

Abstract
Friedreich ataxia is caused by decreased levels of frataxin, a mitochondrial acidic protein that is assumed to act as chaperone in the assembly of Fe–S clusters on the scaffold Isu protein. Frataxin has the in vitro capacity to form iron-loaded multimers, which also suggests an iron storage function. It has been reported that alanine substitution of residues in an acidic ridge of yeast frataxin (Yfh1) elicits loss of iron binding in vitro but has no effect on Fe–S cluster synthesis in vivo. Here, we show that a marked change in the electrostatic properties of a specific region of Yfh1 surface—by substituting two or four acidic residues by lysine or alanine, respectively—impairs Fe–S cluster assembly, weakens the interaction between Yfh1 and Isu1, and increases oxidative damage. Therefore, the acidic ridge is essential for the Yfh1 function and is likely to be involved in iron-mediated protein–protein interactions.
Keywords: acidic ridge, Fe–S cluster, frataxin, Isu1, mitochondria, acidic residue
Introduction
Friedreich ataxia, a progressive recessive neurodegenerative disease, is caused by a deficit in the levels of frataxin, a small acidic protein localized to mitochondria in eukaryotes (Campuzano et al, 1996). Frataxin has a role in the biosynthesis of Fe–S clusters, as suggested by severe alteration of the activity and assembly of Fe–S proteins in frataxin-deficient cells (Rötig et al, 1997; Duby et al, 2002; Mühlenhoff et al, 2002). A link with Fe–S cluster formation was confirmed by independent lines of evidence. In vitro and at low iron to protein ratios, monomeric frataxins from different organisms bind both Fe2+ and Fe3+ ions with a defined stoichiometry (Adinolfi et al, 2002; He et al, 2004; Nair et al, 2004; Cook et al, 2006). Yeast frataxin forms a complex with Isu–IscU, the scaffold protein that assembles Fe–S clusters, and with Nfs1–IscS, the cysteine desulphurase that provides the sulphur atoms (Gerber et al, 2003; Ramazzotti et al, 2004). It is thus widely accepted that frataxin is an iron chaperone, which delivers iron to Isu (Yoon & Cowan, 2003) or IscS (Layer et al, 2006). It has also been proposed that frataxin has an iron storage function. Defects in frataxin increase sensitivity to oxidants and in vitro, at low ionic strength and high iron to protein ratios, frataxins aggregate as large polymeric structures that trap numerous iron atoms (Adamec et al, 2000; Adinolfi et al, 2002).
The three-dimensional structure of frataxin is highly conserved from bacteria to humans and is characterized by a β-sheet that packs against two α-helices (Cho et al, 2000; Dhe-Paganon et al, 2000; Musco et al, 2000; He et al, 2004). The first helix and the edge of the β1-sheet form a semi-conserved acidic ridge. The role of the acidic ridge in iron binding has been shown in vitro by nuclear magnetic resonance spectroscopy and mutant studies (Nair et al, 2004; Cook et al, 2006). A triple E18K/E19K/D22K mutation of CyaY leads to the complete loss of iron-promoted protein aggregation and Fe2+ ion binding to the monomers, indicating that these residues have the highest affinity for this cation (Adinolfi et al, 2002; Nair et al, 2004). In yeast, replacement of several acidic residues of Yfh1 by neutral residues encompassing the two presumed iron-binding sites causes the loss of iron-promoted aggregation (D86A(N)/E90A(Q)/E93A(Q), D78N/D82N/E89Q, D79N/D86N/E90Q, E93A/D97A/E103A) or ferroxidase activity (D79A/D82A). However, the yeast mutants show only a slight increase in cellular oxidative damage and no defect in the biosynthesis of Fe–S clusters (Aloria et al, 2004; Gakh et al, 2006). This has raised the puzzling question of the biological role of the conserved acidic ridge. Here, we show that replacing four of the most conserved residues of the frataxin family—D86, E89, D101 and E103—either by lysines or by alanines severely impairs the metabolism of Fe–S clusters in vivo.
