

Abstract
Myelin protein zero (P0 or P0 glycoprotein), the major integral membrane protein in peripheral nervous system myelin, plays a key role in myelin membrane compaction and stability. While the structure of P0 extracellular domain was determined by crystallography, the paucity of any structural data on the highly positive-charged P0 cytoplasmic domain (P0-cyt) has greatly limited our understanding of the mechanism of P0 function. Here, using circular dichroism and intrinsic fluorescence spectroscopy, we attempted to elucidate the structure of human P0-cyt (hP0-cyt) in membrane mimetic environments composed of detergents or lipid vesicles. We found that the secondary structure of P0-cyt was polymorphic—at the lipid/protein ratio corresponding to that of mature peripheral myelin (∼50:1), hP0-cyt mainly adopted a β-conformation, whereas when the proportion of lipid increased, the structure underwent a β→α transition. By contrast, the secondary structure of the major isoform of myelin basic protein, another myelin protein with a very large positive charge, remained unchanged across a wide range of lipid/protein ratios. We propose that when hP0-cyt is bound at sufficient concentration to lamellar lipid bilayers such as myelin, it folds into a β-conformation; before this threshold lipid/protein ratio is reached, the domain is α-helical. We suggest that the cytoplasmic apposition (major dense line) in compact myelin may be stabilized via the hydrogen-bonding of β-strands formed as a result of local P0-P0 aggregation.
INTRODUCTION
Myelin protein zero (P0), an integral glycoprotein, is the major protein in peripheral nervous system myelin (1). P0 is a single-pass transmembrane protein with a 124-residue disulfide-stabilized VH-like extracellular domain (2–4) and a highly basic, 69-residue cytoplasmic domain (P0-cyt). As a cell adhesion molecule, P0 serves as the structural cement in peripheral nervous system myelin (5), playing a key role in the compaction and stability of both extracellular and cytoplasmic surfaces of the myelin membrane. Mutations in MPZ, the gene for this protein, can cause serious neurological disorders, including Charcot-Marie-Tooth disease type 1B, Dejerine-Sottas syndrome (DSS), and congenital hypomyelination neuropathy (4,6), presumably either by disrupting the normal myelin architecture through misfolding of the myelin-incorporated mutated P0, or by disrupting the normal biosynthesis of the myelin. In either case, myelin membrane structure and intermembrane interactions are altered. Intermembrane interactions are accounted for by a balance between repulsive electrostatic interactions and attractive van der Waals interactions (7) and based on the theory of colloid stability developed by Derjaguin, Landau, Verwey, and Overbeek (referred to as DLVO theory) (8). As examples, the swelling and compaction at the extracellular apposition of peripheral nervous system myelin as a function of pH and ionic strength can be largely explained by such considerations, as can the discontinuous myelin compaction of the extracellular apposition at physiological pH and ionic strength, which may be due to an ionic interaction of the P0 extracellular domain involving His-52 and Arg-45 (9,10). By contrast, the mechanism underlying the invariance of the narrow cytoplasmic apposition of peripheral nervous system myelin to a wide range of pH values and ionic strengths (9,11) remains unexplained.
Solution scattering of full-length P0 in a membrane lipid mimetic (1–8 mg protein/ml in 0.1% sodium dodecylsulfate, SDS) (12) suggests that like the extracellular domain in crystals (that form from protein at ∼30 mg/ml) (10) the entire molecule also assembles tetramerically. Thus, the folded cytoplasmic domain of P0, whose structure at atomic resolution has not yet been solved crystallographically, must conform to the overall tetrameric assembly and also fit into the narrow intermembrane gap at the cytoplasmic apposition.
Using circular dichroism (CD) and intrinsic fluorescence spectroscopies, we carried out systematic studies on human P0-cyt (hP0-cyt) structure in membrane mimetic environments. Our experiments addressed the structure of the protein at concentrations that were ∼10 to 1000-fold less than the concentration at which the tetramers form. We first studied the protein folding in membrane-mimicking detergents including zwitterionic dodecylphosphocholine (DPC), micellar SDS, and a low dielectric organic solvent 2,2,2-trifluoroethanol (TFE). We further simulated the native environment of hP0-cyt by using small unilamellar lipid vesicles (SUVs) made of phosphatidylcholine (PC) and phosphatidylserine (PS). Our results suggest a novel secondary structure change of hP0-cyt as a function of the relative proportion of membrane lipid and protein constituents. In aqueous buffer or at lipid/protein mole ratios <∼50:1 (which is the level in peripheral nervous system myelin), hP0-cyt maintained a β-conformation, whereas when the ratio increased (i.e., concentration of lipid or detergent increases), a β→α transition occurred. By contrast, under similar experimental conditions the 18.5 kDa isoform of bovine myelin basic protein (bMBP), another key protein in the cytoplasmic apposition of myelin sheaths, did not show a similar trend in structural changes. Vesicles made of negative-charged PS or phosphatidylinositol (PI), which were examined to study the charge effect of lipid on the structures of hP0-cyt and bMBP, maintained similar secondary structures as compared with those of the proteins in aqueous buffer.
EXPERIMENTAL PROCEDURES
SDS and TFE were obtained from Sigma (St. Louis, MO). DPC, egg PC, brain PS, bovine liver PI, and cholesterol were obtained from Avanti Polar Lipids (Alabaster, AL). Dephosphorylated 18.5 kDa bMBP was obtained from Upstate (Charlottesville, VA).
Peptide synthesis
The 69-residue-long hP0-cyt peptide was synthesized at the Department of Molecular Biochemistry and Pharmacology, Institute for Pharmacological Research “Mario Negri,” Milan, Italy, using the solid-phase peptide synthesis method (13). The peptide was purified by reverse-phase HPLC, and its integrity determined by MALDI-TOF. The purity of the peptide used in this study was >95%.
Preparation of lipid vesicles
The vesicles were prepared according to published procedures, and are reported to be 40 nm in diameter (14). Briefly, PS, or PI, or a mixture of PC and PS (molar ratio 12:1) or PC, PS, and cholesterol (molar ratio 62:5:33), were dissolved in pure chloroform and dried under a gentle stream of N2 gas to form a lipid film, and further dried under high vacuum overnight. The dried lipid film was hydrated in an aqueous buffer to form a lipid suspension. Small, unilamellar vesicles (SUVs) were produced by sonicating the lipid suspension in an ice-water bath using an ultrasonic homogenizer equipped with a microtip probe (Cole-Parmer Instrument, Chicago, IL) until the solution became translucent. Titanium particles shed by the ultrasonic probe into the solution were removed by centrifugation. Total lipid concentration in lipid vesicles ranged from 0.1–4 mg/ml for CD and fluorescence spectroscopy studies and 25 mg/ml for small-angle x-ray diffraction.
Circular dichroism (CD) spectroscopy
All CD samples were prepared in phosphate-buffered saline (PBS) unless otherwise noted. All data were acquired on an AVIV Model 202 Circular Dichroism Spectropolarimeter (Lakewood, NJ). Samples of vesicles made of PS or PI alone were made in 0.5 mM Tris-HCl, pH 7.4, instead of PBS to avoid turbidity caused by protein-induced vesicle aggregation. Spectra were recorded in a 0.1-cm path-length rectangular quartz cell at 25°C from 260 nm to 195 nm with 1 nm increments and an averaging time of one second per data point. For all CD measurements, protein concentration was 20 μM for hP0-cyt, and 10 μM for myelin basic protein (MBP). Five to seven scans for each sample were averaged, smoothed using the inverse exponential method, and plotted using SigmaPlot 2001 (Systat Software, Richmond, CA). All CD spectra were corrected for buffer, detergents, or lipid vesicle contributions. Ellipticity is reported as mean molar residue ellipticity (MRE) in degree-cm2-dmol−1. The fractional contents of secondary structures, namely α, β, and random coil, were calculated using the software CDPro (15). The wavelength used for the analysis was 195–260 nm, and 29 soluble proteins constituted the reference set. The per residue molar absorption units of circular dichroism Δɛ, which is measured in mdeg-M−1-cm−1, is related to MRE by Δɛ = MRE/3298. The calculated fractions of each secondary structure element from all three programs included in CDPro (CONTIN, SELCON3, and CDSSTR) were first averaged, then the averaged fractions of H(r) (regular α-helix) and H(d) (distorted α-helix), S(r) (regular β-strand) and S(d) (distorted β-strand), and T (turns) and U (unordered) were used to give the overall content of α, β, and random structure (γ), respectively.
