

Abstract
Cardiotrophin-1 (CT-1) was identified as a growth factor for cardiac myocytes, and CT-1 protects myocytes from cell death. Adult CT-1−/− mice exhibit neural deficits including the loss of preganglionic sympathetic neurons, but their autonomic and cardiac parameters have not been examined. We used these mice to determine if the absence of CT-1 or loss of preganglionic sympathetic input altered heart rate, left ventricular pressure, cardiac contractility (dP/dt), or cell death following ischemia-reperfusion. Basal heart rate was increased in CT-1−/− mice, and this difference was abolished by ganglionic block. Left ventricular pressure and dP/dt were unchanged. Dobutamine stimulated similar increases in heart rate and dP/dt in both genotypes, but ventricular pressure was significantly lower in CT-1 nulls. Cardiac expression of interleukin-6 (IL-6) mRNA was increased significantly in CT-1 null mice, while leukemia inhibitory factor (LIF) mRNA was unchanged. Infarct size normalized to area at risk was no different in CT-1 −/− mice (33.8±1.0 % vs. 37.7±3.2 % WT) 24 hours after ischemia-reperfusion. Induction of IL-6 mRNA after infarct was significantly abrogated in CT-1 null mice compared to wild type mice, but LIF mRNA induction remained significant in CT-1 null mice and might contribute to cardiac protection in the absence of CT-1.
Keywords: gp130, cardiac protection, leukemia inhibitory factor, cardiotrophin-1, interleukin-6
1. Introduction
Cardiotrophin-1 (CT-1) is a cytokine originally identified for its ability to stimulate cardiac myocyte hypertrophy in vitro [1, 2]. CT-1 is part of a larger family of cytokines that includes Interleukin-6 (IL-6), Leukemia Inhibitory Factor (LIF), Ciliary Neurotrophic Factor (CNTF), Oncostatin-M (OSM), Interleukin-11 (IL-11), Cardiotrophin-Like Cytokine (CLC), and Neuropoietin/Cardiotrophin-2 (NP/CT-2). All of these cytokines share the common signaling receptor gp130. IL-6 and IL-11 use a gp130 homodimer, while the other family members activate a heterodimer composed of gp130 and the LIF receptor (LIFR) (reviewed by [3–5].
Several lines of evidence suggest that CT-1 and related cytokines play a critical role in cardiac development and injury response. CT-1 is expressed in the developing heart tube and supports the survival of cardiac myocytes [6–9]. Mice lacking gp130 die during embryogenesis and exhibit severe ventricular hypoplasia [10], while ventricular wall thickness is diminished in mice lacking postnatal expression of gp130 [11]. The absence of CT-1 during development does not generate the lethal cardiac malformations seen in mice lacking gp130 [12], but heart size and cardiac function have not been examined in these mice. Exogenous CT-1 prevents cell death during and after cardiac ischemia-reperfusion, resulting in decreased infarct size [9, 13, 14], while endogenous CT-1 and the gp130 receptor are elevated significantly following myocardial infarction [15, 16]. These data suggest that endogenous CT-1 plays an important cardio-protective role following ischemia-reperfusion, but the role of endogenous CT-1 in cardiac protection has not been tested directly.
Although mice lacking CT-1 do not develop severe cardiac malformations, they exhibit a significant loss of at least two classes of neurons. The absence of CT-1 results in the loss of a subset of motoneurons [12, 17], and in a decreased number of preganglionic sympathetic neurons [18]. Other related cytokines are involved in supporting motoneuron survival and function [17], but CT-1 appears to be the only cytokine required for the survival and maintenance of preganglionic sympathetic neurons [18]. Postganglionic sympathetic neurons projecting to the heart increase heart rate, ventricular pressure, and contractility through activation of cardiac beta receptors. Thus, cardiac function in CT-1 null mice could be altered by direct effects on the heart or by the loss of sympathetic transmission.
It is not known if the absence of CT-1, or the subsequent loss of preganglionic sympathetic neurons, alters cardiac function or injury response in adult mice. The aim of this study was to determine if the lack of CT-1 altered cardiac function, cardiac autonomic control, or infarct size after acute myocardial infarction, and to investigate potential compensation by related cytokines.