Results
Mutations D86K, E89K, D101A, D86A/E89A, D86K/E89K, D101A/E103A, D101K/E103K and D86A/E89A/D101A/E103A were obtained by site-directed mutagenesis of the YFH1 gene (Fig 1) and introduced into yeast. These positions are exposed to the solvent in the frataxin structure and therefore should not affect the stability of the three-dimensional protein fold. In CyaY, lysine residues introduced at equivalent positions markedly change the electrostatic potential of the protein without affecting the tertiary structure (Adinolfi et al, 2002; A.P. & C. Pastore, unpublished data).
Figure 1.
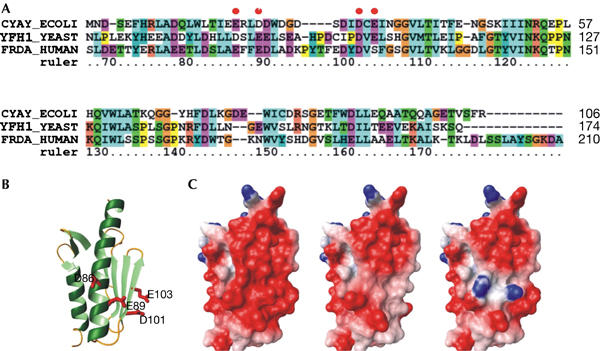
Sequence and structural comparisons. (A) Sequence alignment of the conserved domain of Escherichia coli (CyaY), yeast (Yfh1) and human frataxins. The numbering refers to the Yfh1 sequence. (B) Ribbon representation of the conserved domain of Yfh1 (2ga5). The side chains of the residues mutated in this article are shown in red. (C) Comparison of the electrostatic surfaces of Yfh1 wild type (left), D86A/E89A/D101A/E103A (middle) and D86K/E89K (right).
Cell sensitivity to iron
Iron is toxic to yeast cells deficient in frataxin as a consequence of the profound alteration in iron homeostasis, which leads to the accumulation of iron in mitochondria (Babcock et al, 1997; Foury & Cazzalini, 1997). Moreover, when Δyfh1 cells are grown in high-iron media, mitochondrial DNA is lost. On addition of 7 mM FeSO4 in the culture medium, the cellular growth of mutants in which a single acidic residue was changed into lysine, or two residues into alanine, was not, or only very slightly, affected (Fig 2A). By contrast, the growth of the D86K/E89K, D101K/E103K and D86A/E89A/D101A/E103A yfh1 mutants was severely inhibited in high-iron media. A high loss of mitochondrial DNA promoted by iron was also observed (Fig 2B). All mutants were more sensitive to hydrogen peroxide than wild type (data not shown), but strong oxidative damage was observed only in mutants harbouring two substitutions with lysine or four substitutions with alanine.
Figure 2.
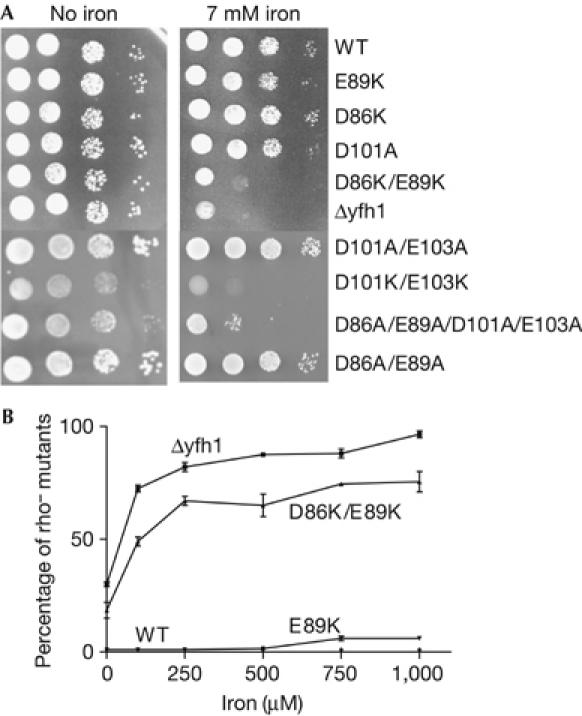
Iron sensitivity of wild-type and mutant cells. (A) Serial dilutions of cell suspensions were spotted onto plates containing glucose minimum medium and different concentrations of FeSO4, and cells were grown at 28°C for 3 days. (B) Cells were grown in liquid glucose-rich medium for 16 h in the presence of increasing concentrations of FeSO4 and spread for single colonies on minimum medium. The small colonies were identified as respiratory-deficient rho− mutants. WT, wild type; Δyfh1, YFH1 gene-deleted strain.
