

Abstract
Calmodulin (CaM) is known to undergo conformational and functional changes on oxidation, allowing CaM to function as an oxidative stress sensor. We report the use of a novel mass spectrometry-based methodology to monitor the structure of apo- and holo-CaM as it undergoes conformational changes as a result of increasing amounts of oxidative damage. The kinetics of oxidation for eight peptides are followed by mass spectrometry, and 12 sites of oxidation are determined by MS/MS. Changes in the pseudo-first-order rate constant of oxidation for a peptide after increasing radiation exposure reveal changes in the accessibility of the peptide to the diffusing hydroxyl radical, indicating conformational changes as a function of increased oxidative damage. For holo-CaM, most sites rapidly become less exposed to hydroxyl radicals as the protein accumulates oxidative damage, indicating a closing of the hydrophobic pockets in the N- and C-terminal lobes. For apo-CaM, many of the sites rapidly become more exposed until they resemble the solvent accessibility of holo-CaM in the native structure and then rapidly become more buried, mimicking the conformational changes of holo-CaM. At the most heavily damaged points measured, the rates of oxidation for both apo- and holo-CaM are essentially identical, suggesting the two assume similar structures.
INTRODUCTION
Calmodulin (CaM) is an important signaling protein in all eukaryotes, modulating the activity of a wide variety of targets (1). CaM is a dumbbell-shaped protein, where two domains, each with two Ca2+-binding EF-hand motifs, are linked together by a region that assumes an unstable, flexible helical secondary structure when CaM is bound to Ca2+ (2–4) and assumes a more disordered conformation when CaM is unbound (5). The x-ray crystal structure of holo-CaM, PDB ID 1CLL (3), and the average NMR structure of apo-CaM, PDB ID 1CFD (6), have been solved (Fig. 1). In the apo-CaM form, all methionines except Met76 are buried in the hydrophobic pocket of one of the two structural lobes. On binding to Ca2+, CaM exposes hydrophobic surfaces, which interact with binding partners. Interestingly, CaM employs a large number of conserved methionines in these hydrophobic surfaces, comprising 46% of the hydrophobic surface area of holo-CaM (7). All of these methionines are susceptible to hydrogen peroxide-induced oxidation to the sulfoxide (8), which increases the size of the amino acid and greatly increases the polarity of the side chain.
FIGURE 1.

High-resolution structures of human CaM. Helix A, red; helix B, pink; helix C, aqua; helix D, yellow; helix E, green; helix F, violet; helix G, blue; helix H, cyan. Methionine side chains are shown in orange, and calcium is shown as green spacefill models. (Top) X-ray crystal structure of holo-CaM (1CLL). (Bottom) Average NMR structure of apo-CaM (1CFD).
The presence of these readily oxidized residues in a functionally important region of CaM results in a protein that is functionally sensitive to oxidative stress. For example, as reactive oxygen species (ROS) accumulate in vivo as a result of a variety of mechanisms (including oxidative phosphorylation), the cellular antioxidant defenses can become overwhelmed, leading to a condition of oxidative stress (9–13). Under these conditions, CaM will become oxidized at one or both of the C-terminal vicinal methionines, Met144 and Met145. After this oxidation, CaM is no longer able to activate the plasma membrane Ca2+-ATPase (PMCA) (14); rather, oxidized CaM binds to PMCA and inhibits its activity. This altered protein-protein interaction (probably in concert with a wide variety of other interactions) may lead to a lower rate of cellular metabolism and a lowered rate of ROS production in vivo (15). As the level of ROS decreases, the cellular repair system, including methionine sulfoxide reductases, are able to begin repairing the oxidative damage, including the methionine oxidation of CaM. Therefore, once oxidative stress conditions have been relieved, oxidized CaM is repaired, and oxidative phosphorylation can again be activated (16). This feedback mechanism may allow the cell to modulate its metabolism in response to oxidative stress using a versatile signaling protein as the oxidative stress sensor (15).
Because of CaM's role as a Ca2+ sensor that is modulated at the biochemical level by reversible modification by ROS, as well as its biological importance, there has been considerable interest in the structural changes from CaM oxidation that leads to the observed functional changes (8,14,16–21). It was determined that the oxidation of Met144 and Met145 at the C-terminus of the protein stabilizes the inhibited state of PMCA in addition to slightly decreasing the calcium affinity of CaM (14). Mutation of the other seven methionines is thought to have minimal effects on the interactions with PMCA, including slightly decreasing the binding affinity for PMCA (19) and decreasing the calcium affinity and binding cooperativity (20). However, a Met→Leu mutation was made at the seven non-C-terminal methionines, resulting in a protein that was almost fully functional in activating PMCA and that retained much of its native structure (19). Met144 and Met145 were selectively oxidized in this L7-CaM mutant, and NMR analyses were reported. These analyses found that oxidation of Met144 and Met145 results in a destabilization and shortening of the C-terminal helix (helix H). It was postulated that this conformational change led to a loss of specific contacts with PMCA, leading to a change in the nature of the interaction and causing oxidized CaM to bind PMCA in a down-regulating manner rather than the activating interaction of unoxidized CaM-PMCA (18). It is important to note that previous studies have indicated that the interactions of CaM to various target proteins probably involve different residues. For example, the M144V mutant of plant CaM does not inhibit the activation of phosphodiesterase or calcineurin but does inhibit NOS activity (22); indeed, experiments using CaM where every methionine except for M144 or M145 was mutated to leucine showed that M144L and M145L mutations affect nNOS and eNOS in quite different manners (23). Therefore, to understand the biochemical effects of CaM oxidation, it is important that all oxidation targets be examined, as oxidation of different residues of CaM may well affect different protein-protein interactions.
A fluorescence-based study of CaM oxidation has also recently been reported. A binding site for a fluorescent probe was engineered into the N-terminal domain of CaM, and the probe was used to examine the structural effects of methionine oxidation on both apo- and holo-CaM. The authors reported that oxidation results in a structural uncoupling between the calcium-binding domains of holo-CaM without causing gross structural changes in helix A. Apo-CaM, however, seems to undergo little structural change in the rotational dynamics of the N-terminal domain; indeed, the authors noted that the overall rotational dynamics were essentially the same for oxidized CaM, regardless of the presence or absence of Ca2+ (17). Although these data are quite valuable for studying the large-scale rotational dynamics of oxidized CaM, they indicate only structural changes directly affecting the fluorophore in helix A and are unable to probe structural changes from the perspective of other regions of CaM.
The biophysical characterization of protein oxidation products is technically challenging. ROS chemistry can be highly complex, leading to a heterogeneous mixture of oxidation products that confound traditional high-resolution structural analysis. CaM, however, has several advantages that have allowed a significant amount of structural data to be gathered. CaM contains nine methionines; the presence of these highly flexible hydrophobic residues is thought to be essential for CaM's ability to bind to a variety of different targets (24). Methionines are the only significant target of oxidative modification in vivo (25) and can be selectively modified in vitro by two-electron oxidation using hydrogen peroxide (8). Heavy oxidation of the methionines of CaM has been shown to cause a dramatic decrease in the helical content of both apo- and holo-CaM as measured by circular dichroism spectroscopy (8). Mutations of each of the methionines to oxidation-resistant leucine do not greatly impact the fold or calcium-binding properties of the protein (19), allowing for sophisticated analyses isolating the influence of each methionine. However, mutation of these methionines to leucine influences the binding of CaM to various partners (26,27), suggesting that these methionines do indeed play an important role in CaM function. The ability to mutate the oxidation sites of CaM without grossly affecting the structure allows more complete and elegant analyses of the structural effects of protein oxidation than are available for most proteins. Even with the variety of tools available for the study of oxidized CaM, many questions are still unanswered regarding the structural effects of CaM oxidation, including the cumulative structural effects of methionine oxidation to wild-type CaM.