2. Materials and Methods
2.1 Animals
CT1−/− mice are fertile [12], and were maintained as homozygous nulls, with C57Bl/6J used as wild type controls.
2.2 Heart weight
Hearts were excised and trimmed of the great vessels and blood was rinsed out of the chambers. The whole heart was weighed and then the atria were removed and the ventricles weighed. Heart weights were compared to body weight of the animals and tibia length. Tibias were cleaned of muscle and connective tissue and measured with calipers.
2.3 Real-Time PCR
Hearts were harvested 24 hours after ischemia-reperfusion and stored immediately in RNAlater. RNA was isolated from the left ventricles using the Qaigen RNAeasy mini kit. Total RNA was quantified by OD260, and 200 ng of total RNA was treated with DNase and reverse transcribed. Each reverse transcription reaction was tested by regular PCR to confirm reverse transcription (RT), and an RNA alone control was included for each sample to test for genomic DNA contamination. Real-time PCR was performed with the ABI TaqMan Fast Universal PCR Master mix in the ABI 7500, using ABI pre-validated TaqMan gene expression assays for mouse IL-6, LIF, neuropoeitin/cardiotrophin-2, and actin as an internal control. For the PCR amplification, 2 μl of RT reactions were used in a total volume of 20 μl, and each sample was assayed in duplicate. Cytokine mRNAs were normalized to actin mRNA in the same sample. Post-infarct cytokine/actin mRNA ratios were compared to unoperated controls of the same genotype to determine the fold-increase following ischemia-reperfusion. Control and post-infarct samples of both genotypes were assayed together. Ischemia-reperfusion did not regulate the expression of actin mRNA (data not shown).
2.4 In situ mouse model of myocardial ischemia-reperfusion
[19] Adult mice were placed in an induction chamber and anesthetized with 4% isoflurane. Once an animal was unconscious it was given pentobarbital 30 mg/kg body weight IP. Animals were placed on a heating pad, the fur on the chest and ventral neck was removed using Nair hair remover, and the skin was wiped with saline and betadine. Mice were intubated, mechanically ventilated, and maintained with 1–2% isoflurane mixed with 100% oxygen (approximately 0.2L/min). End tidal CO2 was continuously monitored to verify adequate minute ventilation. Core body temperature was monitored by a rectal probe and maintained at ~37°C, and a 5-lead ECG was monitored throughout the surgery and experimental protocol using a PowerLab data acquisition system (ADInstruments, Inc., Colorado Springs, CO) on a Macintosh ibook G4.
The mouse was turned to a right lateral decubitus position and a thoracotomy performed in the 2nd or 3rd intercostal space with the aid of a dissecting microscope. A ligature (8-0 nylon mono-filament or equivalent on a taper needle) was placed around a proximal segment of the left anterior descending coronary artery and the ends of the suture were passed through a tube (PE10) with a blunted end to prevent tissue damage. The ligature was tightened to induce regional myocardial ischemia, which was confirmed by ECG changes, regional cyanosis, and wall motion abnormalities. After 45 minutes the coronary ligature was released, and reperfusion confirmed by visible epicardial hyperemia. The ligature was left in place for re-occlusion to delineate risk size. As soon as reperfusion was verified the chest was closed in layers. A small catheter was left in the thorax for 10–20 min to evacuate air and fluids. The mice were then removed from the ventilator, and repositioned every 30 min until able to maintain sternal recumbency (usually less than 15 min). Mice were then returned singly to a cage and the cage was placed half on and half off a heating pad for 30 min before being returned to the animal room.
All surgical procedures were performed under aseptic conditions. All procedures were approved by the Institutional Animal Care and Use Committee, and comply with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication No. 85-23, revised 1996). Control animals did not undergo any surgical procedures.
