

The International Symposium on Neurodegeneration and Neuroprotection was organized by S. Klumpp with the support of an international advisory board, and was held in Muenster, Germany, between 23 and 26 July 2006. This meeting was a continuation of the International Symposium on Pharmacology of Cerebral Ischemia series.
Introduction
At the International Symposium on Neurodegeneration and Neuroprotection in Muenster, Germany, around 50 scientists and clinicians from all over the world presented their most recent data on mechanisms of neuronal cell death after cerebral ischaemia, potential therapeutic strategies and ongoing clinical trials. In addition, emerging topics in this field were discussed, such as the important role of the microvascular unit, brain injury-induced immunodeficiency and reversible phosphorylation. In particular, protein kinases and phosphatases are becoming the focus of stroke research, as reversible phosphorylation has a crucial role in the fine-tuning of many biochemical pathways, including cell death and survival signalling. This symposium reflected the impressive progress that has been made in experimental and clinical research on ischaemic brain damage over the past few years, and provided an encouraging outlook on new targets and therapeutic approaches.
Neurodegeneration after ischaemic stroke
A main focus of the symposium was on the complex mechanisms of ischaemic brain injury, which involve energy failure, excitotoxicity, the accumulation of reactive oxygen species (ROS), programmed cell death and inflammatory processes. Several speakers showed that the biochemical hallmarks of apoptosis can be observed in models of neurodegenerative diseases, including the regulation of apoptotic factors, mitochondrial dysfunction and the release of cytochrome c or apoptosis-inducing factor (AIF), activation of caspases and DNA fragmentation. C. Wasterlain (Los Angeles, CA, USA) showed that neuronal cell death after ischaemia also displayed features of apoptosis and necrosis in the same cell, indicating that ‘programmed necrosis' contributes to infarct development after stroke.
The cytotoxic accumulation of intracellular calcium has been established as a crucial step in ischaemic neuronal cell death (Orrenius et al, 2003). Until recently, it was widely accepted that cytotoxic intracellular calcium overload after ischaemia is mediated mainly through the stimulation of glutamate receptors or voltage-dependent calcium channels. However, R. Simon (Portland, OR, USA) reported on acid-sensing ion channels (ASICs), which form a new class of pH-dependent ion channel-coupled receptors that have been shown to have a prominent role in the disruption of calcium homeostasis after the reduction of extracellular pH. As extracellular pH rapidly drops in ischaemic brain tissue, proton-activated ASICs contribute to ischaemia-induced calcium influx and neuronal cell death. Recent findings presented by Simon indicate that the activities of ASICs—channel activation, inactivation and recovery from desensitization—are tightly regulated by intracellular pH (pHi). Increasing pHi enhances the sensitivity of the receptors and their recovery rate. This is of great importance, as ASICs probably contribute to ischaemic neuronal death in the cortical penumbra region where alkalization of the pHi has been detected.
After cerebral ischaemia, mitochondrial dysfunction can ensue as a result of oxidative stress, energy failure and the disruption of cellular calcium homeostasis; this is considered to be a crucial event that triggers the execution of programmed cell death in neurons (Fig 1). An important regulatory step of this process occurs at mitochondrial membranes, where members of the B-cell leukaemia/lymphoma 2 (Bcl-2) family of proteins either promote (Bax, Bid, Bad and Bim) or prevent (Bcl-2 and Bcl-xL) the formation of outer membrane pores that cause mitochondrial dysfunction and allow the release of pro-apoptotic factors, such as cytochrome c and AIF, from the mitochondrial intermembrane space (Chao & Korsmeyer, 1998). In neurons, the nucleolar phosphoprotein nucleophosmin might contribute to the lethal activation of Bax, thereby linking death signalling from the nucleus and mitochondrial pathways of apoptosis. L. Kerr (Edinburgh, UK) showed that nucleophosmin translocates into mitochondria and interacts with the carboxyl terminus of Bax in apoptotic cells. Nucleophosmin knockout or small-interfering RNA (siRNA)-mediated gene silencing prevented cell death; this suggests that Bax–nucleophosmin interactions are a potential therapeutic target for neuroprotective strategies. It is interesting to note that the tumour suppressor and transcription factor p53 is another potential binding partner of nucleophosmin that has a leading role in neuronal apoptosis upstream of mitochondrial damage. Indeed, p53 is a promising target for stroke therapy, as it is rapidly upregulated in ischaemic brain tissue where it could mediate programmed cell death through the transcription of Bax, p53-upregulated modulator of apoptosis (PUMA) or Noxa, as well as the subsequent mitochondrial damage and activation of caspases (Culmsee & Mattson, 2005). P. Chan (Stanford, CA, USA) showed transcription-independent pro-apoptotic activity of p53 in a rat model of transient global ischaemia, in which cell death occurred in hippocampal CA1 neurons after the mitochondrial translocation of p53 and its interaction with the anti-apoptotic Bcl-xL.
