

Abstract
Peptidic self-assembled nanostructures are said to have a wide range of applications in nanotechnology, yet the mechanistic details of hierarchical self-assembly are still poorly understood. The Phe-Phe recognition motif of the Alzheimer's Aβ peptide is the smallest peptide able to assemble into higher-order structures. Here, we show that the Ile-Phe dipeptide analog is also able to self-associate in aqueous solution as a transparent, thermoreversible gel formed by a network of fibrillar nanostructures that exhibit strong birefringence upon Congo red binding. Besides, a second dipeptide Val-Phe, differing only in a methyl group from the former, is unable to self-assemble. The detailed analysis of the differential polymeric behavior of these closely related molecules provides insight into the forces triggering the first steps in self-assembly processes such as amyloid formation.
INTRODUCTION
Successful synthesis of organized supramolecular assemblies is a fundamental step toward the release of new materials or functional supramolecular devices. The controlled self-assembly of biomolecular structures, preferably from the simplest building blocks possible, is therefore of great interest (1–3). Gels represent new soft biocompatible materials that have numerous potential applications in fields like biomaterials, biosensors, tissue engineering, and drug delivery (4–6). Under appropriate conditions, self-assembled arrays of natural and designed proteins and peptides are often observed to trap bulk solvent and result in the formation of transparent gels (7–9). Peptides have emerged as promising gelling compounds since their self-assembly results from the interplay of several weak interactions, such as hydrogen bonding, electrostatics, and hydrophobic forces, which finally organize the monomeric components and lead to the generation of long, noncovalent, supramolecular assemblies (10). These noncovalent forces are reminiscent of those driving amyloid fibril formation, which result, both in vivo and in vitro, in the formation of highly ordered supramolecular assemblies from initially monomeric species (11,12). Fragments of the major proteins involved in Alzheimer's disease, i.e., Tau and Aβ42 peptide, have been shown to act as gelators in vitro, whereas microscopic characterization of the gels has revealed the presence of fibril networks (13,14).
We and other authors have recently shown that specific short stretches in proteins are responsible for their aggregating behavior (15–17) and, in agreement with this observation, several short peptides of amyloidogenic proteins have been shown to form supramolecular structures, indistinguishable from those formed by the complete polypeptide chains (18,19).
Besides their easy design and synthesis, short peptides are both excellent model systems for the study of biological self-assembly and ideal building blocks for the production of a wide range of biological materials. The dipeptide NH2-Phe-Phe-COOH, described as the smallest peptide able to assemble into higher-order structures (20), corresponds to residues 19 and 20 of the central hydrophobic cluster (CHC) of the highly amyloidogenic peptide Aβ42. Position 19 has been shown to strongly affect the assembly and aggregation of Aβ (21). In a recent work, we substituted Phe19 with the other 19 proteinogenic amino acids and assayed the effect of these single mutations on Aβ42's aggregation (22). All substitutions, with the exception of Phe19Ile, resulted in peptides with decreased aggregation propensities relative to that of the wild-type molecule. Thus, an interest arose to determine the molecular properties of the dipeptide NH2-Ile-Phe-COOH (Fig. 1), an analog of the diphenylalanine element shown to self-assemble in vitro. Here, we show that the Ile-Phe dipeptide self-associates to form a transparent, thermoreversible gel formed by a network of fibrillar nanostructures in water. Besides, a second dipeptide NH2-Val-Phe-COOH, differing only in a methyl group from the former, is unable to self-assemble, in agreement with the lower aggregation propensity reported for Phe19Val Aβ42 relative to that of the Phe19Ile version (22). The detailed analysis of the differential self-association capability of these two molecules provides clues for the understanding of hierarchical self-assembly.
FIGURE 1.

Gelation of Ile-Phe dipeptide. (A) Structures of the dipeptides compared in this work. 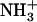 -Val-Phe-COO− (upper) and
-Val-Phe-COO− (upper) and 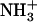 -Ile-Phe-COO− (lower). (B) Photograph of 2% (w/v) dipeptide samples under blue light.
-Ile-Phe-COO− (lower). (B) Photograph of 2% (w/v) dipeptide samples under blue light. 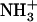 -Ile-Phe-COO− forms a gel (left) whereas
-Ile-Phe-COO− forms a gel (left) whereas 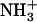 -Val-Phe-COO− remains in solution (right).
-Val-Phe-COO− remains in solution (right).
METHODS
General methods and materials
Lyophilized NH2-Ile-Phe-COOH dipeptide and NH2-Val-Phe-COOH dipeptide with >99% purity were obtained from Bachem (Bubendorf, Switzerland). Dipeptide samples were diluted in 1,1,1,3,3,3 hexafluoro-2-propanol to obtain stocks of 100 mg/ml and 200 mg/ml, which were further diluted to the assay concentration in H2O, except for NMR and Fourier transform infrared (FTIR) assays, where the samples were diluted in deuterium oxide (D2O). 1,1,1,3,3,3 hexafluoro-2-propanol and 2-(p-toluidinylnaphthalene)-6-sulfonate (TNS) were purchased from Sigma (St. Louis, MO). D2O enriched >99.97% in isotope D2O (d = 1.11) was purchased from SDS (13124, Peypin, France).
Light absorbance at 360 nm
The turbidity of the different dipeptide samples at each temperature was measured monitoring the absorbance at 360 nm on a CARY-400 Varian spectrophotometer (Les Ulis, France). To study the dependence of peptide self-assembly on concentration, the turbidity was measured at 293 K. To study the dependence of peptide assembly state on the temperature, each sample was first heated to 333 K or cooled to 283 K before measuring the reassembly or disassembly, respectively. These samples were subsequently cooled or heated in 5-K stages and equilibrated for 15 min before measuring the turbidity at each assayed temperature.
