA replication and therefore is likely is to prove useful for various previously inaccessible applications, for example depletion of damaged mt DNA from non-proliferating cells as a prelude to gene therapy.
Figure 4.

Isolation of rho0 143B cell lines. (A) Clonal cell lines isolated after transient expression of UL12.5 were scored for mt DNA content by staining with PicoGreen and MitoTracker Red. (i) Control 143B cells, (ii) clone E4, which retains mt DNA and (iii,iv) clones A8 and E2, which lack mt DNA. Scale bars, 10 μm. (B) Rho0 143B cells show the nutritional requirements of cells lacking mt DNA. The indicated cell lines were cultured in the presence (+) and absence (−) of supplemented pyruvate and uridine. mt, mitochondrial; rho0, cells depleted of mt DNA.
It seems likely that mt DNA is directly degraded by the DNase activity of UL12.5; consistent with this hypothesis, our ongoing mutational analysis of UL12.5 has yet to uncouple mt DNA depletion from the nuclease activity of this protein (J.A.C., H.A.S. & J.R.S., unpublished data). However, it is less clear how the mt mRNAs are depleted. The simplest possibility is that they decay through normal mt mRNA turnover mechanisms after loss of the mitochondrial genome. However, we note that the mt mRNAs are depleted as rapidly as the mt DNA after HSV infection (Fig 1), an observation that is difficult to reconcile with a simple template loss model for mt mRNA depletion, as mt mRNAs are normally stable, decaying with a half-life of approximately 3 h in uninfected cells (Chandrasekaran et al, 2004). These considerations raise the possibility that mt mRNAs are actively destabilized in HSV-infected cells. In this context, it might be significant that the Kaposi's sarcoma herpesvirus (KSHV) UL12 orthologue triggers degradation of most mRNAs encoded by the nuclear genome (Glaunsinger & Ganem, 2004), a function that can be genetically separated from its DNase activity (Glaunsinger et al, 2005). Further studies are therefore required to determine if UL12.5, or UL12, actively destabilizes mt mRNAs in a manner analogous to the action of its KSHV orthologue on cytoplasmic mRNAs, in addition to eliminating mt DNA.
Mitochondria have key roles in orchestrating cellular antiviral defences, including apoptosis and the type I interferon response (Green, 2005; Hiscott et al, 2006), in addition to their crucial respiratory function. It is therefore tempting to speculate that HSV-induced depletion of mt DNA and/or mt mRNA neutralizes one or more host defence mechanisms. However, our studies have yet to provide any support for this hypothesis and further work is therefore needed to determine whether and how mt DNA depletion benefits HSV. In addition, it will be important to determine if other members of the herpesviridae also provoke mt DNA depletion.
Mt DNA depletion has been linked to a wide range of inherited and acquired degenerative disorders in humans (reviewed by Dimauro, 2004; Wallace, 2005), illustrating the profound impact of impaired mitochondrial function in vivo. It is therefore possible that mt DNA depletion contributes to the pathogenesis of HSV infections. For example, HSV establishes latency in sensory neurons, a type of cell that is highly sensitive to the pathological effects of mt DNA damage (Spinazzola & Zeviani, 2005). Thus, it will be of great interest to determine if mt DNA is depleted from sensory neurons during reactivation from latency and, if so, whether such depletion contributes to the pain and/or other sensory symptoms associated with reactivation episodes. In addition, it is important to determine whether cells that have been depleted of mt DNA by HSV or other herpesviruses are ever able to survive long-term in the intact host. In this context, we note that limited expression of viral lytic phase genes, such as UL12.5, is not invariably lethal to the infected cell (Khanna et al, 2004). Thus, it is conceivable that HSV and/or other herpesviruses might contribute to the aetiology of some of the degenerative conditions that have been associated with somatic mt DNA loss or damage. Further studies are required to examine this possibility.
Methods
Cells, viruses, plasmids and transfection. Cell lines, viral mutants, plasmids and transfection protocols are described in the supplementary information online.
Nucleic acid isolation and hybridization. Total cellular RNA or DNA was analysed for mt mRNA and mt DNA by northern or Southern blot hybridization by using probes for COX2 (IMAGE Consortium clone #3894223), ND6 or the entire mitochondrial genome, as described in the supplementary information online.