Figure 1.
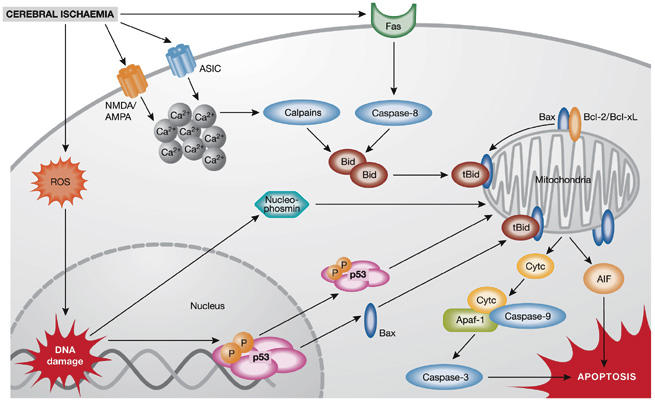
Mechanisms of ischaemic neuronal cell death. Ischaemic neuronal death is mediated by a plethora of intracellular mechanisms, such as increased intracellular calcium levels, reactive oxygen species (ROS), DNA damage and mitochondrial dysfunction. Post-ischaemic cytotoxic calcium influx is mediated through the stimulation of N-methyl-D-aspartate (NMDA), D,L-α-amino-3-hydroxy-5-methyl-isoxazolpropionic acid (AMPA) glutamate receptors or acid-sensing ion channels (ASICs). Increased intracellular calcium, ROS accumulation and activation of Fas death receptors result in activation of caspase-8 or calpains and mediate cleavage of Bid to truncated Bid (tBid), which integrates the different death pathways at the mitochondrial checkpoint of apoptosis. At the mitochondrial membrane tBid interacts with Bax, which is usually neutralized by anti-apoptotic B-cell leukaemia/lymphoma 2 (Bcl-2) family proteins Bcl-2 or Bcl-xL. Dimers of tBid and Bax form pores in the outer mitochondrial membrane, thereby releasing cytochrome c (Cytc) or apoptosis-inducing factor (AIF), which execute caspase-dependent or caspase-independent cell death, respectively. After release into the cytosol, cytochrome c complexes with apoptotic protein-activating factor-1 (Apaf-1) and procaspase-9 to form the apoptosome, which further activates executor caspases, such as caspase-3. By contrast, AIF translocates rapidly to the nucleus where it mediates large-scale DNA fragmentation and cell death in a caspase-independent manner. In addition, nuclear pathways of neuronal cell death are activated in response to DNA damage, for example, through phosphorylation (P) and activation of p53 or mitochondrial translocation of nucleophosmin.
Although caspase activation has been widely documented in ischaemic brain tissue in various stroke models, increasing evidence suggests a substantial role for caspase-independent pathways in ischaemic neuronal cell death, which involve the mitochondrial release of AIF. C. Culmsee and N. Plesnila (Munich, Germany) presented evidence of rapid AIF translocation to the nuclei of injured neurons in experimental models of cerebral ischaemia and brain trauma, respectively. Most notably, reduced AIF protein levels in siRNA-treated neurons or in harlequin (Hq)-mutant mice, which express low levels of AIF (Culmsee et al, 2005), resulted in a pronounced reduction of neuronal cell death in models of brain trauma and cerebral ischaemia. Small-molecule Bid inhibitors prevented mitochondrial release of AIF and neuronal cell death induced by glutamate or oxygen–glucose deprivation in vitro, revealing these compounds to be potential neuroprotective therapeutics (Becattini et al, 2006).
Reversible protein phosphorylation
Reversible protein phosphorylation is under the control of opposing activities of protein kinases and protein phosphatases, and has a crucial role in the regulation of cellular signal transduction in a plethora of neural cell functions, including neurogenesis, differentiation, gene transcription and cell death signalling (Klumpp & Krieglstein, 2002b). During the symposium, expert reviews of research on reversible protein phosphorylation, examples from screening approaches for kinase functions in neurons and studies on particular signalling pathways highlighted the importance of this fast-emerging topic for the understanding of neuronal cell death, and the development of novel neuroprotective strategies.