Intrinsic fluorescence spectroscopy
Fluorescence emission spectra of hP0-cyt Trp residue were obtained using a HORIBA Jobin Yvon Spex Fluorolog II spectrofluorimeter (Edison, NJ) and 1 cm path-length quartz cuvette (Starna Cells, Atascadero, CA,) by exciting samples at 295 nm. Excitation and emission slits were set at 5 mm and data were collected at 0.5 nm increments from 310 nm to 410 nm at 25°C. The control spectra without protein were collected as baselines and were subtracted from the corresponding spectra of samples having protein concentrations of 5 μM. The following equation was used to correct the spectra from SUV samples for the inner-filter effect resulting from the strong absorbance of lipid vesicles at both emission and excitation wavelengths (16):
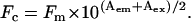 |
Here, Fc is the corrected fluorescence intensity, Fm is the measured fluorescence intensity after background subtraction, and Aem and Aex are the absorbances at the emission and excitation wavelengths, respectively (17,18).
RESULTS
hP0-cyt conformation depended on detergent or lipid/protein mole ratio
hP0-cyt is a highly basic domain (pI = 10.88), containing 21 positive-charged residues (from 9 arginines and 12 lysines) distributed across its entire sequence (Fig. 1 A). Secondary structure predictions (19–21) indicated significant amounts of α-helix (Fig. 1 B). By contrast, our initial far UV-CD spectrum suggested <10% α and >35% β secondary structures when hP0-cyt was in an aqueous buffer (Table 1). When the protein was mixed with different membrane-mimicking detergents (DPC, SDS, and TFE), an increase in ellipticity at both 222 nm and 208 nm in the CD spectra, which is typical of an α-helical structure (22), was observed in a detergent-concentration-dependent manner (Fig. 2, A–C). In 2 mM DPC, which is equal to 100:1 mol ratio of DPC:hP0-cyt, α-helical structure increased to ∼40% and β-structure decreased to <20% (Fig. 3 A), while at the same 100:1 mol ratio of SDS/protein, hP0-cyt underwent a greater conformational change, with close to 60% α and <5% β-structure (Fig. 3 B). At all concentrations tested, TFE also effectively induced protein folding to an α-helical structure and decreased the amount of β-structure (Fig. 3 C). The observed β→α transition of hP0-cyt structure induced by these detergents encouraged us to explore the structural features of hP0-cyt in an environment more closely related to its native conditions—i.e., the myelin membrane.
FIGURE 1.

Sequence of hP0-cyt (A) and its secondary structure (B) predicted according to Chou-Fasman, JPred, and PROF (19–21). ED, P0 extracellular domain; TM, P0 transmembrane domain; Cyt, P0 cytoplasmic domain. hP0-cyt contains 69 residues (151–219) and is highly basic, with a net charge of +15, pl = 10.88. H, α-helix; e, β-strand; t, turns.
TABLE 1.
Calculated secondary structure and α-β equilibrium constants for hP0-cyt (in PBS) at different protein concentrations
| hP0-cyt (μM) in PBS | 20 μM | 50 μM | 100 μM | 250 μM |
|---|---|---|---|---|
| α | 0.06 | 0.10 | 0.21 | 0.21 |
| β | 0.36 | 0.38 | 0.25 | 0.22 |
| γ | 0.58 | 0.52 | 0.54 | 0.57 |
| Kαβ | 6.00 | 3.80 | 1.19 | 1.05 |
| s (mM−1)* | 10 | 6 | 11 | 5 |
The amount of secondary structure was calculated using CDPro.
The value s was calculated according to 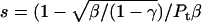 .
.
FIGURE 2.

CD spectra of hP0-cyt at 20 μM in different membrane environments. CD spectra of hP0-cyt in simple membranes made of membrane-mimicking compounds DPC (A), SDS (B), and TFE (C), or in lipid SUVs made of PC/PS (mole ratio 12:1) (D), or PC/PS/cholesterol (mole ratio 62:5:33) (E) are recorded at different compound concentrations or lipid/protein mole ratios. SUV samples of different lipid/protein mole ratios 0, 4:1, 20:1, 50:1, 100:1, and 200:1, are designated as PBS, SUV4, SUV20, SUV50, SUV100, and SUV200, respectively.
FIGURE 3.

Plots of calculated secondary structure of hP0-cyt in different membranes. Secondary structure fractions calculated from CDPro program were plotted as a function of concentration of membrane-mimicking compound or lipid/protein mole ratio. Symbols: ○, α; □, β; ▵, random coil.
As total lipid extracts of myelin contain lipids not present in the cytoplasmic leaflet, we used lipids that would be more akin to the native environment of P0-cyt. We produced lipid SUVs using mixtures of PC/PS, or PC/PS/cholesterol, all of which are thought to be important constituents in the cytoplasmic surface of peripheral nervous system myelin (23). Interestingly, similar to that from protein in aqueous buffer, CD spectra of hP0-cyt in PC/PS vesicles suggested a predominant β-structure at lipid/protein ratios <50:1 (the level of the mature peripheral myelin), but underwent a concentration-dependent β→α transition at lipid/protein ratios >50:1 (Fig. 2, D and E). Noticeably, when cholesterol was added to PC/PS, the transition from β→α structure of hP0-cyt occurred at lower lipid/protein mole ratio and in a more dramatic way than observed in the absence of cholesterol. Specifically, in vesicles made of PC/PS/cholesterol, hP0-cyt showed >20% α and <30% β-structures at 20:1 lipid/protein mole ratio, while at the same lipid/protein ratio, <10% α and >30% β-structures were maintained in PC/PS vesicles lacking cholesterol. Interestingly, when the lipid/hP0-cyt molar ratio is >20:1, both α and β contents of the protein start to decrease in vesicles containing cholesterol, which was not observed in the absence of cholesterol (Fig. 3, D and E). This difference in structural changes might indicate certain functions of cholesterol in hP0-cyt structure in the myelin membrane. Coupled with the conformational change in these different membrane-like environments is a rather limited change in random structure of hP0-cyt (Fig. 3). This indicates that the observed conformational change might be happening primarily between β- and α-conformations rather than involving unstructured regions.
Negative-charged vesicle lipids had little effect on the secondary structures of hP0-cyt
As hP0-cyt is highly basic, vesicles composed of only negative-charged PS were used to investigate whether the vesicle-induced structural change in hP0-cyt might result from electrostatic interactions between the protein and phospholipid headgroups (Fig. 4 A). CD spectra for PS/hP0-cyt mole ratio from 4 to 100:1 revealed a nearly constant protein secondary structure with <10% α, 30–40% β, and ∼60% random coil (Fig. 3 F). Similarly, vesicles made of only PI, another negative-charged lipid on the cytoplasmic surface of peripheral nervous system myelin, had very little effect on the secondary structure of hP0-cyt (Fig. 4 B). These results strongly argue that the negative charge of vesicle lipids might not be the driving force of the observed β→α structural transition at lipid/protein ratios above that in mature peripheral myelin.
FIGURE 4.

CD spectra of hP0-cyt in SUVs made of only negative-charged lipid PS (A) or PI (B), and the 18.5 kDa bovine MBP isoform in lipid SUVs made of PC/PS (mole ratio 12:1) (C), or PC/PS/cholesterol (mole ratio 62:5:33) (D).