NMR
NMR experiments were collected in a 500-MHz Avance Bruker spectrometer (Berlin, Germany) equipped with a triple-resonance TXI probehead. High-resolution 1H NMR spectra were recorded for several Ile-Phe and Val-Phe dipeptide samples at different concentrations and different temperatures. The samples used consisted of 0.05%, 0.1%, 0.5%, 1%, and 2% (w/v) of dipeptide dissolved in D2O from stock solutions. Spectra were also collected in the range 295–330 K to observe the temperature dependence of each individual sample.
Microscopy
Dipeptide samples (1.5%, w/v) were placed on carbon-coated copper grids and left for 5 min. The grids were then stained with 2% (w/v) uranyl acetate for another 2 min before analysis using a Hitachi (Tokyo, Japan) H-7000 transmission electron microscope operating at an accelerating voltage of 75 kV. A sample of 2% (w/v) Ile-Phe gel smeared on a 1-cm slide was allowed to dry at room temperature, followed by gold coating, before being imaged on a Hitachi S-570 scanning electron microscope.
Congo red binding
Congo red (CR) was diluted in a buffer containing 5 mM sodium phosphate and 150 mM NaCl, pH 7.0, to obtain a stock of 100 μM CR. A 2% (w/v) Ile-Phe gel was formed in the presence of 5 μM CR final concentration. A gel sample was placed on a microscope slide and sealed. The CR birefringence was detected under cross-polarized light using an optic microscope (Leica DMRB, Heidelberg, Germany).
Absorption and fluorescent spectra of Phe
The Phe fluorescence emission spectra of the dipeptide samples were recorded in a Perkin–Elmer (Wellesley, MA) 650–40 fluorescence spectrophotometer. The samples were excited at 250 nm and the emission between 260 nm and 400 nm was measured. Both excitation and emission slits were set at 10 nm. The absorption spectrum of Phe was measured between 230 nm and 330 nm on a CARY-400 Varian spectrophotometer.
FTIR
Diluted, gelled, and air-dried dipeptide samples were used for FTIR spectroscopy analysis. Exchangeable hydrogen atoms were replaced by deuterium by dissolving the dipeptide stocks in D2O. Infrared spectra were recorded with an FTS-6000 FT-IR spectrophotometer (BioRad, Hemel Hempstead, UK) equipped with a liquid nitrogen-cooled mercury/cadmium telluride detector and purged with a continuous flow of nitrogen gas or with a Bruker Tensor 27 FT-IR spectrometer. For each spectrum, 200 interferograms were collected and averaged. In every case, the buffer spectrum was subtracted and the baseline corrected. Second derivatives of the spectra were used to determine the frequencies at which the different spectral components were located. To monitor the effect of pH on self-assembly, we measured the pH established by the dipeptides themselves upon dilution in H2O, pH 5.8. This pH was either increased to pH 12.0 by addition of 1 N NaOH or lowered to pH 2.0 by addition of 1 N HCl to test the effect of N- and C-terminal group ionization in Ile-Phe self-assembly.
TNS binding
The fluorescence emission spectra of TNS with the dipeptide samples were recorded at 293 K in a Perkin-Elmer 650-40 fluorescence spectrophotometer. TNS was diluted in H2O to obtain a 1 mM stock solution. The samples were excited at 323 nm and the fluorescence emission was measured between 350 nm and 550 nm. Both excitation and emission slits were set at 10 nm. To follow the Ile-Phe dipeptide kinetic self-assembly by TNS binding, a 2% Ile-Phe sample in a solution containing a final 10 μM TNS concentration was heated to 333 K and then cooled gradually to 278 K. The sample was excited at 323 nm and the fluorescence emission at 423 nm was monitored.
Light scattering
Light scattering of a 2% Ile-Phe sample was measured using a Perkin-Elmer 650-40 fluorescence spectrophotometer. The sample was excited at 360 nm and the emission at the same wavelength was monitored. To follow the Ile-Phe dipeptide kinetic self-assembly, the sample was heated and cooled as in the TNS binding assay.
RESULTS
General observations
Lyophilized dipeptides could be dissolved at very high concentrations (200 mg/ml) in 1,1,1,3,3,3 hexafluoro-2-propanol. Although both peptides appeared to be highly soluble in the organic solvent, a rapid assembly into macroscopic structures was observed visually for the Ile-Phe peptide soon after dilution on H20 at final concentrations >0.5%, w/v. At 1%, w/v, the solution begins to gelate, forming a solid gel above 1.5% (w/v) final concentration (Fig. 1). Surprisingly, the Val-Phe solution remained liquid in all assayed conditions (Fig. 1).
Dependence of dipeptide self-assembly on the concentration
Gelation usually represents a macroscopic manifestation of a molecular self-assembly process (10), thus suggesting the formation of high aspect ratio nanostructures by the Ile-Phe dipeptide. To better quantify the dependence of peptide self-assembly on the concentration, we monitored the changes in light absorbance at 360 nm (Fig. 2 A) and recorded 1H-NMR spectra of solutions of both peptides at different concentrations (Fig. 2 B). The absorbance of Ile-Phe solutions at 360 nm is highly dependent on the peptide concentration, producing a sigmoid curve in which the transition between the soluble and polymerized states occurs at 1.1% (w/v) peptide concentration. No increase in absorbance was detected for the Val-Phe solutions even at high peptide concentrations.
FIGURE 2.

Dependence of peptide self-assembly on the concentration. (A) Turbidity of 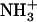 -Ile-Phe-COO− (solid circles) and
-Ile-Phe-COO− (solid circles) and 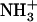 -Val-Phe-COO− (open circles) at different peptide concentrations. (B) 1H NMR spectra of
-Val-Phe-COO− (open circles) at different peptide concentrations. (B) 1H NMR spectra of 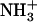 -Ile-Phe-COO− (left) and
-Ile-Phe-COO− (left) and 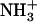 -Val-Phe-COO− (right) at different peptide concentrations (w/v).