Protein kinases have been established as key regulators in many important cellular processes, such as proliferation, maintenance of cell shape, survival signalling and apoptosis. Approximately 500 genes encode members of the kinase family in the human genome, and the predicted human kinome might be further enlarged by splice variants with altered regulation of activity, substrate specificity or subcellular localization. The specific inhibition of defined protein kinases has been considered as a potential therapeutic strategy in many diseases, including cancer and inflammatory and neurodegenerative disorders. S. Loh (Leicester, UK) presented an RNA interference (RNAi)-screening approach to search for protein kinases involved in neurite outgrowth or retraction. On the basis of this screen, several kinases were identified that either promoted or inhibited neurite outgrowth, or mediated neurite retraction after exposure to the Rho activator lysophosphatidic acid (LPA). For example, the screening exposed a potential role of the serine/threonine kinase Rho-associated coiled-coil-containing protein kinase-1 (ROCK-1)—a downstream effector of the Rho GTPase—in neurite retraction after LPA treatment in neurons. Similar screening approaches are underway to identify kinases that have a crucial role in the regulation of neuronal survival in models that are relevant to neurodegenerative diseases.
Protein phosphatases are divided into three main enzyme families: tyrosine phosphatases, serine/threonine phosphatases and histidine phosphatases (Klumpp & Krieglstein, 2002a,b). It is interesting to note that only approximately 150 genes that encode phosphatases have been identified, which must compete with more than 500 protein kinases to regulate protein phosphorylation. Therefore, specific regulation of phosphatase activities in a particular signalling pathway might not only depend on a substrate motif alone but also probably involves other mechanisms, such as subcellular differences in the generation of ROS, which might affect the respective activities of phosphatases in defined cellular compartments through local redox regulation.
In the keynote lecture, N. Tonks (Cold Spring Harbor, NY, USA) reviewed the diverse functions and regulation of the protein tyrosine phosphatase (PTP) superfamily. Reversible tyrosine phosphorylation is regulated by protein tyrosine kinases (PTKs) and PTPs, and has a role in various signalling pathways, including regulation of growth factor signalling through receptor tyrosine kinases (RTKs) in neurons. Approximately 100 human genes belong to the PTP superfamily, which is divided into two main enzyme categories: the classic phospho-tyrosine (pTyr)-specific phosphatases, and the dual-specificity phosphatases (DSPs) that also dephosphorylate serine/threonine residues and inositol phospholipids. In his overview, Tonks showed that reversible inhibition of PTP by ROS is a crucial regulatory mechanism for both enzyme categories (Tonks, 2005). In contrast to the detrimental burst of oxidative stress that kills cells after exposure to lethal stress, he discussed the controlled ROS formation that occurs under physiological conditions. Tonks proposed that this could be a mechanism of cell signalling which controls protein phosphorylation through the inhibition of PTP in defined subcellular compartments. For example, reversible regulation by active-site cysteine oxidation has been shown for PTP1B, which is a pTyr PTP involved in the control of growth factor and insulin RTK signalling that has become a therapeutic target in obesity, diabetes, cancer and neurodegenerative diseases. In particular, PTP inhibition has been shown to be an efficient strategy to enhance neuroprotective growth factor signalling in neurons. Similar to pTyr PTPs, ROS might also regulate the activity of DSPs, for example by oxidation of Fe2+ in the active centre of calcineurin or by transient cysteine oxidation in the phosphatase and tensin homologue (PTEN). P. Downes (Dundee, UK) summarized our understanding of PTEN, which is a tumour suppressor protein involved in the regulation of cell survival, proliferation and migration. The most prominent substrate of PTEN related to cell survival signalling is the lipid phosphoinositide-3-phosphate (PIP3). Dephosphorylation of PIP3 to PIP2 antagonizes the activity of PIP3 kinase (PI3K), which is activated in most growth factor-mediated signalling pathways, and which mediates survival and growth signalling through the activation of protein kinase B/Akt. A pro-apoptotic role for PTEN has also been described in neurons, whereas reduced PTEN levels provide neuroprotection against glutamate toxicity and apoptosis both in vitro and in vivo.