2.5 Heart rate, LVP, dP/dt
Mice were sedated with isoflurane, placed in supine position on a heating pad, and ECG leads connected to monitor heart rate. After basal heart rate was determined, mice were intubated with the outer sheath from a 22G angiocath, and placed on a rodent ventilator. A microtipped pressure transducer (1.4 French, Millar) was inserted into the right carotid artery and advanced into the left ventricle for measurement of left ventricular pressure using a PowerLab data acquisition system. Left ventricular pressure, dP/dt, and heart rate were analyzed using Chart software v5.4. When the animal was stable it was given hexamethonium chloride (5 mg/kg IP) to abolish autonomic reflexes [20]. A small polyvinyl catheter was placed in the left jugular vein for dobutamine administration (20 μg/kg/min for 3 min.).
2.6 Area at risk and infarct size
Twenty-four hours after the onset of reperfusion, the mice were placed in an induction chamber and anesthetized with 4% isoflurane. Once unconscious, mice were given pentobarbital 30 mg/kg body weight IP, placed on a heating pad, re-intubated, and the chest cavity re-opened at the site of the initial incision. The coronary artery was then re-occluded and fluorescent particles (1 mg/ml in saline; Duke Scientific #34-1, 2-8 μm size) were infused through a polyethylene catheter (PE10) with a 30-gauge needle tip in the left ventricle of the heart. Microspheres were infused at a rate of 300 μl/min for 5 minutes to delineate the area at risk. After 2 minutes of infusion the abdominal aorta was cut to release the pressure from increased blood volume preserving the coronary capillary beds. The heart was then excised for infarct size analysis, and cut into transverse slices 1 mm thick using a cutting block. Both sides of the slices were photographed under UV light, and the captured images were saved in Photoshop for measurement of area at risk. The slices were then placed in 2,3,5-triphenyl-tetrazolium chloride solution (TTC, 1% w/v in sodium phosphate buffer at 37ºC, pH 7.4) for 20 minutes. The staining procedure was carried out in the dark to prevent breakdown of the TTC by light. The slices were then placed in 10% neutral buffered formalin for approximately 10 minutes to increase the contrast between stained and unstained tissue. Myocardium that did not stain red was presumed to be infarcted. Both sides of each 1 mm section were photographed under white light, and images saved in Photoshop. Risk and infarct areas (mm2) for each slice were traced and digitized. The volume (in mm3) of myocardium at risk and infarcted myocardium was calculated from the measured areas and slice thickness. Infarct size was normalized as percentage of the area-at-risk. All infarct size analyses were performed in a blinded fashion by two people. The coefficient of variance between the two independent analyses ranged from 0.7–9%, and averaged 6.0±0.8%. The data presented are the average of the two independent determinations of infarct/risk.
3. Results
CT-1 is expressed in the developing heart tube and may be crucial for promoting the survival and hypertrophy of cardiac myocytes during development. If that is the case, one would expect the hearts from adult CT-1 null mice to be smaller than wild type hearts. We found no difference heart weight, or the ratios of heart weight to body weight or heart weight to tibia length (Table 1), between CT-1 −/− mice and wild type controls. Likewise, the ratios of bi-ventricular weight to body weight or tibia length did not differ between genotypes. These data suggest that the absence of CT-1 is not sufficient to replicate the decreased heart size observed in the absence of gp130.
Table 1.
Heart weight/body weight comparison (Mean ± SE)
| Wild Type | CT-1 −/− | |
|---|---|---|
| (n=5) | (n=8) | |
| HW (mg) | 134 ± 3.3 | 139 ± 3.7 |
| BW (g) | 29.2 ± 0.2 | 28.3 ± 1.0 |
| HW/BW (mg/g) | 4.6 ± 0.1 | 4.93 ± 0.12 |
| BVW/BW (mg/g) | 4.4 ± 0.2 | 4.3 ± 0.3 |
| BVW/TL (mg/mm) | 6.8 ± 0.2 | 7.1 ± 0.2 |
HW= heart weight; BW= body weight; BVW= bi-ventricular weight; TL= tibia length
CT-1 is part of a larger family of cytokines that utilize the gp130 receptor and have overlapping functions. Therefore, upregulation of related cytokines might compensate for the loss of CT-1 during cardiac development. The CT-1 related cytokines LIF and IL-6 are expressed in the heart and might compensate for the loss of CT-1. In addition, NP/CT-2 was identified recently as a gp130 family member that is expressed at high levels during embryogenesis [21]. We used real-time PCR to quantify LIF, IL-6 and NP/CT-2 gene expression in adult heart to determine if these cytokines were elevated in CT-1 null mice compared to wild type mice. NP/CT-2 mRNA was not detected in adult heart, although it was amplified from embryonic brain positive control tissue (data not shown). LIF and IL-6 mRNAs were detected in the adult left ventricle, and IL-6 gene expression was elevated approximately 5-fold in CT-1 null mice (Fig. 1A). This suggests that upregulation of IL-6 compensates for the absence of CT-1 during development.