We report the application of dose-dependent protein oxidative surface mapping for examining the local environment of several sites in both apo-CaM and holo-CaM simultaneously as the protein suffers increasing amounts of oxidative damage from diffusing hydroxyl radicals. The first use of dose-dependent protein oxidative surface mapping has been previously reported for the study of structural changes to the sporulation regulatory protein Spo0F in Bacillus subtilis in response to oxidative damage (28). The technique relies on previous work on protein oxidative surface mapping (29–35), including a previous report using the technique to map the CaM-mellitin binding surface (36). To perform dose-dependent oxidative surface mapping, a dilute solution of the analyte protein is γ-irradiated, forming a constant, low concentration of hydroxyl radicals in solution. These hydroxyl radicals oxidize amino acid side chains at a rate depending largely on two factors: the inherent reactivity of the amino acid and the accessibility of the amino acid to the radical (30,32,34,37–41). This phenomenon has been used to qualitatively and semiquantitatively map the surface of proteins and protein-protein complexes, as previously reviewed (42–44). Catalase is included in solution to prevent either direct two-electron oxidation by hydrogen peroxide (45) or metal-catalyzed Fenton-like reactions (40).
This technique takes advantage of the kinetics of oxidation by low-intensity γ-rays. A detailed review of protein-hydroxyl radical chemistry in oxygenated solution is beyond the intended scope of this article and has been previously published (46). Amino acid oxidation is known to occur primarily by second-order kinetics, dependent on the concentrations of hydroxyl radical and oxidation target (47). Under conditions of constant γ-irradiation, the hydroxyl radical concentration is maintained at a constant level (dependent on the radiation intensity and the rate of scavenging by the various components of the buffer, including the hydroxyl radical itself), leading to pseudo-first-order kinetics. The hydroxyl radicals are formed in the dilute solution in a uniform manner. The likelihood of any particular hydroxyl radical reacting with a protein oxidation target nearby rather than another radical, a buffer molecule, or another nonprotein molecule in solution is a direct function of that oxidation target's reactivity (i.e., the rate constant of oxidation). The pseudo-first-order rate constant of oxidation for any protein oxidation target is dependent on the inherent chemical reactivity of the oxidation target (which is not grossly affected by oxidation at other residues) and the accessibility of the target to the radical (which changes as a result of conformational change). Therefore, we can probe the solvent accessibility of each oxidation target as a function of irradiation time (or, in this case, overall absorbed dosage at a constant dose rate).
This technique has been applied in this study to probe the conformation of bovine CaM as a result of oxidative damage in both the Ca2+-bound form and the Ca2+-free form. Pseudo-first-order kinetics were measured simultaneously for 10 different peptides in CaM, representing all 9 methionines as well as at least 3 nonmethionine residues. The kinetics of oxidation were monitored over a range of absorbed dosages from 116.5 Gy to 1398 Gy. The resulting data allow us to describe the conformational changes experienced by each region of the protein in a gross level of detail and to directly compare the differences in the response of apo-CaM and holo-CaM to oxidative damage.
EXPERIMENTAL PROCEDURES
Materials
Lyophilized CaM (isolated from bovine testes and enriched to ≥98%, essentially salt-free), lyophilized catalase from bovine liver (≥10,000 units/mg protein), ammonium bicarbonate, ethylenediaminetetraacetic acid (EDTA), and 96% formic acid were purchased from Sigma-Aldrich (St. Louis, MO). Sequencing grade modified trypsin was obtained from Promega (Madison, WI). Acetonitrile was purchased from Caledon Laboratories (Georgetown, Ontario). Purified water (17.8 MΩ) was obtained from an in-house Hydro Picopure 2 system. All chemicals were used without further purification unless otherwise specified.
CaM oxidation
Two CaM samples were prepared for γ-irradiation. The first sample (apo-CaM) consisted of 10 mM ammonium bicarbonate (pH 7.54), 20 μM CaM, 100 μM EDTA, and 10 nM catalase tetramer. The second sample (holo-CaM) consisted of 10 mM ammonium bicarbonate (pH 7.54), 20 μM CaM, 100 μM calcium carbonate, and 10 nM catalase tetramer. Catalase tetramer was added to prevent indirect two-electron oxidation of methionines from hydrogen peroxide formed during γ-irradiation (45). The samples were allowed to equilibrate overnight. Twenty-six aliquots of 10 μl each were prepared from both the holo-CaM and apo-CaM samples and placed in 500 μl polyethylene microfuge tubes. These samples were placed in a dual-point 127Cs γ-irradiator and rotated at ∼60 rpm. These samples were irradiated at a dosage rate of 23.3 Gray (Gy)/min. The samples were irradiated between 0 and 60 min, with two aliquots of holo-CaM and two aliquots of apo-CaM taken every 5 min.
CaM digestion
Each aliquot of CaM was heated to 65°C for 90 min directly after irradiation to enhance denaturation of the protein, 20 μg of sequencing-grade modified trypsin was dissolved in 40 μl of 50 mM acetic acid, and 1 μl of the trypsin solution was added to each sample aliquot. These samples were incubated at 37°C overnight. Each sample was then diluted with 30 μl of acetonitrile with 0.1% formic acid (v/v) to a final CaM concentration of ∼5 μM.
Nanoelectrospray quadrupole time-of-flight mass spectrometry
Although the samples did include significant nonvolatile salts, we were able to analyze them directly without further purification using a flow injection device (48) to infuse sample into the orthogonal nanoelectrospray source of a Micromass Q-TOF Ultima Global mass spectrometer (nESI-Q-TOF MS). Mass spectra were collected under typical nanoelectrospray conditions, with a capillary potential of 3200 V, a 100-V cone potential, and a 10-V collision cell potential. Mass spectra were collected from m/z 270 to 2500 for 9 min (530 spectra), and the signal was summed to increase signal to noise. A typical resolving power of ∼12,000 full width at half maximum was achieved, with a typical mass accuracy of ∼40 ppm using external calibration only. Peptides were identified by mass measurement and later verified by LC-MS/MS by use of a quadrupole ion trap (described below).
Although oxidized peptides may be separated from unoxidized peptides by reverse-phase HPLC, this separation can also introduce differential ion suppression effects from coeluting ions as well as the inability to compare signal abundance ratios directly to determine the apparent rate of oxidation, which can introduce more error into the analysis than direct infusion. Therefore, only direct infusion mass spectra were used for quantification of the amount of oxidation per peptide. The resulting mass spectra were used to determine the kinetics of oxidation by measuring the signal intensity of the unoxidized version of each peptide and comparing the signal intensity with that of each of the various oxidized versions.