Inflammatory responses and immune deficiency
Cerebral ischaemia triggers a plethora of inflammatory reactions that can progress for days after the onset of the insult, and is initiated by cytokines, adhesion molecules, prostanoid mediators of inflammation and nitric oxide (Iadecola & Alexander, 2001). In particular, there is strong evidence for roles of the enzymes inducible nitric oxide synthase (iNOS) and cyclooxygenase 2 (COX-2) as predominant inflammatory mediators, and promising preclinical data on their respective antagonists have shown cerebroprotective effects. K. Iadecola (New York, NY, USA) discussed the neurotoxic effects of COX-2-derived prostaglandin E2 after cerebral ischaemia, and showed that its EP1 receptors have an essential role in this process. An EP1 receptor inhibitor reduced brain injury when administered 6 h after middle cerebral artery occlusion.
W. Lukiw (New Orleans, LA, USA) presented another promising approach that provides neuroprotection through the inhibition of inflammatory responses. The docosanoid neuroprotectin D1 (NPD1) prevented the infiltration of leucocytes, inhibited an increase in inflammatory markers (such as COX-2 and nuclear factor-κB) in ischaemic brain tissue and reduced brain damage by approximately 50%. Similar protective effects were observed in models of retinal neuron injury and Alzheimer disease, in which NPD1 reduced the regulation of pro-inflammatory genes, for example, tumour necrosis factor-α and COX-2, whereas anti-apoptotic proteins, such as Bcl-2 or Bcl-xL, were upregulated.
In contrast to the enhanced local inflammatory reactions seen in ischaemic brain tissue, the immune system seems to be impaired after stroke, and infections after cerebral ischaemia have a prominent role in the complications and mortality of stroke patients (Meisel et al, 2005). U. Dirnagl (Berlin, Germany) showed that central nervous system injury-induced immunodeficiency syndrome (CIDS) after cerebral ischaemia in mice is caused by overactivation of the sympathetic nervous system and significantly increases the risk of pneumonia, which is the most common cause of death in stroke patients (Prass et al, 2006). According to Dirnagl and also A. Planas (Barcelona, Spain), who investigated such stroke-associated infections in clinical trials, immunodeficiency might be explained by extensive apoptotic loss of T-helper cells type 1 (Th1) and a shift to a Th2 cytokine status that results in elevated blood levels of anti-inflammatory interleukins, such as IL-4 and IL-10. The data on CIDS from experimental and clinical studies have resulted in stroke treatment guidelines that advise immediate treatment of infection, whereas antibiotic prophylaxis is not recommended (Chamorro et al, 2005; Vargas et al, 2006).
The neurovascular unit
Cerebrovascular deregulation is a key feature in stroke pathology and, in particular, in haemorrhagic transformation after ischaemia (del Zoppo & Mabuchi, 2003). Under physiological conditions, the balance between energy demands owing to neural activity and substrate delivery through blood flow is tightly regulated by a functional unit of neurons comprising astrocytes and vascular cells. G. del Zoppo (La Jolla, CA, USA) showed that in ischaemic brain tissue, the microvasculature is destroyed and the extracellular matrix is broken down. Both pathological features apparently contribute to ischaemic neuronal cell death through a dramatic reduction in the blood supply to the brain and the concomitant destruction of the blood–brain barrier that results in oedema formation. The activation of matrix-metalloproteinase 2 (MMP-2) and MMP-9 has been exposed as a main cause of vascular dysfunction, destruction of the extracellular matrix and haemorrhagic transformation after stroke. C. Meyer (Stanford, CA, USA) showed that ROS formation is a primary trigger for MMP-9 regulation, inflammatory responses, and the related detrimental effects in vascular cells, astrocytes and neurons. In addition to specific MMP-9 inhibitors, preservation of the microvasculature and neuroprotection after an ischaemic insult could be achieved with the antibiotic minocycline, which also reduced microglial activation and MMP-9 regulation. Protection of the neurovascular unit is therefore emerging as a potential strategy to preserve the delicate balance of blood supply and energy demand in the microenvironment of the brain, thereby reducing neurological deficits and neuron death after an ischaemic insult.