Figure 1.

LIF and IL-6 mRNA in left ventricle. IL-6 (A) and LIF (B) mRNAs were quantified by real-time PCR in the left ventricles of wild type (WT, filled bars) and CT-1−/− mice (clear bars). Cytokine mRNAs were assayed in duplicate and normalized to actin mRNA in the same sample. Data shown are Mean ± SE (n=5 WT, n=7 CT1−/−, * p< 0.05, t-test).
Cardiac function was largely normal in CT1−/− mice. Basal heart rate was increased modestly in CT-1 −/− compared to wild type mice (WT, 486±9.3; CT-1 −/−, 519.5±10.5; mean±sem, n=5; p<0.05), but ganglionic block (Figure 1) or insertion of the pressure probe into the left ventricle (Table 2) abolished the difference in heart rate between genotypes. Insertion of a pressure probe into the left ventricle increased heart rate in both genotypes (Table 2). Left ventricular pressure (LVP) and cardiac contractility (dP/dt ) were unchanged in CT-1 −/− compared to wild type mice (Table 2).
Table 2.
Baseline hemodynamic parameters before ganglionic block (Mean ± SE)
| Wild type | CT-1 −/− | |
|---|---|---|
| (n=4) | (n=5) | |
| HR (beat/min) | 580.5 ± 9.1 | 584.8 ± 3.9 |
| LVP (mm Hg) | 107.4 ± 3.3 | 102.3 ± 2.2 |
| dP/dtMAX (mmHg/s) | 8171 ± 762 | 7704 ± 483 |
| dP/dtMIN (mmHg/s) | −7004 ± 502 | −6459 ± 516 |
HR, heart rate; LVP, left ventricular pressure; dP/dtMAX, maximal rate of pressure development; dP/dtMIN, maximal rate of pressure decay
The absence of CT-1 might result in subtle changes in ventricular pressure and cardiac contractility that are not apparent under basal conditions, but are visible upon stimulation of the heart. To test this, hexamethonium (5 mg/kg) was administered to block ganglionic transmission, and the β agonist dobutamine (20 μg/kg/min) was infused for 3 minutes to directly stimulate cardiac function. Heart rate following dobutamine treatment did not differ significantly between genotypes (Figure 1A), but average left ventricular pressure (Figure 1C) was decreased slightly in CT-1 −/− mice compared to wild type mice (CT1, 95±1; WT, 104±4 mm Hg, mean±sem, p<0.05). This did not correspond to altered cardiac contractility, since dP/dtMAX and dP/dtMIN were indistinguishable between genotypes before and after stimulation of cardiac β receptors (Figure 1B, D).
Infusion of exogenous CT-1 protects cardiac myocytes from apoptosis following ischemia-reperfusion [9, 13, 14], and endogenous CT-1 and gp130 are elevated in the heart after ischemia-reperfusion [15]. These date suggest a role for endogenous CT-1 in cardiac protection following injury that is distinct from a developmental role. To determine if the loss of endogenous CT-1 increased cell death following ischemia-reperfusion, area-at-risk and infarct size were analyzed in wild type and CT-1 null mice 24 hours after surgery. Surprisingly, the lack of endogenous CT-1 had no effect on infarct size relative to area at risk. In both genotypes, 45 minutes of ischemia followed by 24 hours of reperfusion resulted in an infarct encompassing approximately 30–40% of the area at risk (Figure 3). The absolute risk volume was not significantly different between the groups: WT, 35.2± 4.5 mm3 (mean ± SE, n=7); CT1−/−, 38.0 ± 4.3 mm3 (mean ± SE, n=6). Although exogenous CT-1 decreases cell death after ischemia-reperfusion, our data indicate that the absence of endogenous CT-1 has no significant effect on infarct size during the first 24 hours after ischemia-reperfusion.