-Val-Phe-COO− (right) at different peptide concentrations (w/v).
As expected, well-resolved sharp peaks can be clearly seen in the 1H-NMR spectra of both dipeptides at 0.02%, w/v, in aqueous solution (Fig. 2 B), indicating their monomeric status. However, as peptide concentration increases, the signals in the spectrum of the Ile-Phe solutions progressively broaden and most NMR peaks become unresolved, indicating a decrease in molecular motion due to the formation of the supramolecular structure. In good agreement with the visible (VIS) spectroscopic data, this effect is especially observable at peptide concentrations >0.5%, w/v. The spectra of Val-Phe solutions display well-resolved signals at all concentrations assayed, confirming that the dipeptide is unable to self-associate into higher-order structures in water (Fig. 2 B).
Structure of the Ile-Phe gel
The nanometric structures formed by the Ile-Phe dipeptide correspond to well-ordered, fibrillar, and elongated assemblies as seen by transmission electron microscopy (TEM) analysis with negative staining (Fig. 3), with almost no presence of amorphous aggregates. This is in contrast with other peptide assemblies, such as amyloid fibrils, in which molecules are easily trapped in kinetically stable arrangements of different topology, usually resulting in a mixture of structured and nonordered material (23). The formed structures are highly ordered and homogeneous, without branching. This can be also observed using scanning electron microscopy (SEM) (Fig. 3). The fibrils display a consistent width of ∼55 nm, which is clearly larger than that reported for typical amyloid fibrils but similar in diameter to the amyloid-like self-assembled peptide nanotubular structures described for diphenylalanine (20). In the TEM images the fibrillar structures appear to be quite transparent to the beam of electrons (Fig. 3), which could suggest that they are more or less hollow. The fibrils are very long (several micrometers) and usually appear to be laterally associated. Only a small number of them appear to be slightly twisted, with most remaining linear, suggesting that they do not tend to adopt a twisted helical structure. A similar observation was made with the diphenylalanine nanotubular structure described by Reches and Gazit (20). The pack of fibrils entangles into a supramolecular network, which is expressed macroscopically as the observed gel. No ordered or amorphous aggregated material could be observed in solutions of the Val-Phe dipeptide by TEM analysis (Fig. 3).
FIGURE 3.

Electron microscopy (EM) images of dipeptide samples. (A and B) Transmission EM images of a 1.5% (w/v) 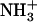 -Ile-Phe-COO− sample at increasing magnification. (C) Transmision EM image of a 1.5% (w/v)
-Ile-Phe-COO− sample at increasing magnification. (C) Transmision EM image of a 1.5% (w/v) 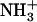 -Val-Phe-COO− sample. (D) Scanning EM image of a gel formed by 2% (w/v)
-Val-Phe-COO− sample. (D) Scanning EM image of a gel formed by 2% (w/v) 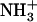 -Ile-Phe-COO−.
-Ile-Phe-COO−.
To further determine the properties of the structures observed by TEM and SEM, Ile-Phe samples (2%, w/v) were stained with the amyloidophilic CR. The structures formed by the dipeptide show a strong green-gold birefringence upon incubation when illuminated under cross-polarized light (Fig. 4), indicating that the Ile-Phe dipeptide already contains all the molecular information needed to self-associate into regular structures that may be somehow similar to those in amyloid fibrils.
FIGURE 4.

Congo red staining and birefringence of 2% (w/v) 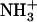 -Ile-Phe-COO−. (A) Dipeptide stained with Congo red and observed at 40× magnification. (B) Same sample observed between crossed polarizers, displaying the green birefringence characteristic of amyloid structures.
-Ile-Phe-COO−. (A) Dipeptide stained with Congo red and observed at 40× magnification. (B) Same sample observed between crossed polarizers, displaying the green birefringence characteristic of amyloid structures.
Dependence of Ile-Phe self-assembly on the temperature
Among other reasons, weak forces are useful for the construction of self-assembled materials because they allow reversibility. This property allows materials to respond to their environment by assembling and disassembling, an important factor in the design of “smart materials”, sensors, or controlled-release devices (24,25). The so-called thermoresponsive materials are an especially interesting kind of nanostructure in which the association state of the building blocks depends on the temperature. To test the temperature dependence of the formation of supramolecular assemblies by the Ile-Phe dipeptide, we monitored the changes in light absorbance at 360 nm and recorded the 1H-NMR spectra at different temperatures (Fig. 5). The nanostructures formed by the dipeptide at 2% (w/v) are sensitive to temperature changes, as indicated by the progressive decrease in absorbance observed when the temperature increases (Fig. 5 A). The melting curve is cooperative and fully reversible, indicating that the assembling and disassembling processes are occurring in a coordinated way, as expected for a self-associated structure in which the noncovalent interactions linking the blocks are progressively gained or lost. It may be observed that the dipeptide sample is solid at 293 K, becomes completely fluid above 313 K, and recovers its initial state upon cooling. The transition temperature depends on the concentration of the dipeptide in the sample, being 304 K for a 2%, w/v, and 299 K for a 1.5%, w/v, sample. The reversible assembly and disassembly of the Ile-Phe in response to changes in temperature was also confirmed by 1H-NMR (Fig. 5 B). The low-temperature NMR signals become progressively better resolved as temperature increases, indicating higher mobility of the building blocks and, thus, disorganization of the supramolecular structures. The signal is broad again upon cooling and the spectrum becomes indistinguishable from the initial one, indicating reassembly of the fibrillar organization. No dependence on the temperature was detected for a 2%, w/v, Val-Phe sample either by VIS-spectroscopy or 1H-NMR spectroscopy.
FIGURE 5.