N-terminal tryptophan microdomain became more folded in membrane-like environments
To investigate whether these membrane mimetics change the tertiary structure of the N-terminal tryptophan micro-domain, we used intrinsic fluorescence spectroscopy. For all membrane conditions tested, we observed a consistent lipid or detergent-concentration-dependent blue shift in the fluorescence emission maximum and an increase in fluorescence intensity compared with the spectrum of hP0-cyt in aqueous bulk phase (Fig. 5). This finding suggests that in membrane environments the N-terminal tryptophan of hP0-cyt is oriented away from the polar, aqueous phase toward a more hydrophobic region—presumably the nonpolar regions of the membrane. This conformational change might have important implications in the folding behavior of hP0-cyt in a native environment.
FIGURE 5.

Fluorescence emission spectra of hP0-cyt at 5 μM in different membrane environments. Trp-specific fluorescence emission spectra of hP0-cyt in the same experimental conditions as shown in Fig. 2 were recorded and lipid/protein mole ratios of SUV samples were designated in the same way. Fluorescence intensity (IF) is presented in arbitrary units (A.U.).
bMBP secondary structure did not change in lipid vesicles
To test whether the observed β→α structural transition of hP0-cyt is a common characteristic of basic proteins in membrane environments, similar CD experiments were conducted using the 18.5 kDa isoform of bMBP, another highly positive-charged myelin protein (calculated pI = 11.4; (23)) that is believed to be important in the compaction of myelin's cytoplasmic apposition. Our data showed that in vesicles composed of either PC/PS or PC/PS/cholesterol, bMBP did not show a similar lipid/protein ratio-dependent β→α conformational change as observed for hP0-cyt, but maintained a similar distribution of secondary structures as bMBP did in aqueous buffer (Fig. 4, C and D; Table 2). Like hP0-cyt, bMBP also showed little change in secondary structures in the presence of only negative-charged lipids, PS or PI (Table 2). These results collectively suggest that the structural characteristics of hP0-cyt in a membrane environment might be a novel feature, and not due simply to a charge effect.
TABLE 2.
Calculated secondary structure of 18.5 kDa bovine MBP in SUVs at different lipid/protein mole ratios
| Calculated structure | PC/PS
|
PC/PS/cholesterol
|
PS
|
PI
|
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PBS | 4 | 20 | 50 | 100 | 4 | 20 | 50 | 100 | 4 | 20 | 50 | 100 | 4 | 20 | 50 | 100 | |
| α | 0.02 | 0.06 | 0.03 | 0.02 | 0.00 | 0.04 | 0.03 | 0.03 | 0.00 | 0.04 | 0.02 | 0.03 | 0.03 | 0.02 | 0.04 | 0.05 | 0.05 |
| β | 0.41 | 0.38 | 0.37 | 0.40 | 0.45 | 0.49 | 0.49 | 0.48 | 0.54 | 0.54 | 0.54 | 0.62 | 0.39 | 0.51 | 0.49 | 0.40 | 0.50 |
| Random | 0.57 | 0.56 | 0.60 | 0.58 | 0.55 | 0.47 | 0.48 | 0.49 | 0.46 | 0.42 | 0.44 | 0.35 | 0.58 | 0.47 | 0.47 | 0.55 | 0.45 |
Lipid/protein mole ratios are 4:1, 20:1, 50:1, and 100:1. The fractional amount of secondary structure was calculated using CDPro.
DISCUSSION
The notion that the highly basic cytoplasmic domain of myelin P0 glycoprotein interacts with acidic lipids at the cytoplasmic apposition in myelin (24) is questioned by our previous x-ray diffraction studies that reveal relative invariance of the intermembrane space at this apposition over a wide range of pH and ionic strength (9,11). Thus, electrostatic interactions cannot solely account for the cytoplasmic apposition in peripheral nervous system myelin. In the current study, which used a membrane-mimetic system, we demonstrated that negative-charged lipids (PS or PI) did not induce significant change in P0-cyt secondary structure, whereas in PC/PS vesicles at lipid/protein ratios at or lower than that of mature peripheral myelin, P0-cyt primarily adopted a β-sheet structure. Such mole ratios resemble that in compact myelin, where the relative P0 content is higher than that during earlier stages of myelin synthesis. Therefore, we suggest that P0-cyt likely adopts a similar β-structure in compact myelin where strong intersheet H-bonding could largely account for the stability of the narrow cytoplasmic apposition.
Structural polymorphism of hP0-cyt dependent on lipid/protein mole ratio
Our results show that in PC/PS vesicles hP0-cyt undergoes a β→α structural transition at lipid/protein mole ratios higher than that of mature peripheral myelin. Curiously, a similar conformational change is observed for amyloidogenic protein Aβ40 with phosphoglycerol, Aβ40 with PC + phosphoglycerol (25), and Aβ40 with ganglioside-containing membranes (26).
The secondary structure contents were calculated as a function of the lipid/protein mole ratio Lt/Pt. The α-β equilibrium Kαβ (see Appendix) was calculated from the secondary structure contents as determined using the CDPro program for hP0-cyt in the absence of lipids. From our observed secondary structure contents of 20 μM hP0-cyt in aqueous buffer, α = 0.06, β = 0.36, and γ = 0.58. Kαβ was thus estimated to be 6.0 (Table 1). The equilibrium constants for different concentrations of hP0-cyt in PBS are given in Table 1.
Concentration-dependent β→α transition in hP0-cyt structure
In our CD measurements, the hP0-cyt structure showed a β→α transition and a nearly constant random structure as a function of the protein concentration in aqueous buffer (Fig. 6 and Table 1). The dramatic increases in α structure of hP0-cyt in DPC, SDS, and TFE indicate that the protein is incorporated into DPC and SDS micelles, or in the lower dielectric environment of TFE where the α-helix is stabilized (27). Therefore, at higher concentration hP0-cyt may interact with neighboring protein molecules in aqueous buffer in a similar way as in detergents (see Appendix).
FIGURE 6.

CD spectra of hP0-cyt at different concentrations in PBS.
Although we did not observe a transition from random coil to β as a function of hP0-cyt concentration from 20–250 μM, such a transition may occur at an even lower concentration as has been observed for Aβ25–35 at pH 4.0 (28). Therefore, depending on its concentration, hP0-cyt may form three different conformations—i.e., random coil → β at 0–20 μM, β→α at >20 μM, and constant random coil content at >20 μM.
Similarity in secondary structures between amyloidogenic peptides and hP0-cyt in lipids
Studies on the interactions between membranes and amyloidogenic proteins—including Alzheimer's β-amyloid protein (25,26,28–35), amylin (36), and calcitonin (37)—have sought to explain the role of biological membranes in mechanisms of amyloid assembly. The interactions between small antimicrobial peptides with membranes have also been studied to investigate therapeutic mechanisms (38). Whereas the immunoglobulin fold (common to certain adhesion proteins) and domain-swapping (which could also underlie adhesion) have been characterized using crystallography (10), the interactions of simple lipid systems with peptide fragments of myelin proteins have not been explored.
For fragments of membrane protein, the peptides can show changes in either conformation or assembly, depending on the lipid/protein mole ratio, the specific lipid species, and the solvent system. Antimicrobial peptides at low concentration adopt either an α-helical or β-strand conformation with helix- or strand-axis parallel to the membrane surface, but the axes become more normal to the surface and transmembrane at higher protein concentration (38). In the presence of lipids, the amyloidogenic peptides Aβ25–35 and Aβ40 show a random coil →β transition and a moderate increase in α-helix as a function of protein concentration (probably due to electrostatic effects). The random coil→β transition occurs at lower lipid/protein ratios, whereas the β→α transition occurs at higher ratios (25,28,30,33,36). As GM1 ganglioside specifically enhances the random coil→β transition, it might be involved in pathological Aβ aggregation (26,29,35). Aβ as an α-helix is observed at higher lipid/protein ratios, and can be either normal to (39) or parallel to (25,26) the membrane surface. The former is consistent with ion channel formation (40). In addition, the amyloidogenic prion protein PrP106-126 exhibits structural polymorphism among random coil, β-sheet, and α-helix depending on the presence of detergents and lipid vesicles (13).