Figure 3.

Infarct size compared to area at risk. A) Area at risk (left) and Infarct (right) in a representative section. The area at risk (“Risk”) is devoid of fluorescent microspheres, while the infarct is yellow/white following TTC staining. Scale bar is 1.25 mm. B) Infarct and risk were quantified 24 hours after ischemia-reperfusion. Horizontal lines represent the mean values of each group. Mean ± SE: WT, 37.7± 3.3, n=7; CT-1 −/−, 33.8± 1.0, n= 6 (p=0.3, t-test).
The cytokines LIF and IL-6 are also elevated in the heart following myocardial infarction [22–26], and compensatory upregulation of these cytokines might protect heart cells in the absence of CT-1. Similarly, NP/CT-2 might be induced following ischemia along with other embryonic genes. Therefore, we used real-time PCR to quantify LIF, IL-6 and NP/CT-2 gene expression in the left ventricle from unoperated and post-infarct wild type and CT-1−/− mice. NP/CT-2 mRNA was not detected in the left ventricle 24 hours after ischemia-reperfusion (data not shown), but LIF and IL-6 mRNA were present in both wild type and CT-1 null mice. Surprisingly, the post-infarct induction of LIF and IL-6 mRNA was impaired, rather than enhanced, in CT-1 null mice (Fig. 4). In wild-type mice cardiac IL-6 mRNA was induced 154-fold following ischemia-reperfusion compared to wild type unoperated mice (154±17.2 fold increase, mean±sem, n=3). Although basal levels of IL-6 mRNA were elevated in CT-1 null mice (Fig. 1), ischemia-reperfusion triggered only a 2.3 -fold increase in IL-6 mRNA compared to unoperated CT1−/− controls (Fig. 4) (fold induction CT1−/− mice: control, 1±0.1, Mean ± SE, n=3 vs. post-infarct, 2.3±0.52 Mean ± SE, n=3; t-test p=0.06). LIF gene expression was increased significantly in CT-1 null mice after infarct (fold induction CT1−/− mice: control, 1±0.24 vs. post-infarct 7.4±0.6, Mean ± SE, n=4, p<0.05), but not to the same extent as in wild type mice (fold induction wild type: control, 1±0.15, n=4 vs. post-infarct 13.7±1.4, n=3, Mean ± SE, p<0.05). These data confirm that the absence of CT-1 alters expression of related cytokines, but reveal unexpected changes in IL-6 and LIF mRNA expression.
Figure 4.
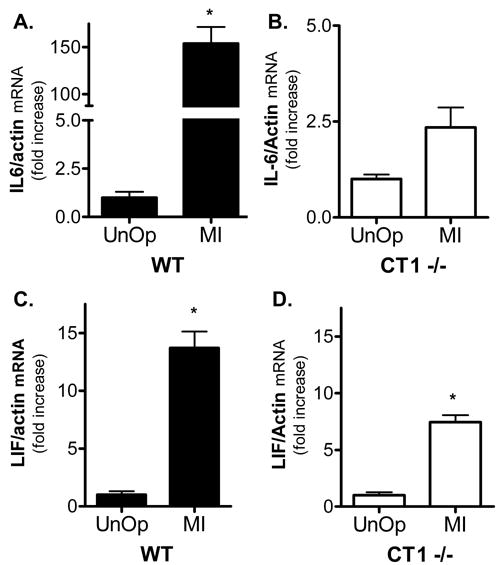
Post-infarct LIF and IL-6 mRNA in left ventricle. IL-6 (A) and LIF (B) mRNAs were quantified by real-time PCR in the left ventricles of wild type (WT) and CT-1−/− mice 24 hours after ischemia-reperfusion (MI). Cytokine mRNAs were normalized to actin mRNA in the same sample, and graphed as fold increase relative to unoperated controls of the same genotype. A) IL-6 is increased significantly after MI in WT mice (* p<0.001), but not in CT1−/− mice (Mean ± SE, n=3 or 4). Data are presented as fold increase (shown in brackets) compared to control levels for each genotype. B) LIF is increased significantly after MI in WT mice (* p<0.05, Mean ± SE, n=3 or 4), and in CT1−/− mice (*p<0.05; Mean ± SE, n= 4). Data are presented as fold increase compared to control levels for each genotype (see Fig. 1 for control values).