Dependence of peptide assembly state on the temperature. (A) Disassembly of 1.5% (w/v) (solid squares) and 2% (w/v) (solid circles) 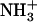 -Ile-Phe-COO− dipeptide upon heating. Reassembly of 2% (w/v)
-Ile-Phe-COO− dipeptide upon heating. Reassembly of 2% (w/v) 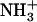 -Ile-Phe-COO−dipeptide upon cooling (open circles). No changes in absorbance were observed upon heating a 2% (w/v)
-Ile-Phe-COO−dipeptide upon cooling (open circles). No changes in absorbance were observed upon heating a 2% (w/v) 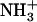 -Val-Phe-COO− sample (solid triangles). (B) 1H NMR spectra of
-Val-Phe-COO− sample (solid triangles). (B) 1H NMR spectra of 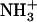 -Ile-Phe-COO− (left) and
-Ile-Phe-COO− (left) and 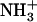 -Val-Phe-COO− (right) at increasing temperatures.
-Val-Phe-COO− (right) at increasing temperatures.
Molecular interactions implied in the assembly process
FTIR spectra were obtained to identify intermolecular interactions in the Ile-Phe gel and solution states (Table 1). At 0.1% (w/v) peptide concentration, an NH band at 3398 cm−1 and an amide I band at 1662 cm−1 corresponding to non-hydrogen-bonded NH and CO functionalities were detected. The absence of a CO stretching band above 1700 cm−1, together with the additional detection of a strong vibrational signal at 1598 cm−1 resulting from the asymmetric stretching of the C-terminal COO− group, indicates that all COO− groups are deprotonated in the dipeptide solution, pH 5.8 (26–28). Surprisingly, the NH and amide I bands are still detected at positions corresponding to non-hydrogen-bonded groups in the polymerized state (2%, w/v), indicating that the amide bond is not involved in self-assembly in aqueous solution. By contrast, the signal at 1598 cm−1 suffers a strong downshift to 1570 cm−1, a change in the position of the C-terminal COO− group that has been previously observed in the vicinity of 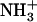 groups (29). Since the dipeptide contains only a single rigid amide bond, the coupling between COO− and
groups (29). Since the dipeptide contains only a single rigid amide bond, the coupling between COO− and 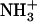 groups suggests a supramolecular structure stabilized by head-to-tail-interactions between dipeptides. According to the above-described observation of a gradual network disintegration into soluble and probably monomeric species upon heating, the COO− band shifts again to higher wavenumbers (1597 cm−1) when the temperature is increased to 323 K, indicating a disruption of the intermolecular interactions formed between the dipeptide molecules. The process is fully reversible and the spectrum recovers its original shape upon cooling. The COO− vibrational downshift is not observed at intermediate peptide concentrations (0.8%, w/v) at which the presence of self-assembled species is already detected by VIS spectroscopy and 1H-NMR, suggesting that the changes in the local environments of COO− groups occur during or after the consolidation of the nanostructures. As expected for a non-self-assembling species, a COO− band at 1598 cm−1 appears in a 2% (w/v) solution of the Val-Phe dipeptide. Lowering the pH of a gel solution to 2.0 by adding HCl or increasing it to 12.0 by adding NaOH further demonstrated the role played by the COO− and
groups suggests a supramolecular structure stabilized by head-to-tail-interactions between dipeptides. According to the above-described observation of a gradual network disintegration into soluble and probably monomeric species upon heating, the COO− band shifts again to higher wavenumbers (1597 cm−1) when the temperature is increased to 323 K, indicating a disruption of the intermolecular interactions formed between the dipeptide molecules. The process is fully reversible and the spectrum recovers its original shape upon cooling. The COO− vibrational downshift is not observed at intermediate peptide concentrations (0.8%, w/v) at which the presence of self-assembled species is already detected by VIS spectroscopy and 1H-NMR, suggesting that the changes in the local environments of COO− groups occur during or after the consolidation of the nanostructures. As expected for a non-self-assembling species, a COO− band at 1598 cm−1 appears in a 2% (w/v) solution of the Val-Phe dipeptide. Lowering the pH of a gel solution to 2.0 by adding HCl or increasing it to 12.0 by adding NaOH further demonstrated the role played by the COO− and 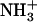 groups in maintaining a highly ordered nanostructure. In these conditions, the carboxyl and amino groups become, respectively, protonated and deprotonated and this results in the disintegration of the network and the formation of numerous amorphous aggregates after 1 −h of incubation (data not shown).
groups in maintaining a highly ordered nanostructure. In these conditions, the carboxyl and amino groups become, respectively, protonated and deprotonated and this results in the disintegration of the network and the formation of numerous amorphous aggregates after 1 −h of incubation (data not shown).
TABLE 1.
Selected FTIR bands of Ile-Phe and Val-Phe dipeptide samples at various concentrations and temperatures
| Peptide | Concentration % (w/v) | Temperature K | NH | Amide I | COO− |
|---|---|---|---|---|---|
| Ile-Phe | 0.1 | 298 | 3398 | 1662 | 1598 |
| Ile-Phe | 0.8 | 298 | 3396 | 1661 | 1598 |
| Ile-Phe | 2 | 298 | 3397 | 1660 | 1570 |
| Ile-Phe | 2 | 323 | 3387 | 1659 | 1597 |
| Val-Phe | 2 | 298 | 3396 | 1663 | 1598 |
| Ile-Phe | 2 | 298 | 3305 | 1618 | 1570 |
| dehydrated | 3403 | 1658 | |||
| 1678 |
Wave numbers are given in cm−1.
The typical amide I peak at ∼1620–1640 cm,−1 usually associated with the presence of β-sheet structures, was not detected in any of the samples analyzed. It has been shown that the loss of bound water in the gel formed by a Tau peptide results in increased formation of β-sheet structure in the gel and, finally, fibrillation (13). The FTIR spectra of a dehydrated gel sample showed no significant shift in the COO− band. In contrast, a strong band at 3305 cm−1 in the NH region corresponding to hydrogen-bonded NHs could be detected, whereas the band corresponding to free NHs was minor. In the amide-I region the presence of additional bands at 1618 cm−1 and 1678 cm−1 is indicative of the formation of new β-sheet-like intermolecular bonds in the supramolecular structure upon loss of water.