We showed that ∼20 μM hP0-cyt in aqueous buffer gave a significant amount of random coil and β-conformation and relatively little α-content, while hP0-cyt at <20 μM might contain more random coil, like the case for amyloidogenic peptides. With an excess of negative-charged lipid (PS), significant amounts of β and little α were found, suggesting that electrostatic effects may stabilize the β-sheet assembly as it does for Aβ amyloid (25). With vesicles made of PC/PS or PC/PS/cholesterol at lipid/protein mole ratios of 50:1 or higher, however, a decrease in β-conformation for hP0-cyt was observed. As the calculated mole ratio for the cytoplasmic surface of intact peripheral nervous system myelin is 54:1 (23), hP0-cyt might be considered to be at a setpoint for conformational transition near this ratio. Moreover, the increase in folding of the N-terminal, tryptophan-containing microdomain in our model membranes indicates that some residues (including the tryptophan) of hP0-cyt may be embedded in the hydrocarbon chain region of the membranes.
hP0-cyt and amyloidogenic proteins appear to have similar β-conformation secondary structures at lipid/protein ratios <∼50:1 (25). Thus, H-bonding between β-chains of hP0-cyt may contribute to the stability of the cytoplasmic apposition between myelin membranes across a wide range of pH and ionic strength.
Implication of structural polymorphism of hP0-cyt in myelin development
The lipid/protein ratio-dependent conformational change in hP0-cyt might indicate a dynamic change in structure during critical developmental stages in myelin formation. In early stages of myelin synthesis when only small amounts of P0 are incorporated into the membranes, the lipid/protein ratio is high and P0-cyt might be primarily α-helical. In the immature myelin environment, an α-helical structure may allow for more flexibility within the cytoplasmic apposition, facilitating the transport of crucial materials within this narrowing space. An α-helical structure for P0-cyt might also promote signal transduction, which is consistent with the study on the phosphorylation of the serine residue in the “RSTK” PKC recognition site in this domain (Fig. 1) (41). At this stage, the N-terminal Trp residue of the cytoplasmic domain together with the acyl chain that is attached to the adjacent Cys residue (42) may serve as anchors that stabilize the helical structure, as shown in recent fluorescence spectroscopy (43,44) and simulation studies (45,46). At later stages of myelination, when more P0 molcules insert into the membrane and there is less relative lipid, P0-cyt would undergo the α→β transition thus facilitating compaction at the cytoplasmic apposition via intersheet H-bonding.
The water content in the cytoplasmic apposition of myelin is ∼30–40% (23), which is considerably less than the excess water in solutions containing unilamellar vesicles. Owing to this difference and to other factors in the myelin microenvironment, the protein conformation may change as the space between apposed membranes narrows during myelination, similar to what we propose here for hP0-cyt. The CD and fluorescence methods in this study necessitated the use of adequate protein and lipid concentrations for measurement but in optically transparent solutions; therefore, the conformation of hP0-cyt at lower water content could not be determined using solution spectroscopy. By changing the lipid/protein ratio, however, we attempted to use a simple paradigm to mimic the dynamic change in the corresponding constituents during myelin development. In early myelination stages, when the cytoplasm of Schwann cells has not yet been expelled from the myelin sheaths, a significant amount of water should remain between cytoplasmic faces in an array of myelin membranes. Therefore, samples having high lipid/protein ratios may, in fact, closely simulate the native conditions.
We propose that hP0-cyt adopts a β-conformation in compact PNS myelin, and that intersheet H-bonding of the apposed hP0-cyt molecules stabilizes the cytoplasmic apposition. This model can account for both the observed narrow cytoplasmic apposition and also the invariance of this apposition at different values of pH and ionic strength. The model also explains the lack of conformational change in hP0-cyt when it interacts with acidic lipids, as observed in our current study.
Implication of cholesterol-induced conformational change in hP0-cyt
The very high level of cholesterol in myelin (47) is thought to limit ion leakage (48) and thus enhance the high resistance and low capacitance of the myelin sheath in its insulative role. Further, cholesterol forms lipid rafts with glycosphingolipids and certain membrane proteins (49), and in peripheral nervous system myelin, it associates with substantial amounts of peripheral myelin protein 22 (PMP22) and P0 in the rafts (50). In our study, we observed an unexpected structural change in hP0-cyt when the protein was mixed with vesicles containing cholesterol—i.e., at lipid/protein mole ratio <20:1, the α-helix secondary structure increased and β and random coil decreased, whereas at higher mole ratios, both α- and β-structures decreased and the random coil content increased (Fig. 3 E). The threshold lipid/protein mole ratio of ∼20 corresponds to cholesterol/protein mole ratio of 6.6 in the vesicle. Therefore, it is likely that cholesterol interacts with hP0-cyt in a concentration-dependent manner. At ratios <6.6, cholesterol favors the folding of hP0-cyt into an α-helical structure, presumably by specifically stabilizing this conformation. The α-helix-inducing property of cholesterol is suggested by our CD data, which show a more rapid β→α conformational change in hP0-cyt compared with the protein in vesicles lacking cholesterol but having the same lipid/protein ratio (Fig. 3 D). This result is also consistent with previous reports suggesting that the addition of 33% cholesterol to artificial membranes induces a β→α transition of Aβ40 (51,52). Intriguingly, in our current report, at mole ratios >6.6, cholesterol seems to unfold the protein into a random coil. Further experiments using lipid vesicles containing varying amounts of cholesterol may help to shed more light on this unique phenomenon.
β-conformation of MBP at cytoplasmic apposition
Unlike hP0-cyt, MBP did not show a β→α conformational change as a function of lipid/protein ratio. Rather, at ratios at least up to 100:1, 50% β and 8% α-helix were always observed. Since the lipid/MBP mole ratio at the cytoplasmic side of central nervous system myelin is estimated to be ∼48 (23), the consistently high β-content of MBP at different lipid/MBP ratios suggests that the β-sheet conformation might be required for this protein to be involved in the membrane apposition. Studies on the MBP-deficient “shiverer” mouse demonstrate that MBP is needed for myelin compaction in the central nervous system (53) but not in the peripheral nervous system (5). This difference may be explained by the presence in peripheral nervous system myelin of P0 whose basic cytoplasmic domain plays a compensatory structural role for the missing MBP. In reconstituted membranes consisting of myelin lipids and basic proteins, x-ray diffraction analysis has localized the proteins in the regions that contain aqueous medium and lipid polar headgroups (54–56). Small-angle x-ray scattering of MBP in solution also indicates that its molecular shape is mainly random coil but with lipids, it converts to a C-shaped structure rich in β-sheet (57–59). A similarly-shaped structure is also shown by electron microscopy (60,61). The preponderance of β-conformation is consistent with earlier molecular modeling (62,63), spectroscopy (64), and with x-ray diffraction from solubilized and dried MBP, which shows typical β-sheet reflections (65). Our current CD findings also support the notion for an adhesive role of β-sheet folded MBP at the cytoplasmic apposition. Unlike hP0-cyt whose conformation depended on the type and relative amount of lipids present, bMBP always showed β-sheet structure independent of lipid concentration (Fig. 4, C and D; Table 2). Molecular modeling indicates that the interstrand H-bonding direction of MBP molecules is likely to be normal to the membrane surfaces (Fig. 7, right).
FIGURE 7.