4. Discussion
The major findings of this study are that the absence of endogenous CT-1 does not decrease heart weight, alter cardiac contractility, or increase infarct size following acute ischemia-reperfusion. The lack of CT-1 resulted in a small decrease in left ventricular pressure that was only observed following dobutamine stimulation, and a small increase in basal heart rate that disappeared after ganglionic block, implicating altered pre-ganglionic transmission rather than a direct effect on the heart. Cardiac IL-6 mRNA was elevated in CT-1 null mice while LIF mRNA was not. Post-infarct induction of IL-6 mRNA was impaired in mice lacking CT-1, but induction of LIF mRNA remained significant.
CT-1 was identified based on its ability to promote hypertrophy in cardiac myocytes [1]. Mice lacking the CT-1 receptor component gp130 die during embryogenesis due in part to severe ventricular hypoplasia [10], and ventricular wall thickness is also diminished in mice lacking postnatal expression of gp130 [11]. The phenotype of gp130 knockout mice, coupled with the expression of CT-1 in the developing heart tube [6], suggested that CT-1 was the gp130 ligand important for cardiac growth and development (reviewed by [27]). CT1 deficient mice are viable and have a normal lifespan, however, suggesting that there are no gross changes in cardiac development [12]. Our data confirm this, indicating that the ratios of heart weight or bi-ventricular weight to body weight or tibia length are similar in CT-1 null mice and wild type mice. Together these data suggest that another gp130 cytokine compensates for the loss of CT-1 during cardiac development.
Functional redundancy is common within the gp130 family of cytokines [3], so we examined the expression of related cytokines to determine if they were increased in CT-1 null mice. Mice lacking LIF [28, 29] or the LIF receptor (LIFR) do not have an obvious cardiac phenotype [30]. This suggests that LIFR-independent members of this cytokine family are sufficient to support normal cardiac development. We found that mRNA encoding IL-6, which signals through a gp130 homodimer, was elevated almost 5-fold in CT-1 null mice. IL-6 alone has little effect on cultured cardiac myocytes [6], but adding soluble IL-6R together with IL-6 stimulates cardiac hypertrophy in vitro [2] and in vivo [31]. A soluble form of the IL-6R is present in plasma, raising the possibility that IL-6 interacts with soluble IL-6R during development to compensate for the absence of CT-1 during development and in the adult heart.
The CT-1 related cytokine Neuropoietin/Cardiotropin-2 was identified recently as a gp130 family member that is expressed at high levels during embryogenesis and not present in adult tissues [21]. NP/CT-2 shares a greater degree of similarity with CT-1 than any of the other gp130 family members, and its robust expression during embryogenesis makes it an interesting candidate to compensate for the loss of CT-1 during development. Although we cannot exclude the possibility that NP/CT-2 compensates for the absence of CT-1 during development, no NP/CT-2 mRNA was detected in the left ventricle of adult WT or CT-1−/− hearts before or after ischemia-reperfusion.
Infusion of exogenous CT-1 into adult animals provokes an acute decrease in blood pressure that stimulates a reflex increase in heart rate [32–34]. These acute effects of CT-1 do not include any changes in ventricular pressure or cardiac contractility. In contrast, chronic infusion of CT-1 for a week increases left ventricular pressure and dP/dtMAX with no effect on heart rate [13]. We expected that the chronic absence of CT-1 might result in decreased ventricular pressure and dP/dtMAX, given the increases in these parameters following chronic CT-1 administration. There was no change, however, in left ventricular pressure or dP/dtMAX in mice lacking CT-1 under basal conditions or after ganglionic block. The only differences in cardiac function observed in CT-1 null mice were a modest increase in basal heart rate and a small deficit in left ventricular pressure following stimulation with the β agonist dobutamine.