Kinetics analysis
Only ions with a signal/noise ratio ≥2 were quantitated. The concentration of unoxidized peptide at t = 0 (M0) was arbitrarily set to 1, and all product concentrations were measured as a fraction of overall products. The average number of oxidation reactions per peptide was generated by the function
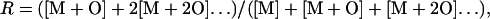 |
(1) |
where [M] is the ion abundance of the unmodified peptide, [M+O] is the abundance of the peptide plus 16 Da, [M+2O] is the abundance of the peptide plus 32 Da, etc. The concentration of unmodified peptide was determined by the function
 |
(2) |
where [M] is the concentration of unmodified peptide, R is the average number of oxidation reactions per peptide, and T is the total number of oxidation targets in the peptide, as determined by MS/MS. This correction for the number of oxidation targets in the peptide allowed us to correct for multiple oxidation events at different targets on a single peptide. The dose-dependent pseudo-first-order rate constant of oxidation k′ was determined using a first-order rate law with the time term t replaced by radiation dosage (in Gray).
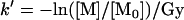 |
(3) |
This nonstandard rate constant unit (Gy−1) was used to better allow the direct comparison of future experiments performed at different dose rates.
Unfortunately, the exact steady-state concentration of hydroxyl radical in solution is unknown. Although the rate of hydroxyl radical formation during γ-irradiation can be calculated as ∼1.09 × 107 mol ·OH liter−1 s−1 (47,49), the steady-state concentration is highly dependent on the buffer composition and cannot be readily calculated when the buffer contains complicated components such as proteins. However, based solely on the rate of hydroxyl radical recombination to form H2O2 (k = 6 × 109 liter mol−1 s−1), the maximum possible concentration of ·OH in solution at the radiation dose rate used here, ignoring the scavenging properties of the buffer and the protein, is 4.26 nM, which is approximately four orders of magnitude lower than the concentration of protein. The radical-scavenging properties of the buffer and protein in solution will lower this concentration even further, allowing one to use an upper limit of [·OH] = 4.26 nM to calculate a limit of k from k′, if desired.
The nonirradiated samples were used to determine any background oxidation, and that amount of oxidation was subtracted from later irradiated samples so the kinetics represented only radiation-induced oxidation. When the signal from the ion of a measured oxidized peptide overlapped with the isotopic distribution of signal from another peptide ion, the contribution of the overlapping isotopic peak was determined from the 0 time point by taking the ratio of the abundance of the minor isotopic component of interest to the most abundant isotopic component in the isotopic packet, and the same measurement was made and subtracted from all later signals to determine only the contribution of the oxidized peptide to the overlapping signal. When multiple charge states could be measured, the charge state that had no overlapping signal with other ion signals was preferentially analyzed to minimize error. When this did not apply, the charge state that showed the lowest variation in the amount of oxidation across all irradiation dosages was used for analysis to minimize experimental error (usually the most abundant ion without overlapping ion signals). No significant difference in peptide cation adduction was detected between oxidized and unoxidized peptides; therefore, only the ratios of the unadducted peptides were measured. The average of the two replicates was plotted. Error bars representing the range of the measurements were also generated for all data points, although in many cases the size of the error bars is smaller than the physical size of the plotted data point. These replicates were designed and performed to isolate the effects of increasing radiation dosage versus the effects of uncontrolled experimental variables among time points (different reaction vessels, different locations in the sample rack, small differences in the mass spectrometric measurements, etc.).
Chip-based HPLC-nanoelectrospray quadrupole ion trap mass spectrometry
Although sufficient signal could be obtained to quantify the amount of oxidation for the peptides, it was insufficient to obtain informative tandem mass spectra (MS/MS) for any of the oxidized peptides to identify the actual sites of oxidation without prior enrichment. To determine these sites of oxidation, we analyzed an aliquot of the apo-CaM irradiated with a 1398-Gy dosage using an Agilent chip-based reverse-phase HPLC-nanoelectrospray device coupled to an Agilent LC/MSD Trap XCT Ultra quadrupole ion trap (QIT). The sample was diluted 5:1 in distilled water to dilute the organic content. An Agilent 1100 series dual HPLC system with an autosampler was used to load 20 μl of the diluted sample into a 40 μl sample loop. This sample was washed onto a C-18 enrichment cartridge for 5 min in 97% Buffer A (water, 0.1% formic acid), 3% Buffer B (acetonitrile, 0.1% formic acid) using a capillary pump at a flow rate of 4 μl/min. A switching valve was then used to bring a nanoflow pump in line with the enrichment cartridge and a 75-μm × 43 mm chip-based column packed with 5 μm C-18 SB-ZX material (Agilent Technologies, Palo Alto, CA). The sample was then eluted at a flow rate of 0.3 μl/min using a 40-min linear gradient from 3% to 55% Buffer B, followed by a 5-min linear gradient from 55% to 80% Buffer B, followed by a column wash. The eluent flowed directly into the built-in chip-based nanoelectrospray emitter and into the QIT. The QIT nanoelectrospray source was operated under typical conditions (1900 V capillary potential, 180 V capillary exit potential, and 40 V skimmer potential), and automated data-dependent MS/MS analyses were performed. The mass spectrometer was set to select only ions for MS/MS that had an m/z ±2 of an oxidized peptide previously detected and quantified in the Q-TOF experiment. MS/MS parameters were set to isolate the ion with a 10 m/z-wide window and to ramp the excitation frequency from 0.3 V to 2 V to induce collision-induced dissociation fragmentation, with fragments scanned from m/z 200 to 2200. All MS/MS spectra were interpreted manually by comparison to in silico fragmentation of the peptide using PROWL (50).
RESULTS AND DISCUSSION
Sites of oxidation of CaM
The sequence of CaM is shown in Fig. 2. Bovine CaM is identical in sequence, posttranslational modification, and structure to human CaM (51). We achieved 100% sequence coverage by direct infusion nESI-Q-TOF MS and identified the previously described N-terminal methionine truncation, N-terminal acetylation, and the trimethylation of Lys115 (51). The digestion was largely complete, with the exception of the 75–77 product (which was considerably more abundant than the 76–77 complete digestion product) and the detection of a low rate of partial digestion at the trimethylated Lys115 (data not shown). Of the 12 peptides identified, 8 had a measurable amount of oxidation (signal/noise ratio >2) in both the apo and the holo form, including the peptides containing all 9 methionine residues. MS/MS experiments enabled assignment of the major site(s) of oxidation for all oxidized peptides detected (e.g., Fig. 3) except for the tripeptide 75–77 (which contains Met76) because of its very small size and the considerable chemical noise in the spectra at m/z <200. Because of the rate of oxidation (discussed below) and the differences between the inherent reactivities of methionine and lysine, this oxidation event almost certainly occurred on Met76. For all other methionine-containing peptides, the major sites of oxidation were also found to be the methionines, which is consistent with the previously reported data indicating that methionine is the second most reactive amino acid (after cysteine, which is not present in CaM) as determined by the measurement of stable oxidation products as detected by electrospray mass spectrometry (52). Unfortunately, MS/MS data cannot be used to quantitatively determine how much oxidation occurred at each site on a peptide with multiple oxidation sites. Side-chain oxidation (especially of methionine) is known to alter both the types of fragment ions observed and their relative abundances in the spectra (53); consequently, only gross generalizations can be made about the amount of oxidation at each site for peptides oxidized at multiple sites.
FIGURE 2.

Sequence of bovine CaM. Peptides detected by mass spectrometry are shown as underlined portions, with solid lines indicating peptides with detected oxidation, and dashed lines indicating peptides with no detected oxidation. Oxidation targets are shown in bold. Lys115 (italicized) is trimethylated in bovine CaM, and the N-terminus is acetylated.
FIGURE 3.