Immunofluorescence microscopy. Vero cells growing on glass coverslips were transfected with pSAK-UL12, pSAK-UL12.5 or the empty vector pcDNA3.1, fixed and stained with antibodies directed against UL12 or cytochrome c, then examined by confocal microscopy as detailed in the supplementary information online.
Imaging of mitochondrial DNA in living cells. HeLa or 143B cells growing in glass-bottomed coverglass slides (NUNC) were stained with PicoGreen (3 μl/ml; Invitrogen, Burlington, ON, Canada) for 1 h at 37°C. In some experiments, MitoTracker Deep Red (1 mM; Invitrogen) was added 30 min after the addition of PicoGreen. Cells were then washed three times with warm medium, and visualized and photographed immediately under a Zeiss LSM510 scanning argon (488 nm) and helium neon (543 and 650 nm) laser confocal microscope, by using a × 40 oil immersion objective.
Cell sorting. 143B cells were co-transfected with pcDNA-mOrange and pcDNA3.1, pSAK-UL12 or pSAK-UL12.5. One day after transfection, cells were trypsinized and sorted by using a FACSAria cell sorter (BD Biosciences, Mississauga, ON, Canada) equipped with standard lasers and detectors. Cells showing red fluorescence were sorted into medium containing 10% FBS supplemented with uridine (50 μg/ml) and sodium pyruvate (100 μg/ml), then cultured. In some experiments, individual cells were sorted into the wells of a 96-well plate to isolate clonal cell lines.
Supplementary information is available at EMBO reports online (http://www.emboreports.org).
Supplementary Material
Supplementary Information
Acknowledgments
We thank R. Maranchuk and D. Rutkowski for technical assistance, and M. Barry, D. Evans, R. Irvin and D. Wilkinson for helpful comments on the manuscript. This research was supported by grants from the Canadian Institutes for Health Research to J.R.S. and Public Health Service grant AI-21747 from the National Institute of Health to S.K.W. J.R.S. holds a Canada Research Chair in Molecular Virology.
References
- Ashley N, Harris D, Poulton J (2005) Detection of mitochondrial DNA depletion in living human cells using PicoGreen staining. Exp Cell Res 303: 432–446 [DOI] [PubMed] [Google Scholar]
- Beal MF (2005) Mitochondria take center stage in aging and neurodegeneration. Ann Neurol 58: 495–505 [DOI] [PubMed] [Google Scholar]
- Chandrasekaran K, Mehrabian Z, Li XL, Hassel B (2004) RNase-L regulates the stability of mitochondrial DNA-encoded mRNAs in mouse embryo fibroblasts. Biochem Biophys Res Commun 325: 18–23 [DOI] [PubMed] [Google Scholar]
- Dimauro S (2004) Mitochondrial medicine. Biochim Biophys Acta 1659: 107–114 [DOI] [PubMed] [Google Scholar]
- Fernandez-Silva P, Enriquez JA, Montoya J (2003) Replication and transcription of mammalian mitochondrial DNA. Exp Physiol 88: 41–56 [DOI] [PubMed] [Google Scholar]
- Fortunato EA, Spector DH (2003) Viral induction of site-specific chromosome damage. Rev Med Virol 13: 21–37 [DOI] [PubMed] [Google Scholar]
- Glaunsinger B, Ganem D (2004) Lytic KSHV infection inhibits host gene expression by accelerating global mRNA turnover. Mol Cell 13: 713–723 [DOI] [PubMed] [Google Scholar]
- Glaunsinger B, Chavez L, Ganem D (2005) The exonuclease and host shutoff functions of the SOX protein of Kaposi's sarcoma-associated herpesvirus are genetically separable. J Virol 79: 7396–7401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein JN, Weller SK (1998) In vitro processing of herpes simplex virus type 1 DNA replication intermediates by the viral alkaline nuclease, UL12. J Virol 72: 8772–8781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DR (2005) Apoptotic pathways: ten minutes to dead. Cell 121: 671–674 [DOI] [PubMed] [Google Scholar]
- Hardwicke MA, Sandri-Goldin RM (1994) The herpes simplex virus regulatory protein ICP27 contributes to the decrease in cellular mRNA levels during infection. J Virol 68: 4797–4810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy WR, Sandri-Goldin RM (1994) Herpes simplex virus inhibits host cell splicing, and regulatory protein ICP27 is required for this effect. J Virol 68: 7790–7799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiscott J, Lin R, Nakhaei P, Paz S (2006) MasterCARD: a priceless link to innate immunity. Trends Mol Med 12: 53–56 [DOI] [PubMed] [Google Scholar]