Neurogenesis and neurorestorative therapy
Hopes were raised for stem-cell directed stroke therapy after it was shown that pluripotent stem cells had protective effects and enhanced endogenous neurogenesis after ischaemic brain injury (Zhang et al, 2005). Presentations by K. Abe (Okayama, Japan), W.-R. Schäbitz (Münster, Germany) and M. Chopp (Detroit, MI, USA) reviewed therapeutic strategies that support adult neurogenesis by enhancing protein levels of fibroblast growth factor-2 (FGF-2), epidermal growth factor (EGF), granulocyte colony-stimulating factor (G-CSF) and brain-derived neurotrophic factor (BDNF). Furthermore, endogenous regeneration and functional improvement were achieved by compounds that are clinically established in other therapeutic categories; for example, erythropoietin, HMG-CoA reductase inhibitors, such as atorvastatin, and phosphodiesterase-5 inhibitors, such as sildenafil, enhanced angiogenesis and neurogenesis in experimental models of stroke, even when applied more than 24 h after the onset of ischaemia.
Transferring neuroprotection to clinical trials
Although several experimental studies originally suggested therapeutic efficacy for many neuroprotective drugs, such as glutamate receptor antagonists and antioxidants, the majority of the corresponding clinical stroke studies failed. The design and quality of the clinical stroke studies were often blamed for these failures; however, recent systematic reviews have also exposed considerable quality deficits in experimental stroke research. Dirnagl, I. Macrae (Glasgow, UK) and A. Buchan (Oxford, UK) discussed problems with experimental studies on neuroprotective drugs, including poor study design, flawed statistical analysis and lack of quality-control mechanisms. In addition to ongoing attempts to improve the quality of clinical trials, progress in experimental methods and quality issues in experimental studies are necessary for the successful transfer of efficient stroke therapies from bench to bedside.
Nevertheless, a few drugs that have been extensively tested in experimental models of cerebral ischaemia have shown promising effects in recent clinical stroke trials. For example, the free-radical scavenger NXY-059 protected brain tissue in different models of cerebral ischaemia, and the recently published results of the Stroke-Acute Ischemic NXY Treatment (SAINT I) trial indicated a small but significant improvement in stroke patients as assessed by the modified Rankin scale 90 days after stroke (Lees et al, 2006). The SAINT II study is enrolling a further 3,200 patients with ischaemic stroke to verify these encouraging results.
M. Ginsberg (Miami, FL, USA) presented promising data from clinical trials with albumin. As shown previously, in extensive experimental work in Ginsberg's laboratory, cerebroprotection by albumin is mediated—with a wide therapeutic time window of several hours after an ischaemic insult—by various mechanisms, including enhanced microvascular perfusion and antioxidant properties. The Albumin in Ischemic Stroke (ALIAS) dose-escalation study showed that high-dose albumin was well-tolerated by patients with acute ischaemic stroke, and this safety profile was not affected by tissue plasminogen activator (tPA; Ginsberg et al, 2006). This is an important finding because tPA therapy, which is the only approved treatment for ischaemic stroke, carries a considerable risk of haemorrhagic transformation, and the ALIAS trial has now shown that high-dose albumin does not accelerate this risk. Moreover, the pilot trial indicated neuroprotective effects of high-dose albumin in stroke patients (Palesch et al, 2006), which are now being addressed in the multicentre ALIAS phase III study.
Conclusions
Inflammatory responses, the neurovascular unit, neurogenesis and angiogenesis, and cell-death pathways after ischaemia are the main targets for stroke therapy. Promising future approaches will combine high-throughput technologies, such as proteomics, genomics and siRNA screening, to investigate further mitochondrial cell-death pathways, systemic immune responses and, in particular, reversible phosphorylation. Stringent criteria in the performance, reporting and analysis of animal data, such as the Stroke Therapy Academic Industry Roundtable (STAIR) criteria, are clearly required for experimental research to identify the most efficient therapeutic strategies for successful transfer into the clinic.

Carsten Culmsee

Josef Krieglstein
Acknowledgments
The symposium was a Biochemical Society Independent Meeting and was generously supported by the Deutsche Forschungsgemeinschaft (DFG), Fonds der Chemischen Industrie, International Society for Neurochemistry, Schwabe, Biotrend, Sanofi-Aventis and PromoCell. We thank all participants for their outstanding presentations and discussions, and S. Klumpp and her team for their excellent organization. Individual reviews of the presentations are published in a special issue of the Biochemical Society Transactions (volume 34, part 6, 2006). C.C. is supported by the Alzheimer Forschungs Initiative. J.K. is supported by the DFG.