Molecular models for hP0-cyt (left) and MBP (right) from the atomic coordinates deposited in PDB data bank (Entry No. 1qcl). In hP0-cyt a short hydrophobic sequence V39LYAM43 is indicated by the stippling. The other β-chain is formed by residues G27KDA30. In both hP0-cyt and MBP molecules, the intersheet H-bonding direction is likely to be normal to the membrane surfaces, i.e., parallel to the membrane-stacking direction. The ribbon representations were generated using PyMOL (74).
Comparison with previous studies on MBP conformation
Previous CD studies of MBP reported random conformation for the protein in aqueous solution, but α- and β-structures with lipids present (66). Convex constraint analysis has been used to calculate the secondary structure content of MBP in aqueous media with or without lipids (66,67). Results from three samples were reported: lipid-free MBP (LF-MBP); LF-MBP with 0.5% CHAPS; and lipid-bound MBP (LB-MBP) with 0.5% CHAPS in 10 mM HEPES buffer at pH 8. LF-MBP and LF-MBP with CHAPS showed 5–9% α, 6–7% β, ∼50% random coil, and ∼30–40% β-turn, whereas LB-MBP with CHAPS showed ∼20% α, ∼20% antiparallel β-sheet, ∼30% random coil, and ∼30% β-turn. From analysis of TLC plates, the MBP-bound lipids contained 26% cholesterol, 6.5% cerebrosides, 8% sulfatides, 32.6% phosphatidylethanolamine, 5.2% PI, 4.3% PS, 15.6% PC, and 3.5% sphingomyelin (SM). The minimum lipid content of MBP after dialysis was 1.5 mg phospholipid/mg protein. Assuming the reported percentage refers to weight content, then the molar contents may be calculated from the molecular masses of the respective lipids. As 1 mg of MBP corresponds to 0.054 μmol, then the molar ratio of total lipid to 18.5 kDa MBP is 2.03:0.054 = 38. This is smaller than the estimated ratio of 48 for lipid/MBP on the cytoplasmic side of CNS myelin (23). The measured molar contents of MBP-bound PS and cholesterol were less than those estimated from the chemical composition data (23). The two different measurements, however, show similar contents of the other lipids.
The CD spectra for native and recombinant 18.5 kDa MBP with or without GM1 and 2-propanol have been determined (66). Native and recombinant MBP in aqueous solution gave ∼10% α and 16–18% β, which were greater than those calculated for LF-MBP (67). GM1 increased the α- and β-contents slightly to ∼13% and ∼17–20%, respectively, while 2-propanol increased them more to ∼28–34%, and ∼21–24%, which are comparable to those of LB-MBP calculated (67).
The secondary structure content of MBP calculated using the convex constraint analysis program (see above) are different from the ones we measured for MBP in PBS solution using the CDPro program, i.e., 2% α, 41% β, and 57% random coil (Table 2). In the previous experiments, MBP was extracted from bovine brain white matter and purified in the lipid-free, denatured form or endogenous lipid-bound form (67); by contrast, we used commercial dephosphorylated bovine 18.5 kDa MBP. The difference in sample preparations may greatly influence the solution conformation of MBP. Alternatively, as PC/PS, PC/PS/cholesterol, and negative-charged PS and PI did not increase the α-content in our study, it may be that other lipids (phosphatidylethanolamine or small amounts of glycolipids) in the tightly MBP-bound form might stabilize the α-conformation of MBP just as 2-propanol increased the α-contents for MBP (66). For Aβ amyloid protein (51) and hP0-cyt (this study), cholesterol appears to increase the α-helical content. As this was not observed for MBP with PC/PS/cholesterol, the interaction between MBP and cholesterol is different from that involving Aβ or P0-cyt.
Comparison of the different reference sets in CDPro program
In the software program CDPro there are different reference sets available, including 29 soluble proteins (SP29), 43 soluble plus five denatured proteins (SDP48), and 43 soluble plus 13 membrane proteins (SMP56) (15,68–70). The original set (SP29; measured wavelength range 178–260 nm) was the only reference set that covered the entire experimental wavelength range of our CD spectra (195–260 nm) and was, therefore, used here for data analysis.
Owing to their large content of random coil, both hP0-cyt and MBP can be regarded as intrinsically disordered proteins. In addition, both hP0-cyt and MBP are membrane-bound proteins. Secondary structures of these types of proteins, therefore, might be better predicted by using reference sets SDP48 or SMP56. We compared the performances of these reference sets in terms of the root mean-square deviation (RMSD) values between the observed and calculated CD spectra for hP0-cyt in PBS, hP0-cyt with PC/PS at 200 lipid/protein ratio, and MBP in PBS (see Table in the Supplementary Material). All three methods in CDPro, CONTINLL, SELCON3, and CDSSTR generated results when SP29 was used as a reference set. For SPD48, SELCON3 did not yield results for hP0-cyt in PBS or for MBP in PBS; and for the SMP56 set, CDSSTR did not give results for MBP. Comparing the RMSD values among the three methods, SELCON3 gave the smallest RMSD values for all three samples using the SP29 set; CDSSTR gave the smallest values for hP0-cyt in PBS and MBP in PBS using the SDP48 set; and CONTINLL gave the smallest value for hP0-cyt in PC/PS vesicles using the SMP56 set. As the SP29 set gave the best overall performance among the three reference sets, then we suggest that, in our current studies, this reference set gave reasonably accurate measurements for the secondary structures of hP0-cyt and MBP.
Possible α- and β-chain domains for hP0-cyt
Secondary structure predictions indicated that most of the residues in the cytoplasmic domain of P0 are in an α-helix and that residues 37–39 (ThrProVal) are in a β-conformation (19–21). This predicted β-sequence is part of a single long stretch of hydrophobic residues (sequence 37–44; TPVLYAML) that is bounded at its N-terminus by Gln-36 and at its C-terminus by Asp-45 (Fig. 1 B). Structure-based homology searching with 3D-PSSM (71) predicted that hP0-cyt might have a compact structure similar to that for bacteriophage-λ gpw (PDB No. 1HYW) in solution (Fig. 7, left). In this study we predicted that the hydrophobic sequence 39–44 (VLYAM) has an extended β-chain, which is conserved for all species except trout and zebrafish. It is likely that this hydrophobic short sequence may nucleate the β-sheet folding for the cytoplasmic domain of P0 when in the appropriate membrane environment. The observed conformational flexibility that we have identified for hP0-cyt may have a crucial role in regulating myelin packing.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Acknowledgments
The authors are grateful to the Chemistry Department of Boston College for providing the circular dichroism spectropolarimeter and fluorescence spectrofluorimeter, and for assistance with the experiments.
M.S. was supported by the European Union within the frame of Neuroprion and Heteroprion networks (contract No. FOOD-CT-2004-506579), Negri-Weizmann Foundation (2006), the Italian Ministry of University and Research (FIRB protocol No. RBNE03PX83, 2005), and Fondazione Cariplo (Project Genoproteomics of Age Related Disorders, 2006). The Kirschner Lab acknowledges institutional support from Boston College.