Role of hydrophobicity in the assembly process
The previous data suggest that head-to-tail interactions between the amino and carboxyl-terminus of dipeptides stabilize the assembly of fibrils. However, the establishment of such intermolecular contacts could not be the initial driving force for Ile-Phe polymerization, as the Val-Phe dipeptide possesses exactly the same groups and remains in solution, most likely in the monomeric state, at high concentrations. For the same reason, the stacking between aromatic rings can be discarded as the main interaction promoting self-assembly in this particular peptide system. This is confirmed by several observations. First, the signals of aromatic protons in 1H-NMR spectra of Ile-Phe are only slightly shifted upfield as the temperature is increased, whereas aromatic signals should be significantly shifted downfield after disassembly of intermolecular aromatic stacking interactions in an assembled peptide. Second, the absorption and fluorescent spectra of Phe in dilute samples (0.02%, w/v) and at near-transition concentration (1%, w/v) display identical shape. Finally, no isosbestic point was observed that could reveal the transition between two spectroscopically different states of Phe when the absorption spectral changes were recorded for a 2% (w/v) sample at variable temperature (results not shown).
It has long been suggested that hydrophobic interactions play an important role in protein and peptide self-assembly (30). The presence of an additional methyl group in the Ile-Phe dipeptide relative to Val-Phe provides it with increased hydrophobicity. We used the polarity-sensitive probe TNS to elucidate whether the self-association process is driven and/or stabilized by hydrophobic interactions. TNS binds with much higher affinity to surfaces or pockets formed by clusters of hydrophobic groups than to solvent-exposed isolated hydrophobic groups, resulting in an increase and blue-shift in the maximum of fluorescence emission compared with the emission of free TNS in aqueous solution. Little binding was detected for a 0.1% (w/v) sample of Ile-Phe (Fig. 6 A), confirming that the hydrophobic side chains of the dipeptides do not associate at low concentrations. In contrast, the probe binds strongly to the macromolecular structures formed in a 2% (w/v) sample, as proven by a large increase in fluorescence and a strong blue-shift of the maximum from 443 nm to 423 nm (Fig. 6 A), indicating the formation of a large hydrophobic environment upon self-assembly. The loss of most of the fluorescence signal upon heating the sample indicates the requirement of an ordered nanostructure for TNS to bind efficiently (Fig. 6 A). The process is again fully reversible and the TNS binding ability is restored upon cooling (data not shown). We took advantage of the reversibility of the process to study whether the self-assembly of the dipeptide and the formation of hydrophobic clusters occur simultaneously. A 2% (w/v) sample was heated to 333 K, and then progressively cooled down to 278 K, simultaneously monitoring the dipeptide self-assembly by light scattering and the formation of hydrophobic regions by TNS fluorescence emission. As can be observed in Fig. 6 B, the light-scattering dependence on the temperature is sigmoid and sharply corresponds to that reported by measuring absorbance at 360 nm. However, the increase in fluorescence emission occurs in two steps. The first, monotonic increase in fluorescence does not coincide with the light-scattering curve and may be interpreted as the hydrophobic interaction-governed self-assembly of the dipeptide in soluble oligomers. The concentration of these soluble assemblies saturates at ∼321 K (after ∼90s), as inferred from the difference between the TNS-binding and light-scattering signals (Fig. 6 B, inset). Accordingly, at these temperatures, NMR signals are well resolved, indicating high mobility of the building blocks and probably absence of rigid supramolecular structures. Several of these small aggregates would probably form a larger aggregate in a second or higher-order reaction and this aggregate would serve as the nucleus for the growth reaction visible from 150 s on, resulting in the rapid formation of larger assemblies detectable by light scattering, with a parallel increase in TNS binding and broadening of the NMR peaks. The data support a nucleation-growth pathway that gives rise to a remarkably high degree of cooperativity. This behavior is reminiscent of the formation of polypeptide aggregates, which usually exhibit a nucleated polymerization reaction in which an initial nucleation event is followed by the extension of newly formed nuclei into larger aggregates, including insoluble fibrils (31).
FIGURE 6.

Role of hydrophobicity on dipeptide self assembly. (A) Fluorescence emission spectra of TNS incubated in the presence of 0.1% (w/v) (dashed lines) and 2% (w/v) (solid lines) 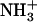 -Ile-Phe-COO− dipeptide. (B) Kinetics of
-Ile-Phe-COO− dipeptide. (B) Kinetics of 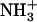 -Ile-Phe-COO− dipeptide self-assembly followed by light scattering (dashed lines) and TNS binding (solid lines). (Inset) Relative amounts of soluble self-assemblies found at the beginning of the polymerization process, as inferred from the difference between the TNS-binding and light-scattering signals.
-Ile-Phe-COO− dipeptide self-assembly followed by light scattering (dashed lines) and TNS binding (solid lines). (Inset) Relative amounts of soluble self-assemblies found at the beginning of the polymerization process, as inferred from the difference between the TNS-binding and light-scattering signals.
DISCUSSION
The dimensions of the fibrillar structures formed by the Ile-Phe dipeptide are similar to those reported for the nanotubes formed by diphenylalanine, the core recognition motif of Alzheimer's β-amyloid polypeptide (20). The crystal x-ray structure of the diphenylalanine peptide, as formed by fast evaporation of an aqueous solution of the peptide at high temperature, showed a crystal packing where the dipeptide forms aligned and elongated channels with a hydrophilic inner surface. The channels are lined with hydrogen-bond donors and acceptors in charged groups (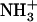 and COO−), with hydrophobic side chains that act as a glue between the cylinders of peptide main chains and promote fiber formation (32,33). This model is compatible with the experimental data for the Ile-Phe fibrillar structures, including the observations that fibrillar structures are somehow transparent to electrons, the absence of β-sheet intermolecular contacts in the gel state, the observed
and COO−), with hydrophobic side chains that act as a glue between the cylinders of peptide main chains and promote fiber formation (32,33). This model is compatible with the experimental data for the Ile-Phe fibrillar structures, including the observations that fibrillar structures are somehow transparent to electrons, the absence of β-sheet intermolecular contacts in the gel state, the observed 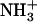 /COO− head-to-tail interactions, and the relevant role played by hydrophobicity in the assembly (Fig. 7).