- Hoffmann PJ, Cheng YC (1978) The deoxyribonuclease induced after infection of KB cells by herpes simplex virus type 1 or type 2. I. Purification and characterization of the enzyme. J Biol Chem 253: 3557–3562 [PubMed] [Google Scholar]
- Khanna KM, Lepisto AJ, Hendricks RL (2004) Immunity to latent viral infection: many skirmishes but few fatalities. Trends Immunol 25: 230–234 [DOI] [PubMed] [Google Scholar]
- King MP, Attardi G (1989) Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science 246: 500–503 [DOI] [PubMed] [Google Scholar]
- Kwong AD, Frenkel N (1987) Herpes simplex virus-infected cells contain a function(s) that destabilizes both host and viral mRNAs. Proc Natl Acad Sci USA 84: 1926–1930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latchman DS (1988) Effect of herpes simplex virus type 2 infection on mitochondrial gene expression. J Gen Virol 69: 1405–1410 [DOI] [PubMed] [Google Scholar]
- Lomonte P, Everett RD (1999) Herpes simplex virus type 1 immediate-early protein Vmw110 inhibits progression of cells through mitosis and from G1 into S phase of the cell cycle. J Virol 73: 9456–9467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez R, Shao L, Bronstein JC, Weber PC, Weller SK (1996a) The product of a 1.9-kb mRNA which overlaps the HSV-1 alkaline nuclease gene (UL12) cannot relieve the growth defects of a null mutant. Virology 215: 152–164 [DOI] [PubMed] [Google Scholar]
- Martinez R, Sarisky RT, Weber PC, Weller SK (1996b) Herpes simplex virus type 1 alkaline nuclease is required for efficient processing of viral DNA replication intermediates. J Virol 70: 2075–2085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez R, Goldstein JN, Weller SK (2002) The product of the UL12.5 gene of herpes simplex virus type 1 is not essential for lytic viral growth and is not specifically associated with capsids. Virology 298: 248–257 [DOI] [PubMed] [Google Scholar]
- Murata T, Goshima F, Daikoku T, Kyoko I-O, Takakuwa H, Kato K, Nishiyama Y (2000) Mitochondrial distribution and function in herpes simplex virus-infected cells. J Gen Virol 81: 401–406 [DOI] [PubMed] [Google Scholar]
- Preston CM (1979) Control of herpes simplex virus type 1 mRNA synthesis in cells infected with wild-type virus or the temperature-sensitive mutant tsK. J Virol 29: 275–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuven NB, Staire AE, Myers RS, Weller SK (2003) The herpes simplex virus type 1 alkaline nuclease and single-stranded DNA binding protein mediate strand exchange in vitro. J Virol 77: 7425–7433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuven NB, Antoku S, Weller SK (2004) The UL12.5 gene product of herpes simplex virus type 1 exhibits nuclease and strand exchange activities but does not localize to the nucleus. J Virol 78: 4599–4608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY (2004) Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol 22: 1567–1572 [DOI] [PubMed] [Google Scholar]
- Shao L, Rapp LM, Weller SK (1993) Herpes simplex virus 1 alkaline nuclease is required for efficient egress of capsids from the nucleus. Virology 196: 146–162 [DOI] [PubMed] [Google Scholar]
- Smiley JR (2004) Herpes simplex virus virion host shutoff protein: immune evasion mediated by a viral RNase? J Virol 78: 1063–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer CA, Dahmus ME, Rice SA (1997) Repression of host RNA polymerase II transcription by herpes simplex virus type 1. J Virol 71: 2031–2040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinazzola A, Zeviani M (2005) Disorders of nuclear–mitochondrial intergenomic signaling. Gene 354: 162–168 [DOI] [PubMed] [Google Scholar]
- Strobel-Fidler M, Francke B (1980) Alkaline, deoxyribonuclease induced by herpes simplex virus type 1: composition and properties of the purified enzyme. Virology 103: 493–501 [DOI] [PubMed] [Google Scholar]
- Wallace DC (2005) A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet 39: 359–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weller SK, Seghatoleslami MR, Shao L, Rowse D, Carmichael EP (1990) The herpes simplex virus type 1 alkaline nuclease is not essential for viral DNA synthesis: isolation and characterization of a lacZ insertion mutant. J Gen Virol 71: 2941–2952 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information