APPENDIX
Quantitation of protein conformational change that is dependent on lipid/protein ratio
Protein and lipid binding isotherms likely depend on the concentrations of protein and lipids. Since there are three conformers, A (α), B (β), and R (random, or γ), and the conformational change is between α and β at given protein and lipid concentrations, then the isotherm may be written as
 |
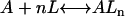 |
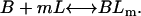 |
Here, A and B are the free form of protein conformations α and β, L is lipid, and ALn and BLm are the lipid-bound forms of the α- and β-conformations, respectively. We assumed that there is no reaction between the lipid-bound forms of proteins having different conformations. The total protein concentration is Pt = [A] + [B] + [ALn] + [BLm] + [R]; and the lipid total concentration is Lt = L + n[ALn] + m[BLm]. The α-content is ([A] + [ALn])/Pt, the β-content is ([B] + [BLm])/Pt, and the random content γ is 1−(α+β). The mole ratio between lipid and protein is φ = Lt/Pt. The α-β equilibrium reaction constants are defined as Kαβ, kαl, and kβl; therefore,
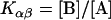 |
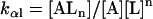 |
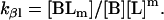 |
Then
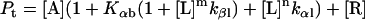 |
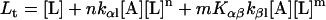 |
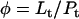 |
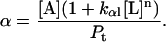 |
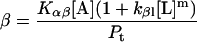 |
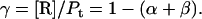 |
[A] and [L] can be derived numerically from the first two equations by using the known Pt, Lt, and reaction constants. The amounts of secondary structure (α, β, and γ) can be calculated from the last three equations. When lipids are not mixed with protein, Lt = L = 0; and the equations simplify to
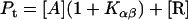 |
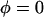 |
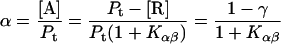 |
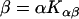 |
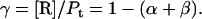 |
Because the α-conformer increased at values above the physiological lipid/protein mole ratio (in our experiments), we assumed that the binding constant kαl >> kβl, kβl = 0, and lipid binding stoichiometry n = 1; therefore,
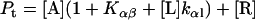 |
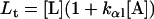 |
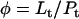 |
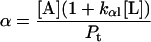 |
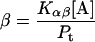 |
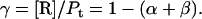 |
[A] and [L] were solved analytically from the first two of these equations.
The constant kαl was determined by systematically changing its value to fit the observed α-content at different concentrations of PC/PS or PC/PS/cholesterol vesicle lipids. The best kαl values chosen were 2.4 mM−1 for PC/PS and 7.6 mM−1 for PC/PS/cholesterol, respectively (Fig. A1). The free energy (ΔG) of lipid binding to an α-helical conformer in vesicles made of PC/PS or PC/PS/cholesterol was calculated to be ∼5 kcal/mol. Note in PC/PS/cholesterol, the calculated curve did not fit the entire range of mole ratios, as the α-content increased and then decreased as a function of lipid/protein mole ratio (Fig. A1, right).
FIGURE A1.

(Left) Observed secondary structure contents (α (○) and β (□)) derived from CDPro and the calculated ones (α (solid line) and β (dashed line)), as a function of the PCPS/hP0-cyt mole ratio. The input parameters for the calculated curves included Kαβ = 6.0, kαl = 2.4 mM−1, n = 1, kβl = 0, and random structure content γ = 0.58. The k value was an average of the best-fit observed α-contents at 100:1 and 200:1 lipid/protein mole ratios. (Right) Secondary structure as a function of PCPS-cholesterol/hP0-cyt mole ratio. The input kαl was 7.6 mM−1 and other parameters were the same as those in PC/PS. The k value was an average of those best-fit observed α-contents at lipid/protein mole ratios of 20:1 and 50:1.
Quantitation of protein aggregation and protein conformational change based on protein concentration
To quantify the concentration-dependent β→α transition of hPO-cyt in aqueous buffer, we used a formulation similar to that for protein aggregation (33,72,73), in which the α-conformer is treated as an aggregate. The total concentration of the protein Pt is
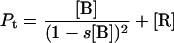 |
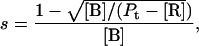 |
where [B] is the concentration of the β-form, s is the binding constant for the addition of the β-form to the self-aggregate, and [R] is the concentration of the random form. The α-content is
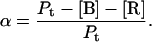 |
We assumed that the content of random coil was constant for different total protein concentrations. From these equations, the contents of the secondary structures were calculated as a function of protein concentration. For example, the binding constant s for the respective concentration was estimated (Table 1), and the average value was found to be 8 mM−1, which corresponds to a free energy ΔG = −5 kcal/mol for addition of β-conformer. The calculated curve was derived from the average values of s = 8 mM−1, and the random structure content 0.55 (Fig. A2).
FIGURE A2.

Measured secondary structure content (α (○) and β (□)) using the CDPro program, and the calculated ones (α, solid line and β, dashed line), as a function of hP0-cyt concentration. The input parameter included the binding constant for monomer addition s = 8 mM−1, and random structure content γ = 0.55 (which was an average of the measured random contents at different hP0-cyt concentrations). At this given γ, s was searched to give the same α- and β-contents at different P0 concentrations. The value of 8 mM−1 was an average of these s values.
At <20 μM, the secondary structure content of hP0-cyt could be accounted for by the binding of monomer (in a random conformation) to aggregate (in a β-conformation). By comparison, the binding constant measured for Aβ25–35 in the noncooperative model is 18 mM−1 (28), which is similar to the one we measured here for the β→α transition of hP0-cyt.
References
- 1.Greenfield, S., S. Brostoff, E. H. Eylar, and P. Morell. 1973. Protein composition of myelin of the peripheral nervous system. J. Neurochem. 20:1207–1216. [DOI] [PubMed] [Google Scholar]
- 2.Lemke, G., E. Lamar, and J. Patterson. 1988. Isolation and analysis of the gene encoding peripheral myelin protein zero. Neuron. 1:73–83. [DOI] [PubMed] [Google Scholar]
- 3.Uyemura, K., M. Suzuki, Y. Sakamoto, and S. Tanaka. 1987. Structure of P0 protein: homology to immunoglobulin superfamily. Biomed. Res. 8:353–357. [Google Scholar]
- 4.Uyemura, K., Y. Takeda, H. Asou, and K. Hayasaka. 1994. Neural cell adhesion proteins and neurological diseases. J. Biochem. (Tokyo). 116:1187–1192. [DOI] [PubMed] [Google Scholar]
- 5.Kirschner, D. A., and A. L. Ganser. 1980. Compact myelin exists in the absence of basic protein in the shiverer mutant mouse. Nature. 283:207–210. [DOI] [PubMed] [Google Scholar]
- 6.Warner, L. E., M. J. Hilz, S. H. Appel, J. M. Killian, E. H. Kolodry, G. Karpati, S. Carpenter, G. V. Watters, C. Wheeler, D. Witt, A. Bodell, E. Nelis, C. Van Broeckhoven, and J. R. Lupski. 1996. Clinical phenotypes of different MPZ (P0) mutations may include Charcot-Marie-Tooth type 1B, Dejerine-Sottas, and congenital hypomyelination. Neuron. 17:451–460. [DOI] [PubMed] [Google Scholar]
- 7.Ninham, B. W., and V. A. Parsegian. 1971. Electrostatic potential between surfaces bearing ionizable groups in ionic equilibrium with physiologic saline solution. J. Theor. Biol. 31:405–428. [DOI] [PubMed] [Google Scholar]
- 8.Verwey, E. J. W., and J. T. G. Overbeek. 1948. Theory of the Stability of Lyophobic Colloids. Elsevier, New York. [DOI] [PubMed]
- 9.Inouye, H., J. Karthigasan, and D. A. Kirschner. 1989. Membrane structure in isolated and intact myelins. Biophys. J. 56:129–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shapiro, L., J. P. Doyle, P. Hensley, D. R. Colman, and W. A. Hendrickson. 1996. Crystal structure of the extracellular domain from P0, the major structural protein of peripheral nerve myelin. Neuron. 17:435–449. [DOI] [PubMed] [Google Scholar]
- 11.Inouye, H., and D. A. Kirschner. 1988. Membrane interactions in nerve myelin. I. Determination of surface charge from effects of pH and ionic strength on period. Biophys. J. 53:235–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Inouye, H., H. Tsuruta, J. Sedzik, K. Uyemura, and D. A. Kirschner. 1999. Tetrameric assembly of full-sequence protein zero myelin glycoprotein by synchrotron x-ray scattering. Biophys. J. 76:423–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Gioia, L., C. Selvaggini, E. Ghibaudi, L. Diomede, O. Bugiani, G. Forloni, F. Tagliavini, and M. Salmona. 1994. Conformational polymorphism of the amyloidogenic and neurotoxic peptide homologous to residues 106–126 of the prion protein. J. Biol. Chem. 269:7859–7862. [PubMed] [Google Scholar]
- 14.Kodama, M., T. Miyata, and Y. Takaichi. 1993. Calorimetric investigations of phase transitions of sonicated vesicles of dimyristoylphosphatidylcholine. Biochim. Biophys. Acta. 1169:90–97. [PubMed] [Google Scholar]
- 15.Sreerama, N., and R. W. Woody. 2004. On the analysis of membrane protein circular dichroism spectra. Protein Sci. 13:100–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lakowicz, J. R. 2006. Principles of Fluorescence Spectroscopy. Springer, New York.