Frataxin levels in wild type and mutants
In a previous report, we showed that Yfh1 levels were low in several temperature-sensitive mutants (Ramazzotti et al, 2004). This probably results from protein degradation by the mitochondrial quality control protease machinery that has recognized folding defects (Wagner et al, 1994). By contrast, Yfh1 levels in all mutants of this study were similar to those of wild type (Fig 3), providing confirmation that the Yfh1 fold is unaffected by the lysines. The electrophoretic mobility of D101A and D101K/E103K Yfh1 in SDS–PAGE was only very slightly increased compared with that of wild-type Yfh1, which migrates as a 20-kDa protein (Branda et al, 1999), whereas D86K, E89K, D86A/E89A, D86K/E89K and D86A/E89A/D101A/E103A Yfh1 migrated roughly as expected for a 14-kDa protein. A similar modification of the electrophoretic mobility was reported for D79A/D82A Yfh1 by Gakh et al (2006), who verified that the molecular weight was identical for wild-type and D79A/D82A Yfh1 by using mass spectrometry.
Figure 3.
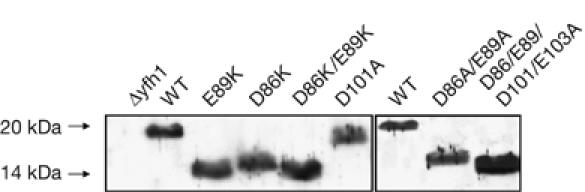
Electrophoretic mobility of wild-type and mutant Yfh1 in SDS–polyacrylamide gels. A 40 μg portion of mitochondrial proteins was loaded onto 14% polyacrylamide gels. Yfh1 was detected by western blot analysis using a polyclonal Yfh1 antibody. D86/E89/D101/E103A is D86A/E89A/D101A/E103A. WT, wild type.
Defect of Fe–S cluster assembly in isolated mitochondria
Fe–S cluster assembly was followed by measuring the incorporation of a 2Fe-2S cluster into 35S-radiolabelled apo-Yah1 ferredoxin, synthesized in vitro and imported into isolated energized mitochondria (Fig 4; Duby et al, 2002). The kinetics of conversion of apo- to holo-Yah1 were not affected in D86K, and only slightly affected in the E89K yfh1 mutant. However, the conversion rate to the holo-form was substantially decreased in the D86K/E89K yfh1 mutant. These data show the synergistic effect of the double substitution on Fe–S cluster assembly.
Figure 4.
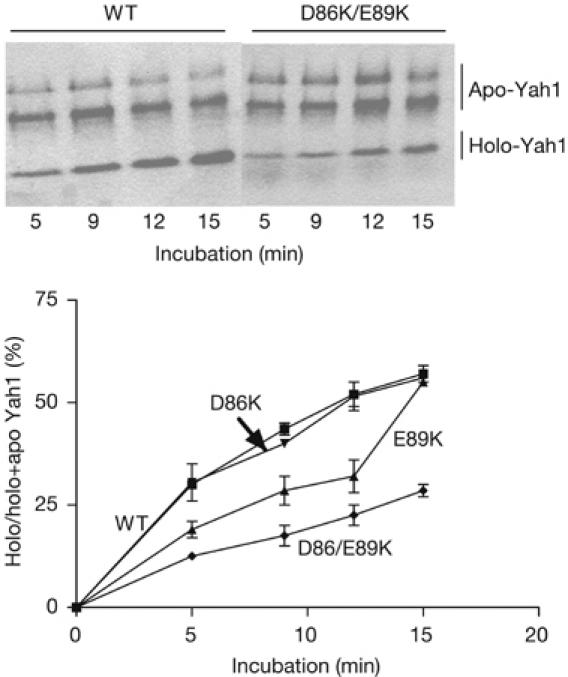
Conversion of 35S-radiolabelled apo- to holo-Yah1 in isolated energized mitochondria from wild-type and mutant strains. In native gel electrophoresis, the acidic mature form of Yah1 gives a fast migrating holo-form and two slowly migrating reduced and oxidized apo-forms (Leibrecht & Kessler, 1997). Exposure time of the autoradiography was 10 days. The experiment was carried out twice for each strain. WT, wild type.