References
- Becattini B, Culmsee C, Leone M, Zhai D, Zhang X, Crowell KJ, Rega MF, Landshamer S, Reed JC, Plesnila N, Pellecchia M (2006) Structure–activity relationships by interligand NOE-based design and synthesis of antiapoptotic compounds targeting Bid. Proc Natl Acad Sci USA 103: 12602–12606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamorro A, Horcajada JP, Obach V, Vargas M, Revilla M, Torres F, Cervera A, Planas AM, Mensa J (2005) The Early Systemic Prophylaxis of Infection After Stroke study: a randomized clinical trial. Stroke 36: 1495–1500 [DOI] [PubMed] [Google Scholar]
- Chao DT, Korsmeyer SJ (1998) BCL-2 family: regulators of cell death. Annu Rev Immunol 16: 395–419 [DOI] [PubMed] [Google Scholar]
- Culmsee C, Mattson MP (2005) p53 in neuronal apoptosis. Biochem Biophys Res Commun 331: 761–777 [DOI] [PubMed] [Google Scholar]
- Culmsee C, Zhu C, Landshamer S, Becattini B, Wagner E, Pellecchia M, Blomgren K, Plesnila N (2005) Apoptosis-inducing factor triggered by poly(ADP-ribose) polymerase and Bid mediates neuronal cell death after oxygen–glucose deprivation and focal cerebral ischemia. J Neurosci 25: 10262–10272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Zoppo GJ, Mabuchi T (2003) Cerebral microvessel responses to focal ischemia. J Cereb Blood Flow Metab 23: 879–894 [DOI] [PubMed] [Google Scholar]
- Ginsberg MD, Hill MD, Palesch YY, Ryckborst KJ, Tamariz D (2006) The ALIAS Pilot Trial: a dose-escalation and safety study of albumin therapy for acute ischemic stroke. I: Physiological responses and safety results. Stroke 37: 2100–2106 [DOI] [PubMed] [Google Scholar]
- Iadecola C, Alexander M (2001) Cerebral ischemia and inflammation. Curr Opin Neurol 14: 89–94 [DOI] [PubMed] [Google Scholar]
- Klumpp S, Krieglstein J (2002a) Phosphorylation and dephosphorylation of histidine residues in proteins. Eur J Biochem 269: 1067–1071 [DOI] [PubMed] [Google Scholar]
- Klumpp S, J Krieglstein (2002b) Serine/threonine protein phosphatases in neuronal apoptosis. Curr Opin Pharmacol 2: 458–462 [DOI] [PubMed] [Google Scholar]
- Lees KR, Zivin JA, Ashwood T, Davalos A, Davis SM, Diener HC, Grotta J, Lyden P, Shuaib A, Hardemark HG, Wasiewski WW: Stroke-Acute Ischemic NXY Treatment (SAINT I) Trial Investigators (2006) NXY-059 for acute ischemic stroke. N Engl J Med 354: 588–60016467546 [Google Scholar]
- Meisel C, Schwab JM, Prass K, Meisel A, Dirnagl U (2005) Central nervous system injury-induced immune deficiency syndrome. Nat Rev Neurosci 6: 775–786 [DOI] [PubMed] [Google Scholar]
- Orrenius S, Zhivotovsky B, Nicotera P (2003) Regulation of cell death: the calcium–apoptosis link. Nat Rev Mol Cell Biol 4: 552–565 [DOI] [PubMed] [Google Scholar]
- Palesch YY, Hill MD, Ryckborst KJ, Tamariz D, Ginsberg MD (2006) The ALIAS Pilot Trial: a dose-escalation and safety study of albumin therapy for acute ischemic stroke. II: Neurologic outcome and efficacy analysis. Stroke 37: 2107–2114 [DOI] [PubMed] [Google Scholar]
- Prass K, Braun JS, Dirnagl U, Meisel C, Meisel A (2006) Stroke propagates bacterial aspiration to pneumonia in a model of cerebral ischemia. Stroke 37: 2607–2612 [DOI] [PubMed] [Google Scholar]
- Tonks NK (2005) Redox redux: revisiting PTPs and the control of cell signaling. Cell 121: 667–670 [DOI] [PubMed] [Google Scholar]
- Vargas M, Horcajada JP, Obach V, Revilla M, Cervera A, Torres F, Planas AM, Mensa J, Chamorro A (2006) Clinical consequences of infection in patients with acute stroke: is it prime time for further antibiotic trials? Stroke 37: 461–465 [DOI] [PubMed] [Google Scholar]
- Zhang RL, Zhang ZG, Chopp M (2005) Neurogenesis in the adult ischemic brain: generation, migration, survival, and restorative therapy. Neuroscientist 11: 408–416 [DOI] [PubMed] [Google Scholar]