- 17.Raghuraman, H., and A. Chattopadhyay. 2004. Interaction of melittin with membrane cholesterol: a fluorescence approach. Biophys. J. 87:2419–2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kruger, R. G., P. Dostal, and D. G. McCafferty. 2004. Development of a high-performance liquid chromatography assay and revision of kinetic parameters for the Staphylococcus aureus sortase transpeptidase SrtA. Anal. Biochem. 326:42–48. [DOI] [PubMed] [Google Scholar]
- 19.Chou, P. Y., and G. D. Fasman. 1974. Prediction of protein conformation. Biochemistry. 13:222–245. [DOI] [PubMed] [Google Scholar]
- 20.Cuff, J. A., M. E. Clamp, A. S. Siddiqui, M. Finlay, and G. J. Barton. 1998. JPred: a consensus secondary structure prediction server. Bioinformatics. 14:892–893. [DOI] [PubMed] [Google Scholar]
- 21.Rost, B., G. Yachdav, and J. Liu. 2004. The PredictProtein server. Nucleic Acids Res. 32:321–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Berova, N., K. Nakanishi, and R. Woody. 2000. Circular Dichroism: Principles and Applications. Wiley-VCH, New York.
- 23.Inouye, H., and D. A. Kirschner. 1988. Membrane interactions in nerve myelin. II. Determination of surface charge from biochemical data. Biophys. J. 53:247–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ding, Y., and K. R. Brunden. 1994. The cytoplasmic domain of myelin glycoprotein P0 interacts with negatively charged phospholipid bilayers. J. Biol. Chem. 269:10764–10770. [PubMed] [Google Scholar]
- 25.Terzi, E., G. Holzemann, and J. Seelig. 1997. Interaction of Alzheimer β-amyloid peptide (1–40) with lipid membranes. Biochemistry. 36:14845–14852. [DOI] [PubMed] [Google Scholar]
- 26.Kakio, A., S. Nishimoto, K. Yanagisawa, Y. Kozutsumi, and K. Matsuzaki. 2002. Interactions of amyloid beta-protein with various gangliosides in raft-like membranes: importance of GM1 ganglioside-bound form as an endogenous seed for Alzheimer amyloid. Biochemistry. 41:7385–7390. [DOI] [PubMed] [Google Scholar]
- 27.Roccatano, D., G. Colombo, M. Fioroni, and A. E. Mark. 2002. Mechanism by which 2,2,2-trifluoroethanol/water mixtures stabilize secondary-structure formation in peptides: a molecular dynamics study. Proc. Natl. Acad. Sci. USA. 99:12179–12184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Terzi, E., G. Holzemann, and J. Seelig. 1994. Reversible random coil-β-sheet transition of the Alzheimer beta-amyloid fragment (25–35). Biochemistry. 33:1345–1350. [DOI] [PubMed] [Google Scholar]
- 29.Choo-Smith, L. P., W. Garzon-Rodriguez, C. G. Glabe, and W. K. Surewicz. 1997. Acceleration of amyloid fibril formation by specific binding of Aβ-(1–40) peptide to ganglioside-containing membrane vesicles. J. Biol. Chem. 272:22987–22990. [DOI] [PubMed] [Google Scholar]
- 30.Kakio, A., S. I. Nishimoto, K. Yanagisawa, Y. Kozutsumi, and K. Matsuzaki. 2001. Cholesterol-dependent formation of GM1 ganglioside-bound amyloid beta-protein, an endogenous seed for Alzheimer amyloid. J. Biol. Chem. 276:24985–24990. [DOI] [PubMed] [Google Scholar]
- 31.Kurganov, B., M. Doh, and N. Arispe. 2004. Aggregation of liposomes induced by the toxic peptides Alzheimer's Aβs, human amylin and prion (106–126): facilitation by membrane-bound GM1 ganglioside. Peptides. 25:217–232. [DOI] [PubMed] [Google Scholar]
- 32.McLaurin, J., D. Yang, C. M. Yip, and P. E. Fraser. 2000. Review: modulating factors in amyloid-beta fibril formation. J. Struct. Biol. 130:259–270. [DOI] [PubMed] [Google Scholar]
- 33.Terzi, E., G. Holzemann, and J. Seelig. 1994. Alzheimer β-amyloid peptide 25–35: electrostatic interactions with phospholipid membranes. Biochemistry. 33:7434–7441. [DOI] [PubMed] [Google Scholar]
- 34.Terzi, E., G. Holzemann, and J. Seelig. 1995. Self-association of beta-amyloid peptide (1–40) in solution and binding to lipid membranes. J. Mol. Biol. 252:633–642. [DOI] [PubMed] [Google Scholar]
- 35.Yanagisawa, K., A. Odaka, N. Suzuki, and Y. Ihara. 1995. GM1 ganglioside-bound amyloid beta-protein (Aβ): a possible form of preamyloid in Alzheimer's disease. Nat. Med. 1:1062–1066. [DOI] [PubMed] [Google Scholar]
- 36.Jayasinghe, S. A., and R. Langen. 2005. Lipid membranes modulate the structure of islet amyloid polypeptide. Biochemistry. 44:12113–12119. [DOI] [PubMed] [Google Scholar]
- 37.Wang, S. S., T. A. Good, and D. L. Rymer. 2005. The influence of phospholipid membranes on bovine calcitonin secondary structure and amyloid formation. Protein Sci. 14:1419–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang, H. W. 2000. Action of antimicrobial peptides: two-state model. Biochemistry. 39:8347–8352. [DOI] [PubMed] [Google Scholar]
- 39.Mason, R. P., R. F. Jacob, M. F. Walter, P. E. Mason, N. A. Avdulov, S. V. Chochina, U. Igbavboa, and W. G. Wood. 1999. Distribution and fluidizing action of soluble and aggregated amyloid beta-peptide in rat synaptic plasma membranes. J. Biol. Chem. 274:18801–18807. [DOI] [PubMed] [Google Scholar]
- 40.Arispe, N., H. B. Pollard, and E. Rojas. 1993. Giant multilevel cation channels formed by Alzheimer disease amyloid beta-protein [Aβ P-(1–40)] in bilayer membranes. Proc. Natl. Acad. Sci. USA. 90:10573–10577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu, W., D. Manichella, H. Jiang, J. M. Vallat, J. Lilien, P. Baron, G. Scarlato, J. Kamholz, and M. E. Shy. 2000. Absence of P0 leads to the dysregulation of myelin gene expression and myelin morphogenesis. J. Neurosci. Res. 60:714–724. [DOI] [PubMed] [Google Scholar]
- 42.Bizzozero, O. A., K. Fridal, and A. Pastuszyn. 1994. Identification of the palmitoylation site in rat myelin P0 glycoprotein. J. Neurochem. 62:1163–1171. [DOI] [PubMed] [Google Scholar]
- 43.de Foresta, B., L. Tortech, M. Vincent, and J. Gallay. 2002. Location and dynamics of tryptophan in transmembrane α-helix peptides: a fluorescence and circular dichroism study. Eur. Biophys. J. 31:185–197. [DOI] [PubMed] [Google Scholar]
- 44.Ladokhin, A. S., and S. H. White. 2001. Alphas and taus of tryptophan fluorescence in membranes. Biophys. J. 81:1825–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jensen, M. O., O. G. Mouritsen, and G. H. Peters. 2004. Simulations of a membrane-anchored peptide: structure, dynamics, and influence on bilayer properties. Biophys. J. 86:3556–3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Johnston, J. M., G. A. Cook, J. M. Tomich, and M. S. Sansom. 2006. Conformation and environment of channel-forming peptides: a simulation study. Biophys. J. 90:1855–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morell, P., and H. Jurevics. 1996. Origin of cholesterol in myelin. Neurochem. Res. 21:463–470. [DOI] [PubMed] [Google Scholar]