Aconitase activity and mitochondrial iron load
Cells were grown in a synthetic medium supplemented with raffinose, a good respiratory carbon source. When no extra iron is added to the medium, iron does not accumulate in mitochondria even in a null yfh1 strain (Foury, 1999), and thus cellular oxidative damage is reduced to a minimal level. Under these conditions, the activity of aconitase—an enzyme requiring a 4Fe–4S cluster for its activity—was decreased only in D86K/E89K, D101K/E103K and D86A/E89A/D101A/E103A mutants (Table 1, No iron). However, when 5 μM FeSO4 was added to the medium (Table 1, 5 μM iron), aconitase activity decreased even in the least severe mutants, although to different extents. Although the mean values of the mitochondrial iron concentrations from several independent experiments, carried out with wild-type and mild mutants, were not statistically different (except for E89K), in each separate experiment taken, the iron concentration was always higher in mutants than in wild type (data not shown). This small disturbance in iron homeostasis is probably the cause of the decrease in the activity of aconitase by losing its external iron atom, which is extremely sensitive to free radicals.
Table 1.
Aconitase activity and mitochondrial iron content
| Strain | No iron* | 5 μM iron* | |||
|---|---|---|---|---|---|
| Aconitase† | Mitochondrial iron‡ (nmol/mg protein) | Aconitase† | |||
| Wild type | 100 | 15±7 | 100 | ||
| D86K | 93±3 | 27±2 | 70±7 | ||
| E89K | 81±5 | 49±4 | 55±8 | ||
| D101A | 95±2 | 21±5 | 73±8 | ||
| D86A/E89A | 90±2 | 22±2 | 75±3 | ||
| D86K/E89K | 31±8 | 82±10 | 18±6 | ||
| D101K/E103K | 34±6 | 75±8 | 20±5 | ||
| D86A/E89A/D101A/E103A | 30±6 | 81±7 | 17±6 | ||
| Δyfh1 | 19±5 | 82±8 | 14±6 | ||
*Assays were performed with mitochondrial extracts from cells grown in raffinose synthetic medium in the absence or in the presence of 5 μM FeSO4. The data are averages obtained from several cultures of two independently isolated transformants.
†Aconitase activity was normalized to isocitrate dehydrogenase activity and expressed as a percentage of the control value of each experiment.
‡Free iron was measured using bathophenantroline sulphonate in the presence of dithionite (Foury & Cazzalini, 1997).
Weakened association of Yfh1 and Isu1 in mutants
We prepared soluble cell extracts from yeast strains overexpressing Yfh1 and Isu1, and used affinity chromatography with an antiserum raised against Yfh1 to detect a physical interaction between Yfh1 and Isu1. Wild-type Yfh1 was quantitatively bound by the Yfh1 antibody, and a substantial fraction of Isu1 (30–50%) was also bound to the resin and co-eluted with Yfh1 at low pH (Fig 5A). When a preimmune serum was used instead of the anti-Yfh1 serum (Fig 5A), no detectable Yfh1 or Isu1 was recovered in the bound fraction. When EDTA was added during the washing steps, Yfh1 was recovered in the bound fraction but Isu1 was released (Fig 5B). These data show that the interaction between Isu1 and Yfh1 requires a metal, probably iron as previously reported (Gerber et al, 2003). When the same experiment was performed with D86K/E89K, no significant Isu1 binding was detected (Fig 5C). The fraction of bound Isu1 was slightly higher for the E89K and D86A/E89A/D101A/E103A mutants but remained very low compared with wild type (Fig 5C,D). These data show that the loss of negative charges at the surface of Yfh1 weakens the interaction between Isu1 and Yfh1. Considering the rather slight defects of the E89K yfh1 mutant in Fe–S cluster assembly, we were expecting a higher fraction of Isu1 to be associated with Yfh1. However, the complex formed by E89K Yfh1 and Isu1 is likely to be less stable and to dissociate during the preparation of cell extracts. It is also possible that although decreased, the levels of the E89K Yfh1 and Isu1 complex are not significantly limiting for the synthesis of Fe–S clusters. Consistently, no decrease in Fe–S cluster assembly has been observed in cells with low levels of frataxin (Aloria et al, 2004; Ramazzotti et al, 2004).