/COO− head-to-tail interactions, and the relevant role played by hydrophobicity in the assembly (Fig. 7).
FIGURE 7.

Molecular model of Ile-Phe self-assembled structures. The model is based on the crystal x-ray structure of the diphenylalanine peptide (33). Dipeptide backbone and hydrophobic side chains are shown as stick representations. 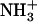 and COO− terminal groups establishing head-to-tail interactions in the central channel are shown in spacefill representation.
and COO− terminal groups establishing head-to-tail interactions in the central channel are shown in spacefill representation.
Overall, the data herein allow us to propose a mechanism for the self-assembly of the Ile-Phe dipeptide into the observed nanostructures and to explain the self-association incompetence of the Val-Phe version. The initial establishment of intermolecular hydrogen bonds or electrostatic interactions between the extremes of dipeptide molecules or amide bonds is likely to be highly disfavored in water due to strong competitive solvent effects. At this stage, the establishment of intermolecular hydrophobic interactions between the side chains of Ile-Phe would drive the formation of sufficiently large primary soluble assemblies, which may act as nuclei or scaffolds for subsequent bonding. Such assemblies would further organize into nanofibrils by head-to-tail interactions between dipeptides. This hierarchical pathway for the self-assembly of Ile-Phe into fibrillar structures would explain the observed curves upon cooling a solution of molecularly dissolved monomers at high temperature. The fact that no specific FTIR signal, relative to that of the soluble monomer, could be detected at the temperature at which the small aggregates maximally populate (Fig. 6 B and Table 1) indicates that they more likely lack an ordered structure and specific bonding, pointing to hydrophobicity as the main driving force in the first stages of polymerization. It would be the subsequent establishment of specific, oriented, noncovalent contacts that might permit the formation of a highly ordered fibrillar superstructure. The lower hydrophobicity of the Val-Phe dipeptide would prevent the formation of the initial assemblies, thus aborting the subsequent nucleation and polymerization events. Alternatively, if the nucleation event occurs, but no specific contacts can be established thereafter, the result is the formation of nonordered amorphous aggregates probably stabilized by nonspecific hydrophobic interactions, as observed here for the Ile-Phe dipeptide at low or high pH.
The proposed dominating role of hydrophobic interactions at the beginning of the process may also explain the self-association properties of different dipeptides in the literature. For instance, whereas Phe-Phe has been shown to form nanotubes (20) and Phg-Phg spherical structures (20), the more polar Trp-Phe, Trp-Trp, and Trp-Tyr dipeptides were unable to self-assemble under the same conditions (20). These observations belie the role of aromatic stacking as a main assembly-driving force and point to the higher hydrophobicity of Phe and Phg as the mechanism responsible for the initial assembly reaction. According to this, Phe is the aromatic residue more commonly found in amyloid-forming peptides (34). Along with our data, and in contrast to previous assumptions, it has been shown that in the natural amyloid-forming peptide amylin the presence of an aromatic residue in the core is not necessary for amyloid formation and a large aliphatic residue performs equally well, whereas substitution of the aromatic residue by an Ala results in very reduced aggregation (35). Also, a recent study of the aggregation of several mutants of human muscle acylphosphatase, in which aromatic residues were substituted with nonaromatic ones, shows that the changes in aggregation rates upon mutation arise predominantly from variations in hydrophobicity and intrinsic β-sheet propensity (36). Interestingly enough, the computational comparison of the binding propensities and the amyloid formation preferences of natural amino acids also revealed that Ile is the least structurally conserved residue in protein binding and at the same time has a high propensity for amyloid formation. This suggests that nature tends to avoid Ile conservation in protein-protein interactions to limit amyloid formation (37). Importantly, in the first study on the effects of mutation on the nucleation step of Aβ, it was shown that for position 18 of this peptide Ile is precisely the residue that promotes the fastest nucleation reaction (38). These observations are in full agreement with the significant correlations between aggregation and both hydrophobicity and β-sheet propensity that we found in the adjacent position 19 of the Aβ42 peptide (22), suggesting that Phe promotes aggregation because of these factors rather than for its aromaticity. Nevertheless, aromatic-aromatic interactions could still play a very important role in allowing specific contacts that dictate either the structure of the assembly, its stability, or the kinetics of self-assembly in peptide-derived nanostructures and amyloid fibrils.
Although the growth of fibrillar structures typically requires nucleation, the nature and properties of the nuclei and first soluble aggregates are still largely unknown due to the difficulty involved in characterizing them. The hydrophobic recruitment mechanism reported here could be of relevance to understanding the fibrillogenesis pathway of peptides, such us Aβ42. Recent studies show that Alzheimer's peptide fibril formation starts with the formation of globular amyloid-derived diffusible ligands (39) rather than with direct assembly of short protofibrils. Fibrils, and probably protofibrils, are stabilized by specific interactions (40), but if, as shown here for dipeptides, these interactions cannot efficiently trigger a self-assembly process in aqueous environment, it seems reasonable to propose that self-association begins with the formation of sufficiently large primary soluble globular structures (amyloid-derived diffusible ligands) driven by more or less unspecific hydrophobic contacts. As for dipeptides, their structural reorganization by specific interactions, including aromatic stacking, could turn them into short protofibrillar scaffolds instead of amorphous aggregates, with the ability to recruit and orientate new peptide units, acting as seeds of the fibrillogenic process. Understanding the details of the first steps of the aggregation/fibrillation mechanism at the molecular level is central to developing strategies for treatment or possible prevention of amyloid-deposition diseases. As shown here, in addition to their biotechnological applications, short peptides can also serve as ideal model systems for such studies.