- 48.Haines, T. H. 2001. Do sterols reduce proton and sodium leaks through lipid bilayers? Prog. Lipid Res. 40:299–324. [DOI] [PubMed] [Google Scholar]
- 49.Brown, D. A., and E. London. 2000. Structure and function of sphingolipid- and cholesterol-rich membrane rafts. J. Biol. Chem. 275:17221–17224. [DOI] [PubMed] [Google Scholar]
- 50.Erne, B., S. Sansano, M. Frank, and N. Schaeren-Wiemers. 2002. Rafts in adult peripheral nerve myelin contain major structural myelin proteins and myelin and lymphocyte protein (MAL) and CD59 as specific markers. J. Neurochem. 82:550–562. [DOI] [PubMed] [Google Scholar]
- 51.Ji, S. R., Y. Wu, and S. F. Sui. 2002. Cholesterol is an important factor affecting the membrane insertion of β-amyloid peptide (Aβ 1–40), which may potentially inhibit the fibril formation. J. Biol. Chem. 277:6273–6279. [DOI] [PubMed] [Google Scholar]
- 52.Micelli, S., D. Meleleo, V. Picciarelli, and E. Gallucci. 2004. Effect of sterols on β-amyloid peptide (AβP 1–40) channel formation and their properties in planar lipid membranes. Biophys. J. 86:2231–2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Privat, A., C. Jacque, J.-M. Bourre, P. Dupouey, and N. A. Baumann. 1979. Absence of the major dense line in myelin of the mutant mouse “shiverer”. Neurosci. Lett. 12:107–112. [DOI] [PubMed] [Google Scholar]
- 54.MacNaughtan, W., K. A. Snook, E. Caspi, and N. P. Franks. 1985. An x-ray diffraction analysis of oriented lipid multilayers containing basic proteins. Biochim. Biophys. Acta. 818:132–148. [DOI] [PubMed] [Google Scholar]
- 55.Mateu, L., V. Luzzati, Y. London, R. M. Gould, and F. G. Vosseberg. 1973. X-ray diffraction and electron microscope study of the interactions of myelin components. The structure of a lamellar phase with a 150 to 180 Å repeat distance containing basic proteins and acidic lipids. J. Mol. Biol. 75:697–709. [DOI] [PubMed] [Google Scholar]
- 56.Murthy, N. S., D. D. Wood, and M. A. Moscarello. 1984. X-ray scattering studies of a model complex of lipid and basic protein of myelin. Biochim. Biophys. Acta. 769:493–498. [DOI] [PubMed] [Google Scholar]
- 57.Epand, R. M., M. A. Moscarello, B. Zierenberg, and W. J. Vail. 1974. The folded conformation of the encephalitogenic protein of the human brain. Biochemistry. 13:1264–1267. [DOI] [PubMed] [Google Scholar]
- 58.Haas, H., C. L. Oliveira, I. L. Torriani, E. Polverini, A. Fasano, G. Carlone, P. Cavatorta, and P. Riccio. 2004. Small angle x-ray scattering from lipid-bound myelin basic protein in solution. Biophys. J. 86:455–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Krigbaum, W. R., and T. S. Hsu. 1975. Molecular conformation of bovine A1 basic protein, a coiling macromolecule in aqueous solution. Biochemistry. 14:2542–2546. [DOI] [PubMed] [Google Scholar]
- 60.Beniac, D. R., M. D. Luckevich, G. J. Czarnota, T. A. Tompkins, R. A. Ridsdale, F. P. Ottensmeyer, M. A. Moscarello, and G. Harauz. 1997. Three-dimensional structure of myelin basic protein. I. Reconstruction via angular reconstitution of randomly oriented single particles. J. Biol. Chem. 272:4261–4268. [DOI] [PubMed] [Google Scholar]
- 61.Ridsdale, R. A., D. R. Beniac, T. A. Tompkins, M. A. Moscarello, and G. Harauz. 1997. Three-dimensional structure of myelin basic protein. II. Molecular modeling and considerations of predicted structures in multiple sclerosis. J. Biol. Chem. 272:4269–4275. [DOI] [PubMed] [Google Scholar]
- 62.Martenson, R. E. 1986. Possible hydrophobic region in myelin basic protein consisting of an orthogonally packed beta-sheet. J. Neurochem. 46:1612–1622. [DOI] [PubMed] [Google Scholar]
- 63.Stoner, G. L. 1990. Conservation throughout vertebrate evolution of the predicted beta-strands in myelin basic protein. J. Neurochem. 55:1404–1411. [DOI] [PubMed] [Google Scholar]
- 64.Harauz, G., N. Ishiyama, C. M. Hill, I. R. Bates, D. S. Libich, and C. Fares. 2004. Myelin basic protein-diverse conformational states of an intrinsically unstructured protein and its roles in myelin assembly and multiple sclerosis. Micron. 35:503–542. [DOI] [PubMed] [Google Scholar]
- 65.Sedzik, J., and D. A. Kirschner. 1992. Is myelin basic protein crystallizable? Neurochem. Res. 17:157–166. [DOI] [PubMed] [Google Scholar]
- 66.Bates, I. R., P. Matharu, N. Ishiyama, D. Rochon, D. D. Wood, E. Polverini, M. A. Moscarello, N. J. Viner, and G. Harauz. 2000. Characterization of a recombinant murine 18.5-kDa myelin basic protein. Protein Expr. Purif. 20:285–299. [DOI] [PubMed] [Google Scholar]
- 67.Polverini, E., A. Fasano, F. Zito, P. Riccio, and P. Cavatorta. 1999. Conformation of bovine myelin basic protein purified with bound lipids. Eur. Biophys. J. 28:351–355. [DOI] [PubMed] [Google Scholar]
- 68.Sreerama, N., S. Y. Venyaminov, and R. W. Woody. 2000. Estimation of protein secondary structure from circular dichroism spectra: inclusion of denatured proteins with native proteins in the analysis. Anal. Biochem. 287:243–251. [DOI] [PubMed] [Google Scholar]
- 69.Sreerama, N., and R. W. Woody. 2000. Estimation of protein secondary structure from circular dichroism spectra: comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal. Biochem. 287:252–260. [DOI] [PubMed] [Google Scholar]
- 70.Sreerama, N., and R. W. Woody. 2004. Computation and analysis of protein circular dichroism spectra. Methods Enzymol. 383:318–351. [DOI] [PubMed] [Google Scholar]
- 71.Kelley, L. A., R. M. MacCallum, and M. J. Sternberg. 2000. Enhanced genome annotation using structural profiles in the program 3D-PSSM. J. Mol. Biol. 299:499–520. [DOI] [PubMed] [Google Scholar]
- 72.Oosawa, F., and S. Asakura. 1975. Thermodynamics of the Polymerization of Protein. Academic Press, New York.
- 73.Inouye, H., and D. A. Kirschner. 2000. A β-fibrillogenesis: kinetic parameters for fibril formation from Congo red binding. J. Struct. Biol. 130:123–129. [DOI] [PubMed] [Google Scholar]
- 74.DeLano, W. L. 2002. The PyMOL Molecular Graphics System. DeLano Scientific, San Carlos, CA.