Figure 5.
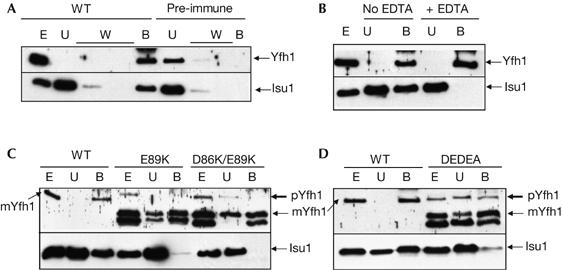
Physical association of Yfh1 and Isu1 in wild-type and mutant cell extracts, using immunoaffinity chromatography with a polyclonal antibody raised against Yfh1. Cell extracts (E), unbound (U) and bound (B) fractions were analysed by SDS–polyacrylamide gel electrophoresis and western blotting using antibodies against Yfh1 and Isu1. (A) Interaction between Yfh1 and Isu1 (wild type) and replacement of Yfh1 antibody by the pre-immune serum (pre-immune). (B) EDTA (5 mM) was added during the washing steps. (C, D) The interaction between Yfh1 and Isu1 is weak in yfh1 mutants. pYfh1 (precursor) and mYfh1 (mature) are shown for the mutants and only mYfh1 is shown for the wild type. In the mutants, the lower band corresponds to a degradation product of Yfh1. DEDEA, D86A/E89A/D101A/E103A; W, washing step fractions; WT, wild type.
Discussion
It was shown previously that substitutions of several acidic residues in helix 1 and strand 1 of Yfh1 by alanines result in loss of the in vitro capacity to form multimeric structures binding iron (Aloria et al, 2004; Gakh et al, 2006). In the yeast mutants, there was only a slight increase in sensitivity to oxidative stress, but no effect on Fe–S cluster assembly. It was thus suggested that frataxin had an iron storage function relevant to anti-oxidant defence, which is distinct from the chaperone function (Gakh et al, 2006). It must be noted, however, that experiments were performed in vitro without the biological partners of frataxin, such as Isu1 and Nfs1, and it can be expected that iron-binding properties within the complex formed by Nfs1, Isu1 and Yfh1 in vivo are different from those of Yfh1 alone in vitro.
Analysing another set of mutations, we show here that the acidic ridge is crucial for Yfh1 function and is involved in Fe–S cluster assembly. Loss of function, however, requires a marked change in the electrostatic properties of the Yfh1 surface, which is obtained by replacing either two acidic residues by lysines or four residues by alanines, whereas single mutants have a mild phenotype. In the D86K/E89K, D101K/E103K and D86A/E89A/D101A/E103A yfh1 mutants, the assembly of Fe–S clusters is severely altered, the interaction between Yfh1 and Isu1 is markedly weakened, and the cellular sensitivity to iron and hydrogen peroxide is increased. By contrast, a mild phenotype is observed in single lysine or double alanine mutants. An appropriate acidic environment is therefore required for the function of frataxin in Fe–S cluster assembly.
Our data do not support the idea that sensitivity to oxidative damage and defects in Fe–S cluster assembly proceed independently: there is a strict correlation between the degree of sensitivity to oxidative agents and defects in Fe–S cluster assembly, D86K/E89K, D101K/E103K and D86A/E89A/D101A/E103A exhibiting both the greatest sensitivity to oxidative agents and the least efficient assembly of Fe–S clusters. We propose that the least severe mutants have a defect in Fe–S cluster synthesis that is under the detection limits of our standard assays. It must be emphasized that an in vivo phenotype can only be detected when the function becomes limiting for the metabolism of the cell. We, and others, have previously found that under standard culture conditions, low Yfh1 levels are not a limiting factor for Fe–S cluster synthesis in yeast (Aloria et al, 2004; Ramazzotti et al, 2004). Therefore, mild Yfh1 defects can remain ‘silent' in vivo during Fe–S cluster synthesis. These subtle defects are, however, sensed by the cell, which responds by increasing the transcription of the genes involved in iron uptake (F.F., unpublished data). This leads to mitochondrial iron overload in iron-replete culture media and an increase in oxidative damage, in a manner that is proportional to the Fe–S cluster metabolism defect. These latter traits are commonly shared by the mutants in other components of the mitochondrial Fe–S cluster synthesis machinery. Therefore, we propose that a defect in the assembly of Fe–S clusters is the primary event in our acidic mutants, whereas increased oxidative damage might be secondary to the abnormal iron status resulting from the defect in Fe-S cluster assembly (Rouault & Tong, 2005). It must also be mentioned that in mammals the iron storage property is assumed by a distinct mitochondrial ferritin (Levi & Arosio, 2004).