Quadrupole ion trap MS/MS spectrum of the +3 charge state of peptide 127–148 with the addition of one oxygen (m/z 836.7). Y-type ions contain the C-terminus and are counted from C-terminus to N-terminus. B-type ions contain the N-terminus and are counted from N-terminus to C-terminus. All oxidation occurred on Met144; no oxidation was detected on Met145. (Inset) A detailed view of the y4 ion (the C-terminal MTAK). Only the unmodified fragment is detected; no signal for the modified fragment is detected.
The chemical nature of methionine oxidation by hydroxyl radical can, however, be determined by analysis of the mass spectra. In no case was any methionine-containing peptide observed to have accumulated more oxygen additions than the number of methionines present on that peptide. Additionally, in all MS/MS fragmentation spectra of multiply oxidized peptides, the oxygen additions were found to be the result of conversion of multiple methionines to methionine sulfoxide rather than the conversion of a single methionine to methionine sulfone (data not shown). This is in agreement with previous studies of the products of hydroxyl radicals with a model methionine-containing peptide, which found that the formation of the sulfone occurs much more slowly than the formation of the sulfoxide (54), as well as previous studies of chemically oxidized CaM that reported the exclusive formation of methionine sulfoxide (21,25). The fact that multiple oxidations did not occur on the same residue largely validates the assumption that the inherent reactivity of each oxidation target did not change, as no previous study has found a substantial effect of oxidation at a distal site affecting the inherent reactivity of an amino acid to hydroxyl radicals, thereby assuring that changes in the rate constant of oxidation will be indicative of a conformational change (42,43).
In addition to determining the sites and nature of oxidation, the order of oxidation for the two C-terminal methionines, Met144 and Met145, could be determined. MS/MS analysis of the singly oxidized peptide 127–148 from apo-CaM after 1398 Gy radiation dosage showed all detectable oxidation to have occurred on Met144 (Fig. 3). Only when two oxygens are added to the peptide is Met145 found to be oxidized. This indicates that in apo-CaM, hydroxyl-radical-mediated oxidation of Met144 occurs before oxidation of Met145. In holo-CaM, it was previously shown that Met145 is oxidized before Met144 (55), suggesting that structural differences between apo- and holo-CaM strongly influence the order of oxidation of the structurally important C-terminal methionines. This difference may be of biological importance, as oxidation of Met145 was shown to significantly alter the tertiary structure of CaM as well as to increase the rate at which CaM was degraded by the proteasome, whereas oxidation of Met144 had no effect on the rate of proteasome degradation and little effect on the CD spectrum (55).
The radiation-induced addition of two oxygens was observed at the lowest radiation dosage (116.5 Gy) for holo-CaM and at the second lowest radiation dosage (233 Gy) for apo-CaM. The relative abundance of the 127–148+2O ion remained very comparable to the relative abundance of the 127–148+1O ion across most radiation dosages for both apo- and holo-CaM (Table 1). If the oxidation of Met144 and Met145 were independent, one would expect that the amount of the 127–148+1O peptide would increase detectably faster than the amount of the 127–148+2O peptide, as the rate of oxidation of Met144 in apo-CaM (Fig. 3) and Met145 in holo-CaM (55) is greater than the rate of oxidation of the neighboring methionine. However, we find that the ratio of M+O to M+2O remains similar throughout most of the time course of the experiment, with the exception of the early timepoints for apo-CaM, where the ratio of singly oxidized peptide to doubly oxidized peptide is actually lower than at later time points. This observation indicates that the oxidation of one C-terminal methionine causes the neighboring methionine to become significantly more solvent accessible and, therefore, to become oxidized relatively rapidly. Such a model would be in agreement with NMR data, which show that on oxidation, the segment of helix H containing both methionines becomes disordered (18). It is also possible that the generation of a highly polar sulfoxide at one C-terminal methionine causes an increase in the inherent reactivity of the neighboring methionine independent of an increase in solvent accessibility. Although there does exist some evidence for field-inductive effects of formal charges to influence the rate of oxidation at aliphatic sites (56), no systematic study of the effects of adjacent polar residues on the rate of oxidation of methionines has been published. However, no such effect has been previously documented in protein oxidative surface-mapping studies; therefore, because of a combination of these data and the previous NMR data, we conclude that the initial oxidation of one of the two C-terminal methionines leads to a destabilization of part of helix H, causing the neighboring methionine to become considerably more exposed.
TABLE 1.
Ratios of singly oxidized to doubly oxidized peptide 128–149
| Holo-CaM
|
Apo-CaM
|
|
|---|---|---|
| Dose (Gy) | [M+O]/[M+2O]* | [M+O]/[M+2O]* |
| 116.5 | 1.29 ± 0.83 | ND |
| 233 | 0.90 ± 0.83 | 0.52 ± 0.30 |
| 349.5 | 1.30 ± 0.24 | 0.65 ± 0.01 |
| 466 | 0.82 ± 0.48 | 0.99 ± 0.55 |
| 582.5 | 1.12 ± 0.50 | 1.17 ± 0.22 |
| 699 | 1.14 ± 0.12 | 1.31 ± 0.006 |
| 815.5 | 1.07 ± 0.02 | 1.33 ± 0.14 |
| 932 | 0.95 ± 0.10 | 1.31 ± 0.10 |
| 1048.5 | 1.01 ± 0.04 | 1.35 ± 0.09 |
| 1165 | 0.90 ± 0.14 | 1.21 ± 0.09 |
| 1281.5 | 0.96 ± 0.04 | 1.37 ± 0.02 |
| 1398 | 1.02 ± 0.25 | 1.40 ± 0.12 |
Error is listed as 2 SD.
Kinetics of oxidation
As stated previously, the kinetics of oxidation were measured for 8 of 12 peptides that accumulated measurable amounts of oxidative damage, and pseudo-first-order rate constants were derived as a function of radiation dosage. These kinetics were determined by comparison of the abundance of the unmodified peptide ion with the abundance of the oxidized peptide ion, with subtraction of effects from overlapping signals when necessary. An example of the data gathered is given in Fig. 4. Fig. 4 A shows the intact mass spectra of apo-CaM at 0 Gy and at 1398 Gy; it can be clearly seen that the mass spectra are quite complex and show many changes after irradiation. Detailed views of a single peptide at different radiation dosages are shown in Fig. 4 B. These mass spectra represent the oxidation of peptide 31–37 of apo-CaM after different amounts of radiation exposure. The 0-Gy dosage point control shows a signal from an unidentified ion with a +3 charge state at an m/z overlapping the signal from the expected oxidation product. The natural isotopic distribution from the +3 charge state allows this signal to be readily differentiated from an overlapping oxidized peptide 31–37. Therefore, the contribution of the unidentified +3 ion to the signal from the oxidized peptide 31–37 can be calculated and subtracted, leaving only the signal from the oxidized peptide 31–37. The apparent rates as a function of radiation dosage for these eight peptides are shown in Fig. 5. Examination of the kinetics of oxidation of the methionine-containing peptides (Fig. 5, C, D, E, G, and H) reveals an interesting theme. First, the initial rate constants (measured at 116.5 Gy) for both the holo and the apo forms correspond very well with the measured solvent accessibility from the x-ray crystal structure of holo-CaM and the average NMR structure of apo-CaM as calculated by the program GETAREA 1.1 (57), shown in Table 2. When the measured high-resolution structure indicates an oxidation target that is more accessible in holo-CaM than in apo-CaM, the initial rate constant for oxidation of that target is higher in holo-CaM than apo-CaM, and vice versa. The only exception to this trend is for Met76, which is the only detected oxidation site in the linker helix between the two lobes of CaM. In examining these calculations, one must take care to realize that the calculated solvent accessibility for each sulfur atom is only an estimate. Protein oxidative surface mapping measures the average solvent accessibility of the oxidation target over time across the entire population of protein molecules; any static model of the protein is only going to serve as an estimate of the solvent accessibility of the dynamic protein. However, our data agree well with the estimate in most cases.