Acknowledgments
We thank Dr. Esteve Padros, Dr. Josep Cladera, and their group for FTIR facilities.
This work was supported by grants BIO2004-05879 and CTQ2006-01080 from the Ministerio de Ciencia y Tecnología (MCYT) Spain, by the Centre de Referència en Biotecnologia, and grants SGR2005-00037 and SGR2005-01037 (Generalitat de Catalunya, Spain).
References
- 1.Hartgerink, J. D., E. Beniash, and S. I. Stupp. 2001. Self-assembly and mineralization of peptide-amphiphile nanofibers. Science. 294:1684–1688. [DOI] [PubMed] [Google Scholar]
- 2.Zhang, S. 2003. Fabrication of novel biomaterials through molecular self-assembly. Nat. Biotechnol. 21:1171–1178. [DOI] [PubMed] [Google Scholar]
- 3.Du, C., G. Falini, S. Fermani, C. Abbott, and J. Moradian-Oldak. 2005. Supramolecular assembly of amelogenin nanospheres into birefringent microribbons. Science. 307:1450–1454. [DOI] [PubMed] [Google Scholar]
- 4.Yoshimura, I., Y. Miyahara, N. Kasagi, H. Yamane, A. Ojida, and I. Hamachi. 2004. Molecular recognition in a supramolecular hydrogel to afford a semi-wet sensor chip. J. Am. Chem. Soc. 126:12204–12205. [DOI] [PubMed] [Google Scholar]
- 5.Haines, L. A., K. Rajagopal, B. Ozbas, D. A. Salick, D. J. Pochan, and J. P. Schneider. 2005. Light-activated hydrogel formation via the triggered folding and self-assembly of a designed peptide. J. Am. Chem. Soc. 127:17025–17029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kisiday, J., M. Jin, B. Kurz, H. Hung, C. Semino, S. Zhang, and A. J. Grodzinsky. 2002. Self-assembling peptide hydrogel fosters chondrocyte extracellular matrix production and cell division: Implications for cartilage tissue repair. Proc. Natl. Acad. Sci. USA. 99:9996–10001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aggeli, A., M. Bell, N. Boden, J. N. Keen, P. F. Knowles, T. C. McLeish, M. Pitkeathly, and S. E. Radford. 1997. Responsive gels formed by the spontaneous self-assembly of peptides into polymeric β-sheet tapes. Nature. 386:259–262. [DOI] [PubMed] [Google Scholar]
- 8.Wang, C., R. J. Stewart, and J. Kopecek. 1999. Hybrid hydrogels assembled from synthetic polymers and coiled-coil protein domains. Nature. 397:417–420. [DOI] [PubMed] [Google Scholar]
- 9.Lyon, R. P., and W. M. Atkins. 2001. Self-assembly and gelation of oxidized glutathione in organic solvents. J. Am. Chem. Soc. 123:4408–4413. [DOI] [PubMed] [Google Scholar]
- 10.Rajagopal, K., and J. P. Schneider. 2004. Self-assembling peptides and proteins for nanotechnological applications. Curr. Opin. Struct. Biol. 14:480–486. [DOI] [PubMed] [Google Scholar]
- 11.Gazit, E. 2005. Mechanisms of amyloid fibril self-assembly and inhibition. Model short peptides as a key research tool. FEBS J. 272:5971–5978. [DOI] [PubMed] [Google Scholar]
- 12.Westermark, P. 2005. Aspects on human amyloid forms and their fibril polypeptides. FEBS J. 272:5942–5949. [DOI] [PubMed] [Google Scholar]
- 13.Juszczak, L. J. 2004. Comparative vibrational spectroscopy of intracellular Tau and extracellular collagen I reveals parallels of gelation and fibrillar structure. J. Biol. Chem. 279:7395–7404. [DOI] [PubMed] [Google Scholar]
- 14.Shen, C. L., M. C. Fitzgerald, and R. M. Murphy. 1994. Effect of acid predissolution on fibril size and fibril flexibility of synthetic β-amyloid peptide. Biophys. J. 67:1238–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Esteras-Chopo, A., L. Serrano, and M. L. de la Paz. 2005. The amyloid stretch hypothesis: Recruiting proteins toward the dark side. Proc. Natl. Acad. Sci. USA. 102:16672–16677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ventura, S., J. Zurdo, S. Narayanan, M. Parreno, R. Mangues, B. Reif, F. Chiti, E. Giannoni, C. M. Dobson, F. X. Aviles, and L. Serrano. 2004. Short amino acid stretches can mediate amyloid formation in globular proteins: the Src homology 3 (SH3) case. Proc. Natl. Acad. Sci. USA. 101:7258–7263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ivanova, M. I., M. R. Sawaya, M. Gingery, A. Attinger, and D. Eisenberg. 2004. An amyloid-forming segment of β2-microglobulin suggests a molecular model for the fibril. Proc. Natl. Acad. Sci. USA. 101:10584–10589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Madine, J., A. J. Doig, A. Kitmitto, and D. A. Middleton. 2005. Studies of the aggregation of an amyloidogenic alpha-synuclein peptide fragment. Biochem. Soc. Trans. 33:1113–1115. [DOI] [PubMed] [Google Scholar]
- 19.Leffers, K. W., H. Wille, J. Stohr, E. Junger, S. B. Prusiner, and D. Riesner. 2005. Assembly of natural and recombinant prion protein into fibrils. Biol. Chem. 386:569–580. [DOI] [PubMed] [Google Scholar]
- 20.Reches, M., and E. Gazit. 2003. Casting metal nanowires within discrete self-assembled peptide nanotubes. Science. 300:625–627. [DOI] [PubMed] [Google Scholar]