The severe defects of the D86K/E89K, D101K/E103K and D86A/E89A/D101A/E103A yfh1 mutants highlight the crucial role of the sites centred around the strongly conserved D86/E89 and D101/E103 residues, respectively. By analogy with the data obtained for the E19K/D22K CyaY mutant, which shows complete loss of Fe2+ ion binding to monomers whereas Fe3+ ion binding around sites 29–33 is retained (Nair et al, 2004), it might be proposed that D86 and E89—the yeast equivalent residues of E19 and D22—host the first populated iron-binding site, whereas D101 and E103—the equivalent residues of D31 and E33—might be part of a secondary site. Our data show that the four residues fulfil their role in the frataxin function cooperatively, as neutralization of all four acidic residues is required to abrogate this function. On the basis of the role of Yfh1 carboxylate groups in iron binding in vitro (Cook et al, 2006), the observation that loss of two negative charges is not deleterious was unexpected. However, there are examples of metal-binding domains that can tolerate a change of acidic residues to alanine or glycine. A comparison of the calcium-binding sites of the EF-hand protein family shows that loss of the carboxyl oxygen-providing aspartates at positions 1, 3 or 12 abolishes calcium binding, whereas substitutions at positions 5 and 9 of the EF loop by glycine or alanine residues are tolerated (Kawasaki et al, 1998). In certain cases, these small residues allow metal coordination by a water molecule. By contrast, bulky basic residues, which drastically change the surface properties of a protein, are never tolerated (Kawasaki et al, 1998). In our yfh1 mutants, positive charges at one site (D86/E89 or D101/E103) are likely to prevent access of Yfh1 ligands such as iron and/or Isu1 or Nfs1 to both sites.
In conclusion, our in vivo approach has identified four acidic residues in two regions of the acidic ridge that act cooperatively in the interaction between Yfh1 and Isu1. A three-dimensional structure of the complex is now required to understand more precisely the mechanisms mediating iron binding and Yfh1/Isu1 interaction.
Methods
Strains, plasmids and media. The parental strain was W303-1BΔyfh1 (MATalpha ade2 ura3 his3 leu2 trp1 yfh1Δ∷KanMX4 rho+; Foury & Cazzallini, 1997). This strain was transformed with the pFL39 centromeric plasmid harbouring wild-type YFH1 or mutated yfh1 genes in a DNA fragment encompassing the two HindIII sites, upstream and downstream of YFH1, respectively. For analysis of Yfh1 and Isu1 interactions, W303-1BΔyfh1 was co-transformed with two multi-copy plasmids as follows. The ISU1 gene was harboured by the pRS425 plasmid, and wild-type or mutated yfh1 genes were expressed under the control of the GAL1 promoter in the Yeplac195 plasmid (pGAL195). GAL1 promoter was inserted into the EcoRI and BamHI sites of the polylinker of Yeplac195, and the YFH1 (yfh1) open-reading frames were amplified by PCR using the oligonucleotides CTAAGCGAGAAGATCTAGTGTAGCAATGA and GTTCAGCTGAGTTTCTTTATAGATGACGTTG, and cloned into the BamHI and SalI sites of pGAL195 using artificial BglII and SalI sites created in the oligonucleotides.
Incorporation of a 2Fe–2S cluster in apo-Yah1 in isolated energized mitochondria. Mitochondria were prepared from raffinose-grown cells collected at the end of the exponential phase. In vitro synthesis of 35S-labelled apo Yah1, import into mitochondria, proteinase K treatment, lysis of mitochondria by Triton X-100, native polyacrylamide gel electrophoresis and autoradiography were carried out as reported previously (Duby et al, 2002).