FIGURE 4.

(A) Mass spectrum of the peptides from apo-CaM after 0-Gy and 1398-Gy radiation exposure. (B) Mass spectra of peptide 31–37 from apo-CaM and its major oxidation product as a function of radiation dosage. The triply charged ion found at lower radiation dosages is from an unidentified source.
FIGURE 5.

Kinetics of oxidation for the eight oxidized peptides detected, corrected for preirradiation oxidation caused by protein purification and storage. The abscissa is the radiation dosage, and the ordinate is the apparent pseudo-first order rate constant. If there are pseudo-zero-order kinetics and no alteration in solvent accessibility, the data points should form a line with a slope of 0. An increase in the rate reflects an increase in the solvent accessibility of the oxidation target, whereas a decrease in the rate reflects either a decrease in the solvent accessibility or saturation of the target. ▪, holo-CaM; ♦, apo-CaM. (A) Peptide 1–13; (B) peptide 14–22; (C) peptide 31–37; (D) peptide 38–74; (E) peptide 75–77; (F) peptide 95–106; (G) peptide 107–126; (H) peptide 127–148.
TABLE 2.
Native solvent accessibility of detected oxidation sites
| Domain | Oxidation target | Solvent accessibility Apo-CaM (Å2) | Solvent accessibility Holo-CaM (Å2) |
|---|---|---|---|
| 1 | Ile9 | 60.46 | 73.58 |
| 1 | Phe16 | 3.25 | 12.95 |
| 1 | Met36 | 0.00 | 8.45 |
| 1 | Met51 | 12.96 | 12.77 |
| 1 | Met71 | 2.45 | 7.66 |
| 1 | Met72 | 0.00 | 1.26 |
| Linker | Met76 | 31.39 | 33.41 |
| 2 | Ser101 | 37.33 | 29.64 |
| 2 | Met109 | 0.00 | 1.93 |
| 2 | Met124 | 0.00 | 10.27 |
| 2 | Met144 | 2.52 | 24.57 |
| 2 | Met145 | 0.00 | 23.34 |
The initial rate constant for Met76 (Fig. 5 E) is lower for apo-CaM than for holo-CaM, even though the estimated solvent accessibility of the two are similar (Table 2). This is probably caused by the increased dynamic nature of the two domains and the linker region of apo-CaM (6). Protein oxidative surface mapping measures the average solvent accessibility of the oxidation target over the time course of the experiment. The increased stability of the central helix of holo-CaM causes Met76 to be more solvent exposed on average, whereas the increased flexibility of apo-CaM (because of the increased disorder of both the linker region and the C-terminal lobe) more freely allows the protein to assume conformations that shield Met76 from solvent, increasing the amount of time that Met76 is shielded from solvent and decreasing the average solvent accessibility. These MS data are also in agreement with previous partial enzymatic digestion studies, which found that Lys77 is a preferential cleavage site for trypsin in holo-CaM but not in apo-CaM (58). All other methionines are considerably more exposed in holo-CaM than in apo-CaM; similarly, the initial rate constants for all of these methionine-containing peptides are significantly higher for holo-CaM than for apo-CaM (Fig. 5, C, D, G, and H). These measurements serve as internal validation that the dosage-dependent protein oxidative surface-mapping technique is a valid approach for this protein and that no unusual electronic structure is causing rampant radical transfer within CaM.
Additional analyses of the dose-dependent rate constants reveal an interesting response to oxidative damage. With slight oxidative damage, the rate constants of oxidation of methionines in holo-CaM begin to drop. The rate constants drop rapidly for Met36, Met109, Met124, and Met144-Met145 between 116.5 and 349.5 Gy (Fig. 5). Because of large experimental errors at low dose rates, it is not possible to accurately determine the amount of decline in the rate constant of oxidation for Met51, Met71, Met72, and Met76 (Fig. 5, D and E). The early dramatic decreases in the rate constants for oxidation of Met36, Met109, Met124, Met144, and Met145 suggest a cooperative conformational change that buries the sulfur of the affected methionines, indicating a closing of the hydrophobic clefts in the C-terminal lobe of the protein, and quite probably at least a partial closing of the N-terminal cleft in the protein, as based on the data for Met36.
At very high radiation dosages, the rate constants of oxidation for Met51, Met71, Met72, Ser101, Met144, and Met145 experience a slow but statistically significant decline. This later slow decline in the rate of oxidation indicates that the sites of oxidation are continuing to become less exposed to the hydroxyl radical as a result of noncooperative conformational changes resulting from further oxidation. Noncooperative changes in the rate constants of oxidation suggest either a noncooperative unfolding event of a collapsed, nonnative tertiary structure (indicative of a molten globule-like state) or a heterogeneous collection of different folding conformations with similar unfolding transitions that occlude the observation of a single defined cooperative unfolding event. In any case, the gradual decline is indicative of a further average closing of the hydrophobic clefts of heavily oxidized CaM within the ensemble of conformations present in solution.
Examination of the rate constants of oxidation for apo-CaM yields a significantly different result from holo-CaM. All methionine-containing peptides exhibit a significant increase in the rate constants between 116.5 and 233 Gy with the exception of Met76, which is already highly exposed in the native structure in the linker region between the two lobes of CaM. This is quite interesting, as it shows that the methionine-containing hydrophobic clefts of apo-CaM undergo exposure to solvent at very mild levels of oxidation. Even more interesting is the fact that, for all methionine-containing peptides except for Met76, the rate constants at 233 Gy are very similar to the initial rate constants of the peptide in holo-CaM. Continued comparison of apo-CaM and holo-CaM shows that the two continue to have roughly the same changes in the rate constants at each methionine during both the early cooperatively folding transitions and the later, molten globule-like transitions, suggesting that they both assume a similar collection of local conformations around the methionine-containing clefts. These data suggest a model in which slight oxidative damage of apo-CaM causes the methionine-containing hydrophobic pockets to open into a local conformation similar to that of unoxidized holo-CaM, although probably not identical. From this point, the methionine clefts of both apo-CaM and holo-CaM then proceed through roughly the same series of local conformational changes as they become more heavily oxidized, both entering either a molten globule-like conformation or a suite of conformations that reduce the average solvent accessibility of the oxidation targets after heavy oxidative damage.
The rate constant of oxidation for Met76 in apo-CaM actually increases through 582.5 Gy (Fig. 5 E). This result indicates that, as apo-CaM undergoes oxidative damage, the conformations that occlude Met76 are destabilized, causing Met76 to become more exposed on average. However, it never reaches the rate constant measured for Met76 in holo-CaM, indicating that the local conformation of the linker region is significantly different between oxidized apo-CaM and oxidized holo-CaM at all levels of oxidation, consistent with the large-scale dynamics reported from previous fluorescence studies (17) and suggesting that, even if the local conformations of the calcium-binding lobes are similar between oxidized apo-CaM and oxidized holo-CaM, the overall structure remains significantly different.