- 21.Bitan, G., S. S. Vollers, and D. B. Teplow. 2003. Elucidation of primary structure elements controlling early amyloid β-protein oligomerization. J. Biol. Chem. 278:34882–34889. [DOI] [PubMed] [Google Scholar]
- 22.de Groot, N. S., F. X. Aviles, J. Vendrell, and S. Ventura. 2006. Mutagenesis of the central hydrophobic cluster in Aβ42 Alzheimer's peptide. Side-chain properties correlate with aggregation propensities. FEBS J. 273:658–668. [DOI] [PubMed] [Google Scholar]
- 23.Zurdo, J., J. I. Guijarro, J. L. Jimenez, H. R. Saibil, and C. M. Dobson. 2001. Dependence on solution conditions of aggregation and amyloid formation by an SH3 domain. J. Mol. Biol. 311:325–340. [DOI] [PubMed] [Google Scholar]
- 24.Muni, N. J., H. Qian, N. M. Qtaishat, R. A. Gemeinhart, and D. R. Pepperberg. 2006. Activation of membrane receptors by neurotransmitter released from temperature-sensitive hydrogels. J. Neurosci. Methods. 151:97–105. [DOI] [PubMed] [Google Scholar]
- 25.Hokugo, A., M. Ozeki, O. Kawakami, K. Sugimoto, K. Mushimoto, S. Morita, and Y. Tabata. 2005. Augmented bone regeneration activity of platelet-rich plasma by biodegradable gelatin hydrogel. Tissue Eng. 11:1224–1233. [DOI] [PubMed] [Google Scholar]
- 26.Venyaminov, S., and N. N. Kalnin. 1990. Quantitative IR spectrophotometry of peptide compounds in water (H2O) solutions. I. Spectral parameters of amino acid residue absorption bands. Biopolymers. 30:1243–1257. [DOI] [PubMed] [Google Scholar]
- 27.Kalnin, N. N., I. A. Baikalov, and S. Venyaminov. 1990. Quantitative IR spectrophotometry of peptide compounds in water (H2O) solutions. III. Estimation of the protein secondary structure. Biopolymers. 30:1273–1280. [DOI] [PubMed] [Google Scholar]
- 28.Venyaminov, S., and N. N. Kalnin. 1990. Quantitative IR spectrophotometry of peptide compounds in water (H2O) solutions. II. Amide absorption bands of polypeptides and fibrous proteins in α-, β-, and random coil conformations. Biopolymers. 30:1259–1271. [DOI] [PubMed] [Google Scholar]
- 29.Eker, F., X. Cao, L. Nafie, and R. Schweitzer-Stenner. 2002. Tripeptides adopt stable structures in water. A combined polarized visible Raman, FTIR, and VCD spectroscopy study. J. Am. Chem. Soc. 124:14330–14341. [DOI] [PubMed] [Google Scholar]
- 30.Deechongkit, S., E. T. Powers, S. L. You, and J. W. Kelly. 2005. Controlling the morphology of cross β-sheet assemblies by rational design. J. Am. Chem. Soc. 127:8562–8570. [DOI] [PubMed] [Google Scholar]
- 31.Thirumalai, D., D. K. Klimov, and R. I. Dima. 2003. Emerging ideas on the molecular basis of protein and peptide aggregation. Curr. Opin. Struct. Biol. 13:146–159. [DOI] [PubMed] [Google Scholar]
- 32.Gorbitz, C. H. 2001. Nanotube formation by hydrophobic dipeptides. Chemistry (Easton). 7:5153–5159. [DOI] [PubMed] [Google Scholar]
- 33.Gorbitz, C. H. 2006. The structure of nanotubes formed by diphenylalanine, the core recognition motif of Alzheimer's β-amyloid polypeptide. Chem. Commun. (Camb.). 2332–2334. [DOI] [PubMed]
- 34.Gazit, E. 2002. A possible role for pi-stacking in the self-assembly of amyloid fibrils. FASEB J. 16:77–83. [DOI] [PubMed] [Google Scholar]
- 35.Tracz, S. M., A. Abedini, M. Driscoll, and D. P. Raleigh. 2004. Role of aromatic interactions in amyloid formation by peptides derived from human Amylin. Biochemistry. 43:15901–15908. [DOI] [PubMed] [Google Scholar]
- 36.Bemporad, F., N. Taddei, M. Stefani, and F. Chiti. 2006. Assessing the role of aromatic residues in the amyloid aggregation of human muscle acylphosphatase. Protein Sci. 15:862–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ma, B., and R. Nussinov. 2006. Trp/Met/Phe hot spots in protein-protein interactions: potential targets in drug design. Curr. Top. Med. Chem. In press. [DOI] [PubMed]
- 38.Christopeit, T., P. Hortschansky, V. Schroeckh, K. Guhrs, G. Zandomeneghi, and M. Fandrich. 2005. Mutagenic analysis of the nucleation propensity of oxidized Alzheimer's β-amyloid peptide. Protein Sci. 14:2125–2131.15987892 [Google Scholar]
- 39.Lambert, M. P., A. K. Barlow, B. A. Chromy, C. Edwards, R. Freed, M. Liosatos, T. E. Morgan, I. Rozovsky, B. Trommer, K. L. Viola, P. Wals, C. Zhang, C. E. Finch, G. A. Krafft, and W. L. Klein. 1998. Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA. 95:6448–6453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Petkova, A. T., Y. Ishii, J. J. Balbach, O. N. Antzutkin, R. D. Leapman, F. Delaglio, and R. Tycko. 2002. A structural model for Alzheimer's β-amyloid fibrils based on experimental constraints from solid state NMR. Proc. Natl. Acad. Sci. USA. 99:16742–16747. [DOI] [PMC free article] [PubMed] [Google Scholar]