Co-purification of Yfh1 and Isu1. Nab protein A Spin Chromatography kit (PIERCE; Perbio Science; UK Ltd, Northumberland, UK) was used for co-purification. Raffinose-grown cells harbouring pGAL195-YFH1 and pRS425-ISU1 plasmids were inoculated in 2% galactose-rich medium and incubated at 28°C for 5 h. Two grams of cells were lysed in 4 ml of buffer A (20 mM Hepes pH 7.4, 50 mM NaCl and 2 mM phenylmethyl sulphonyl fluoride (PMSF)) by vortexing for 3 min with 4 g of glass beads. After centrifugation at low speed to eliminate cell debris, the supernatant was centrifuged at 100,000g for 45 min. The soluble extract (200 μl) was diluted twofold with 200 μl of buffer B (50 mM sodium phosphate and 75 mM NaCl, pH 7.4) and mixed on a rocker at 4°C for 3 h with 100 μl of the anti-Yfh1 antibody affinity resin. The bound material was washed twice with 400 μl of buffer B and eluted with 400 μl of 0.1 M glycine pH 2.5, which releases immunoglobulins from the resin-immobilized protein A. The eluate was neutralized with 40 μl of 1 M Tris–HCl pH 8.0.
Supplementary information is available at EMBO reports online (http://www.emboreports.org).
Supplementary Material
Supplementary Information
Acknowledgments
This work was funded by the Belgian National Fund for Scientific Research (F.F.) and by the Muscular Dystrophy Association and the Friedreich's Ataxia Research Alliance foundations (A.P.).
References
- Adamec J, Rusnak F, Owen WG, Naylor S, Benson LM, Gacy AM, Isaya G (2000) Iron-dependent self assembly of recombinant yeast frataxin: implications for Friedreich ataxia. Am J Hum Genet 67: 549–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adinolfi S, Trifuoggi M, Politou AS, Martin S, Pastore A (2002) A structural approach to understanding the iron-binding properties of phylogenetically different frataxins. Hum Mol Genet 11: 1865–1877 [DOI] [PubMed] [Google Scholar]
- Aloria K, Schilke B, Andrew A, Craig EA (2004) Iron-induced oligomerization of yeast frataxin homologue Yfh1 is dispensable in vivo. EMBO Rep 5: 1096–1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babcock M, de Silva D, Oaks R, Davis-Kaplan S, Jiralerspong S, Montermini L, Pandolfo M, Kaplan J (1997) Regulation of mitochondrial iron accumulation by Yfh1p, a putative homolog of frataxin. Science 276: 1709–1712 [DOI] [PubMed] [Google Scholar]
- Branda SS, Cavadini P, Adamec J, Kalousek F, Taroni F, Isaya G (1999) Yeast and human frataxin are processed to mature form in two sequential steps by the mitochondrial processing peptidase. J Biol Chem 274: 22763–22769 [DOI] [PubMed] [Google Scholar]
- Campuzano V et al. (1996) Friedreich's ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 271: 1423–1427 [DOI] [PubMed] [Google Scholar]
- Cho SJ, Lee MG, Yang JK, Lee JY, Song HK, Suh SW (2000) Crystal structure of Escherichia coli CyaY protein reveals a previously unidentified fold for the evolutionarily conserved frataxin family. Proc Natl Acad Sci USA 97: 8932–8937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook JD, Bencze KZ, Jankovic AD, Crater AK, Busch CN, Bradley PB, Stemmler AJ, Spaller MR, Stemmler TL (2006) Monomeric yeast frataxin is an iron-binding protein. Biochemistry 46: 7767–7777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhe-Paganon S, Shigeta R, Chi YI, Ristow M, Shoelson SE (2000) Crystal structure of human frataxin. J Biol Chem 275: 30753–30756 [DOI] [PubMed] [Google Scholar]
- Duby G, Foury F, Ramazzotti A, Herrmann J, Lutz T (2002) A non-essential function for yeast frataxin in iron–sulphur cluster assembly. Hum Mol Gene 11: 2635–2643 [DOI] [PubMed] [Google Scholar]