Examination of the peptides oxidized at amino acids other than methionine reveals a similar story. Peptide 2–14 (Fig. 5 A) was shown by MS/MS to be oxidized primarily at Ile9, although a minor amount of oxidation was detected at Phe12, and MS/MS evidence also suggested potential unidentified minor oxidation sites elsewhere along the peptide. The amount of oxidation of this peptide is below our limits of detection until 466-Gy radiation exposure. From this point throughout the remainder of the experiment, both apo-CaM and holo-CaM oxidize with about the same rate constant, suggesting a similar local conformation.
Peptide 14–21 (Fig. 5 B) is also consistent with this model. Based on the MS/MS data, the sole major oxidation site for this peptide appears to be Phe16, which is more exposed in the native structure of holo-CaM than apo-CaM (Table 2). The initial amount of oxidation is below our limits of detection; however, the oxidation of holo-CaM rises above our limits of detection well before the rate of apo-CaM, suggesting that the initial rate constant of Phe16 was higher for holo-CaM than for apo-CaM. After 466-Gy radiation exposure, the rate constant of holo-CaM appears to drop, although because of the relatively high amount of experimental error for the low-signal-to-noise holo-CaM data points, we cannot state with much certainty the degree of the decline. However, at radiation dosages of 582.5 Gy and greater, we observe a similar rate constant for both apo-CaM and holo-CaM, suggesting a similar local conformation around Phe16 after this amount of oxidation.
Of the nonmethionine oxidized residues, Ser101 was oxidized most rapidly, giving the best signal/noise ratios and allowing for the most accurate measurements of oxidation kinetics. Examination of the kinetics of oxidation of Ser101 (Fig. 5 F) reveals a story similar to the one revealed by the methionine kinetics. The β-carbon of Ser101, which is the oxidation target atom (46), is essentially fully exposed in both apo-CaM and holo-CaM; appropriately, they have identical rate constants. Interestingly, they also clearly show that the residue experiences what may appear to be a cooperative conformation change between the 233-Gy and 349.5-Gy dosages that buries the β-carbon; after more extensive oxidative damage, the rate constant of oxidation for Ser101 gradually declines, indicating that the molten globule-like state also buries Ser101 more extensively. The rate of decrease in the rate constant of Ser101 is identical for apo-CaM and holo-CaM, again suggesting that the oxidized versions of both proteins assume similar conformations local to Ser101.
One possible explanation of the observed similarity in the kinetics of oxidation of damaged apo-CaM and holo-CaM is that modest oxidation of holo-CaM lowers the Ca2+ binding affinity of CaM sufficiently that holo-CaM is stripped of its calcium ions, becoming apo-CaM. Measurements of calcium affinity of wheat germ CaM that has been oxidized at a C-terminal methionine have been reported. These results showed that oxidized CaM fully assumed the calcium-bound form in the presence of 100 μM free calcium (14). Although this measurement was performed only with a CaM oxidized at one residue, previous results indicate that oxidation of this C-terminal methionine causes the majority of functional changes (19) as well as significant changes in the tertiary structure (18). Additionally, a fluorescently labeled tetracysteine mutant of CaM in which all nine methionines were chemically oxidized to methionine sulfoxide was shown to be in the calcium-bound form at a concentration of 200 μM free Ca2+ (17), indicating that CaM should be fully calcium bound under our conditions for holo-CaM. Therefore, we conclude that the similarity in oxidation kinetics between apo-CaM and holo-CaM does indeed indicate a similarity in local conformation as a result of oxidative damage and not the conversion of holo-CaM to apo-CaM as a result of a loss of calcium-binding affinity.
One point of significant interest is the lack of data for amino acids in the central linker between the two lobes of CaM. Met76 (Fig. 5 E) is part of this unstable helix in holo-CaM and part of the highly flexible portion of the central linker in apo-CaM. However, Met76 is fully exposed in both the apo-CaM and holo-CaM native structures (Table 2), making it useless for determining local unfolding (although it is a very good probe for detecting local folding). No other oxidation targets were detected in this central helix region. The fact that the rate constant changes in response to oxidative damage to Met76 in holo-CaM are not mimicked by apo-CaM suggests that the conformations assumed by the linker region of oxidized holo-CaM are not identical to those assumed by apo-CaM, even though previous reports have shown that methionine oxidation leads to structural uncoupling of the opposing domains of holo-CaM similar to that observed for apo-CaM (17). These data also indicate that there is a structural difference between apo-CaM and oxidized apo-CaM in the linker region, as the rate constant of oxidation of Met76 in apo-CaM is seen to rapidly increase on initial oxidative damage, followed by a noncooperative decrease.
CONCLUSIONS
In summary, oxidation of both apo-CaM and holo-CaM leads to a change in the local conformation of most of the oxidation targets examined. The general pattern of oxidation suggests a model in which the initial oxidation of apo-CaM causes the methionine-containing hydrophobic pockets to open up and assume a local conformation at least somewhat similar to that of unoxidized holo-CaM, as indicated by the very similar rate constants of oxidation for all methionines in the hydrophobic clefts. This open conformation then begins to close as further oxidative damage is experienced, burying the methionines in both lobes (although the effect seems more dramatic for the methionines in the C-terminal lobe, as may be expected based on previous data). The early dramatic decline in the rate constants of several oxidation targets suggests a cooperative conformation change that buries these methionine pockets. Previous fluorescence spectroscopy measurements have shown that oxidation of CaM does not lead to increases in the hydrodynamic radius (55); therefore, the decrease in the rate constants of oxidation is not caused by a multimerization of the protein but rather by conformational changes within the CaM monomer. The gradual decline in the rate constant of various oxidation targets after heavy oxidative damage appears to be caused by a noncooperative switch of CaM structure to a molten globule-like local conformation or a collection of conformations at the calcium-binding lobes that further buries the oxidation targets. Such a model would be in general agreement with the model proposed by Chen et al., where both oxidized apo-CaM and oxidized holo-CaM have significant domain mobility (17), potentially allowing transient domain-domain interactions that occlude some of the oxidation targets.
Acknowledgments
The authors thank Dr. Daniel M. Roberts of the University of Tennessee at Knoxville for his helpful insights and critical analysis of our results.
This research was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Environmental Health Sciences.
References
- 1.Yap, K. L., J. Kim, K. Truong, M. Sherman, T. Yuan, and M. Ikura. 2000. Calmodulin target database. J. Struct. Funct. Genomics. 1:8–14. [DOI] [PubMed] [Google Scholar]
- 2.Babu, Y. S., C. E. Bugg, and W. J. Cook. 1988. Structure of calmodulin refined at 2.2 A resolution. J. Mol. Biol. 204:191–204. [DOI] [PubMed] [Google Scholar]
- 3.Chattopadhyaya, R., W. E. Meador, A. R. Means, and F. A. Quiocho. 1992. Calmodulin structure refined at 1.7 A resolution. J. Mol. Biol. 228:1177–1192. [DOI] [PubMed] [Google Scholar]
- 4.Barbato, G., M. Ikura, L. E. Kay, R. W. Pastor, and A. Bax. 1992. Backbone dynamics of calmodulin studied by 15N relaxation using inverse detected two-dimensional NMR spectroscopy: the central helix is flexible. Biochemistry. 31:5269–5278. [DOI] [PubMed] [Google Scholar]
- 5.Zhang, M., T. Tanaka, and M. Ikura. 1995. Calcium-induced conformational transition revealed by the solution structure of apo calmodulin. Nat. Struct. Biol. 2:758–767. [DOI] [PubMed] [Google Scholar]
- 6.Kuboniwa, H., N. Tjandra, S. Grzesiek, H. Ren, C. B. Klee, and A. Bax. 1995. Solution structure of calcium-free calmodulin. Nat. Struct. Biol. 2:768–776. [DOI] [PubMed] [Google Scholar]
- 7.O'Neil, K. T., and W. F. DeGrado. 1990. How calmodulin binds its targets: sequence independent recognition of amphiphilic alpha-helices. Trends Biochem. Sci. 15:59–64. [DOI] [PubMed] [Google Scholar]
- 8.Sun, H., J. Gao, D. A. Ferrington, H. Biesiada, T. D. Williams, and T. C. Squier. 1999. Repair of oxidized calmodulin by methionine sulfoxide reductase restores ability to activate the plasma membrane Ca-ATPase. Biochemistry. 38:105–112. [DOI] [PubMed] [Google Scholar]
- 9.Bergamini, C. M., S. Gambetti, A. Dondi, and C. Cervellati. 2004. Oxygen, reactive oxygen species and tissue damage. Curr. Pharm. Des. 10:1611–1626. [DOI] [PubMed] [Google Scholar]
- 10.Droge, W. 2003. Oxidative stress and aging. Adv. Exp. Med. Biol. 543:191–200. [DOI] [PubMed] [Google Scholar]
- 11.Dalle-Donne, I., D. Giustarini, R. Colombo, R. Rossi, and A. Milzani. 2003. Protein carbonylation in human diseases. Trends Mol. Med. 9:169–176. [DOI] [PubMed] [Google Scholar]
- 12.Gutteridge, J. M., and J. Mitchell. 1999. Redox imbalance in the critically ill. Br. Med. Bull. 55:49–75. [DOI] [PubMed] [Google Scholar]
- 13.Stadtman, E. R., and B. S. Berlett. 1998. Reactive oxygen-mediated protein oxidation in aging and disease. Drug Metab. Rev. 30:225–243. [DOI] [PubMed] [Google Scholar]
- 14.Yao, Y., D. Yin, G. S. Jas, K. Kuczer, T. D. Williams, C. Schoneich, and T. C. Squier. 1996. Oxidative modification of a carboxyl-terminal vicinal methionine in calmodulin by hydrogen peroxide inhibits calmodulin-dependent activation of the plasma membrane Ca-ATPase. Biochemistry. 35:2767–2787. [DOI] [PubMed] [Google Scholar]
- 15.Squier, T. C., and D. J. Bigelow. 2000. Protein oxidation and age-dependent alterations in calcium homeostasis. Front. Biosci. 5:D504–D526. [DOI] [PubMed] [Google Scholar]
- 16.Bigelow, D. J., and T. C. Squier. 2005. Redox modulation of cellular signaling and metabolism through reversible oxidation of methionine sensors in calcium regulatory proteins. Biochim. Biophys. Acta. 1703:121–134. [DOI] [PubMed] [Google Scholar]
- 17.Chen, B., M. U. Mayer, and T. C. Squier. 2005. Structural uncoupling between opposing domains of oxidized calmodulin underlies the enhanced binding affinity and inhibition of the plasma membrane Ca-ATPase. Biochemistry. 44:4737–4747. [DOI] [PubMed] [Google Scholar]
- 18.Anbanandam, A., R. J. Bieber Urbauer, R. K. Bartlett, H. S. Smallwood, T. C. Squier, and J. L. Urbauer. 2005. Mediating molecular recognition by methionine oxidation: conformational switching by oxidation of methionine in the carboxyl-terminal domain of calmodulin. Biochemistry. 44:9486–9496. [DOI] [PubMed] [Google Scholar]
- 19.Bartlett, R. K., R. J. Bieber Urbauer, A. Anbanandam, H. S. Smallwood, J. L. Urbauer, and T. C. Squier. 2003. Oxidation of Met144 and Met145 in calmodulin blocks calmodulin dependent activation of the plasma membrane Ca-ATPase. Biochemistry. 42:3231–3238. [DOI] [PubMed] [Google Scholar]
- 20.Lafitte, D., P. O. Tsvetkov, F. Devred, R. Toci, F. Barras, C. Briand, A. A. Makarov, and J. Haiech. 2002. Cation binding mode of fully oxidised calmodulin explained by the unfolding of the apostate. Biochim. Biophys. Acta. 1600:105–110. [DOI] [PubMed] [Google Scholar]
- 21.Gao, J., D. H. Yin, Y. Yao, H. Sun, Z. Qin, C. Schoneich, T. D. Williams, and T. C. Squier. 1998. Loss of conformational stability in calmodulin upon methionine oxidation. Biophys. J. 74:1115–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kondo, R., S. B. Tikunova, M. J. Cho, and J. D. Johnson. 1999. A point mutation in a plant calmodulin is responsible for its inhibition of nitric-oxide synthase. J. Biol. Chem. 274:36213–36218. [DOI] [PubMed] [Google Scholar]
- 23.Montgomery, H. J., R. Bartlett, B. Perdicakis, E. Jervis, T. C. Squier, and J. G. Guillemette. 2003. Activation of constitutive nitric oxide synthases by oxidized calmodulin mutants. Biochemistry. 42:7759–7768. [DOI] [PubMed] [Google Scholar]
- 24.Gellman, S. H. 1991. On the role of methionine residues in the sequence-independent recognition of nonpolar protein surfaces. Biochemistry. 30:6633–6636. [DOI] [PubMed] [Google Scholar]
- 25.Gao, J., D. Yin, Y. Yao, T. D. Williams, and T. C. Squier. 1998. Progressive decline in the ability of calmodulin isolated from aged brain to activate the plasma membrane Ca-ATPase. Biochemistry. 37:9536–9548. [DOI] [PubMed] [Google Scholar]
- 26.Zhang, M., M. Li, J. H. Wang, and H. J. Vogel. 1994. The effect of Met→Leu mutations on calmodulin's ability to activate cyclic nucleotide phosphodiesterase. J. Biol. Chem. 269:15546–15552. [PubMed] [Google Scholar]
- 27.Edwards, R. A., M. P. Walsh, C. Sutherland, and H. J. Vogel. 1998. Activation of calcineurin and smooth muscle myosin light chain kinase by Met-to-Leu mutants of calmodulin. Biochem. J. 331:149–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sharp, J. S., D. M. Sullivan, J. Cavanagh, and K. B. Tomer. 2006. Measurement of multisite oxidation kinetics reveals an active site conformational change in Spo0F as a result of protein oxidation. Biochemistry. 45:6260–6266. [DOI] [PubMed] [Google Scholar]
- 29.Wong, J. W., S. D. Maleknia, and K. M. Downard. 2003. Study of the ribonuclease-S-protein-peptide complex using a radical probe and electrospray ionization mass spectrometry. Anal. Chem. 75:1557–1563. [DOI] [PubMed] [Google Scholar]