- Foury F (1999) Low iron concentration and aconitase deficiency in a yeast frataxin homologue deficient strain. FEBS Lett 456: 281–284 [DOI] [PubMed] [Google Scholar]
- Foury F, Cazzalini O (1997) Deletion of the yeast homologue of the human gene associated with Friedreich's ataxia elicits iron accumulation in mitochondria. FEBS Lett 411: 373–377 [DOI] [PubMed] [Google Scholar]
- Gakh O, Park S, Liu G, Macomber L, Imlay JA, Ferreira GC, Isaya G (2006) Mitochondrial iron detoxification is a primary function of frataxin that limits oxidative damage and preserves cell longevity. Hum Mol Genet 15: 467–479 [DOI] [PubMed] [Google Scholar]
- Gerber J, Mühlenhoff U, Lill R (2003) An interaction between frataxin and Isu1/Nfs1 that is crucial for Fe/S cluster synthesis on Isu1. EMBO Rep 4: 906–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Alam SL, Proteasa SV, Zhang Y, Lesuisse E, Dancis A, Stemmler TL (2004) Yeast frataxin solution structure, iron binding, and ferrochelatase interaction. Biochemistry 43: 16254–16262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki H, Nakayama S, Kretsinger RH (1998) Classification and evolution of EF-hand proteins. Biometals 11: 277–295 [DOI] [PubMed] [Google Scholar]
- Layer G, Ollagnier de Choudens S, Sanakis Y, Fontecave M (2006) Iron–sulphur cluster biosynthesis: characterization of Escherichia coli cyay as an iron donor for the assembly of [2Fe–2S] clusters in the scaffold ISCU. J Biol Chem 281: 16256–16263 [DOI] [PubMed] [Google Scholar]
- Leibrecht I, Kessler D (1997) A novel L-cysteine/cystine C-S-lyase directing 2Fe–2S cluster formation of Synechocystis ferredoxin. J Biol Chem 272: 10442–10447 [DOI] [PubMed] [Google Scholar]
- Levi S, Arosio P (2004) Mitochondrial ferritin. Int J Biochem Cell Biol 36: 1887–1889 [DOI] [PubMed] [Google Scholar]
- Mühlenhoff U, Richhardt N, Ristow M, Kispal G, Lill R (2002) The yeast frataxin homolog Yfh1p plays a specific role in the maturation of cellular Fe/S proteins. Hum Mol Gene 11: 2025–2036 [DOI] [PubMed] [Google Scholar]
- Musco G, Stier G, Kolmerer B, Adinolfi S, Martin S, Frenkiel T, Gibson T, Pastore A (2000) Towards a structural understanding of Friedreich's ataxia: the solution structure of frataxin. Struc Fol Des 8: 695–707 [DOI] [PubMed] [Google Scholar]
- Nair M, Adinolfi S, Pastore C, Kelly G, Temussi P, Pastore A (2004) Solution structure of the bacterial frataxin ortholog, CyaY: mapping the iron binding sites. Structure 12: 2037–2048 [DOI] [PubMed] [Google Scholar]
- Ramazzotti A, Vanmansart V, Foury F (2004) Mitochondrial functional interactions between frataxin and Isu1p, the iron–sulphur cluster scaffold protein, in Saccharomyces cerevisiae. FEBS Lett 16: 215–220 [DOI] [PubMed] [Google Scholar]
- Rötig A, de Lonlay P, Chretien D, Foury F, Koenig M, Sidi D, Munnich A, Rustin P (1997) Aconitase and mitochondrial iron–sulphur protein deficiency in Friedreich ataxia. Nat Genet 17: 215–217 [DOI] [PubMed] [Google Scholar]
- Rouault TA, Tong WH (2005) Iron–sulphur cluster biogenesis and mitochondrial iron homeostasis. Nat Rev Mol Cell Biol 6: 345–351 [DOI] [PubMed] [Google Scholar]
- Wagner I, Arlt H, van Dyck L, Langer T, Neupert W (1994) Molecular chaperones cooperate with PIM1 protease in the degradation of misfolded proteins in mitochondria. EMBO J 13: 5135–5145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon T, Cowan JA (2003) Iron-sulphur cluster biosynthesis. Characterization of frataxin as an iron donor for assembly of [2Fe–2S] clusters in ISU-type proteins. J Am Chem Soc 125: 6078–6084 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information