- 30.Sharp, J. S., J. M. Becker, and R. L. Hettich. 2004. Analysis of protein solvent accessible surfaces by photochemical oxidation and mass spectrometry. Anal. Chem. 76:672–683. [DOI] [PubMed] [Google Scholar]
- 31.Maleknia, S. D., M. Brenowitz, and M. R. Chance. 1999. Millisecond radiolytic modification of peptides by synchrotron x-rays identified by mass spectrometry. Anal. Chem. 71:3965–3973. [DOI] [PubMed] [Google Scholar]
- 32.Aye, T. T., T. Y. Low, and S. K. Sze. 2005. Nanosecond laser-induced photochemical oxidation method for protein surface mapping with mass spectrometry. Anal. Chem. 77:5814–5822. [DOI] [PubMed] [Google Scholar]
- 33.Maleknia, S. D., and K. Downard. 2001. Radical approaches to probe protein structure, folding, and interactions by mass spectrometry. Mass Spectrom. Rev. 20:388–401. [DOI] [PubMed] [Google Scholar]
- 34.Hambly, D. M., and M. L. Gross. 2005. Laser flash photolysis of hydrogen peroxide to oxidize protein solvent-accessible residues on the microsecond timescale. J. Am. Soc. Mass Spectrom. 16:2057–2063. [DOI] [PubMed] [Google Scholar]
- 35.Shum, W. K., S. D. Maleknia, and K. M. Downard. 2005. Onset of oxidative damage in alpha-crystallin by radical probe mass spectrometry. Anal. Biochem. 344:247–256. [DOI] [PubMed] [Google Scholar]
- 36.Wong, J. W., S. D. Maleknia, and K. M. Downard. 2005. Hydroxyl radical probe of the calmodulin-melittin complex interface by electrospray ionization mass spectrometry. J. Am. Soc. Mass Spectrom. 16:225–233. [DOI] [PubMed] [Google Scholar]
- 37.Kiselar, J. G., S. D. Maleknia, M. Sullivan, K. M. Downard, and M. R. Chance. 2002. Hydroxyl radical probe of protein surfaces using synchrotron x-ray radiolysis and mass spectrometry. Int. J. Radiat. Biol. 78:101–114. [DOI] [PubMed] [Google Scholar]
- 38.Chance, M. R. 2001. Unfolding of apomyoglobin examined by synchrotron footprinting. Biochem. Biophys. Res. Commun. 287:614–621. [DOI] [PubMed] [Google Scholar]
- 39.Goldsmith, S. C., J. Q. Guan, S. Almo, and M. Chance. 2001. Synchrotron protein footprinting: a technique to investigate protein-protein interactions. J. Biomol. Struct. Dyn. 19:405–418. [DOI] [PubMed] [Google Scholar]
- 40.Sharp, J. S., J. M. Becker, and R. L. Hettich. 2003. Protein surface mapping by chemical oxidation: structural analysis by mass spectrometry. Anal. Biochem. 313:216–225. [DOI] [PubMed] [Google Scholar]
- 41.Sharp, J. S., J. T. Guo, T. Uchiki, Y. Xu, C. Dealwis, and R. L. Hettich. 2005. Photochemical surface mapping of C14S-Sml1p for constrained computational modeling of protein structure. Anal. Biochem. 340:201–212. [DOI] [PubMed] [Google Scholar]
- 42.Takamoto, K., and M. R. Chance. 2006. Radiolytic protein footprinting with mass spectrometry to probe the structure of macromolecular complexes. Annu. Rev. Biophys. Biomol. Struct. 35:251–276. [DOI] [PubMed] [Google Scholar]
- 43.Guan, J. Q., and M. R. Chance. 2005. Structural proteomics of macromolecular assemblies using oxidative footprinting and mass spectrometry. Trends Biochem. Sci. 30:583–592. [DOI] [PubMed] [Google Scholar]
- 44.Maleknia, S. D., J. W. Wong, and K. M. Downard. 2004. Photochemical and electrophysical production of radicals on millisecond timescales to probe the structure, dynamics and interactions of proteins. Photochem. Photobiol. Sci. 3:741–748. [DOI] [PubMed] [Google Scholar]
- 45.Xu, G., J. Kiselar, Q. He, and M. R. Chance. 2005. Secondary reactions and strategies to improve quantitative protein footprinting. Anal. Chem. 77:3029–3037. [DOI] [PubMed] [Google Scholar]
- 46.Garrison, W. M. 1987. Reaction-mechanisms in the radiolysis of peptides, polypeptides, and proteins. Chem. Rev. 87:381–398. [Google Scholar]
- 47.Buxton, G. V., C. L. Greenstock, W. P. Helman, A. B. Ross, and W. Tsang. 1988. Critical-review of rate constants for reactions of hydrated electrons, hydrogen-atoms and hydroxyl radicals (·OH/·O−) in aqueous solution. J. Phys. Chem. Ref. Data. 17:513–886. [Google Scholar]
- 48.Deterding, L. J., M. A. Moseley, K. B. Tomer, and J. W. Jorgenson. 1989. Coaxial continuous flow fast atom bombardment in conjunction with tandem mass spectrometry for the analysis of biomolecules. Anal. Chem. 61:2504–2511. [DOI] [PubMed] [Google Scholar]
- 49.LaVerne, J. A. 2000. OH radicals and oxidizing products in the gamma radiolysis of water. Radiat. Res. 153:196–200. [DOI] [PubMed] [Google Scholar]
- 50.Fenyo, D., W. Zhang, B. T. Chait, and R. C. Beavis. 1996. Internet-based analytical chemistry resources: A model project. Anal. Chem. 68:721A–726A. [Google Scholar]
- 51.Watterson, D. M., F. Sharief, and T. C. Vanaman. 1980. The complete amino acid sequence of the Ca2+-dependent modulator protein (calmodulin) of bovine brain. J. Biol. Chem. 255:962–975. [PubMed] [Google Scholar]
- 52.Xu, G., and M. R. Chance. 2005. Radiolytic modification and reactivity of amino acid residues serving as structural probes for protein footprinting. Anal. Chem. 77:4549–4555. [DOI] [PubMed] [Google Scholar]
- 53.Grunert, T., K. Pock, A. Buchacher, and G. Allmaier. 2003. Selective solid-phase isolation of methionine-containing peptides and subsequent matrix-assisted laser desorption/ionisation mass spectrometric detection of methionine- and of methionine-sulfoxide-containing peptides. Rapid Commun. Mass Spectrom. 17:1815–1824. [DOI] [PubMed] [Google Scholar]
- 54.Xu, G., and M. R. Chance. 2005. Radiolytic modification of sulfur-containing amino acid residues in model peptides: fundamental studies for protein footprinting. Anal. Chem. 77:2437–2449. [DOI] [PubMed] [Google Scholar]
- 55.Sacksteder, C. A., J. E. Whittier, Y. Xiong, J. Li, N. Galeva, M. E. Jacoby, S. Purvine, T. D. Williams, M. C. Rechsteiner, D. J. Bigelow, and T. C. Squier. 2006. Tertiary structural rearrangements upon oxidation of Methionine145 in calmodulin promotes targeted proteasomal degradation. Biophys. J. 91:1480–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sharp, J. S., and K. B. Tomer. 2006. The effects of anion proximity in peptide primary sequence on the rate and mechanism of leucine oxidation. Anal. Chem. 78:4885–4893. [DOI] [PubMed] [Google Scholar]
- 57.Fraczkiewicz, R., and W. Braun. 1998. Exact and efficient analytical calculation of the accessible surface areas and their gradients for macromolecules. J. Comput. Chem. 19:319–333. [Google Scholar]
- 58.Newton, D. L., M. D. Oldewurtel, M. H. Krinks, J. Shiloach, and C. B. Klee. 1984. Agonist and antagonist properties of calmodulin fragments. J. Biol. Chem. 259:4419–4426. [PubMed] [Google Scholar]