The increased heart rate observed in CT-1 nulls was surprising due to their deficit in preganglionic sympathetic neurons [18]. Sympathetic transmission stimulates heart rate and cardiac contractility through the release of norepinephrine [35], and the amount of norepinephrine release in the heart is regulated by the activation of sympathetic pre-ganglionic nerves [36, 37]. Therefore, a 20% loss in preganglionic inputs to cardiac sympathetic neurons would be expected to either decrease heart rate or cause no change. A larger (50%) loss of pre-ganglionic projections to the adrenal medulla in CT-1 null mice did not result in decreased norepinephrine or epinephrine production [18], however, suggesting that other compensatory processes bring neurotransmitter synthesis to normal levels. Compensatory changes in norepinephrine production and cardiac nerve activity might account for the increased basal heart rate that disappears upon ganglionic block.
Perhaps the most surprising result of our study is that the lack of endogenous CT-1 does not lead to increased infarct size following acute ischemia-reperfusion. CT-1 protects cardiac myocytes from cell death both in vitro and in vivo [6–9, 38]. CT-1 is elevated in the heart and plasma within 24 hours of myocardial infarction [15, 16, 39], and addition of exogenous CT-1 decreases infarct size whether it is added before ischemia or during reperfusion [9]. It seemed likely that the absence of CT-1 would be detrimental and increase infarct size. However, no difference in infarct size relative to area at risk was observed 24 hours after ischemia-reperfusion in mice lacking CT-1 compared to wild type mice. Although differences might appear over a longer time frame, these acute data raise the possibility that other cytokines compensate for the absence of CT-1 during ischemia-reperfusion.
LIF, IL-6, and IL-6R protein are elevated in cardiac myocytes following myocardial infarction [22–24, 26]. These cytokines prevent apoptosis and stimulate cardiac hypertrophy in a manner essentially indistinguishable from the actions of CT-1. All three cytokines stimulate myocyte hypertrophy through activation of the Jak-STAT (Signal Transducers and Activators of Transcription) pathway, and prevent apoptosis through activation of ERK1/2 (Extracellular signal Regulated protein Kinases 1 & 2) and the phosphatidylinositol 3-OH kinase (PI3 kinase)/Akt pathways [7–9, 40–44]. LIF is elevated to a greater degree after infarct in mice lacking IL-6 compared to wild type mice [42], and we expected that one or both of these cytokines would be elevated to a greater extent after ischemia-reperfusion in CT-1 −/− mice than in control mice. Contrary to our expectations, induction of IL-6 mRNA was significantly abrogated in CT-1 null mice and induction of LIF mRNA was lower than in wild type mice. Post-infarct induction of LIF mRNA remained significant, however, and may provide a compensatory mechanism for protection of cardiac myocytes in the absence of CT-1. Although we did not quantify protein levels, previous studies found that the increase in cytokine mRNA after ischemia-reperfusion is accompanied by elevated cytokine production [22, 24, 25]. The receptors for these cytokines are also elevated in the heart after ischemia-reperfusion [15, 22]. Since infarct size is similar in CT-1 null mice and wild type mice, increased expression of LIF and its receptors may be sufficient to protect cardiac myocytes from cell death in the absence of CT-1.
The gp130 cytokine family is characterized by functional redundancy, and the phenotypes of mice lacking individual cytokines [12, 17, 28, 29, 45] are much less severe than the phenotypes of mice lacking their shared receptors [10, 30, 46]. Nonetheless, the early and robust expression of CT-1 in the developing heart tube, and the sustained expression of CT-1 in the adult heart, suggested that CT-1 played a unique role in promoting cardiac myocyte survival and hypertrophy during development and after injury. Although CT-1 is important for the developmental survival of a subset of motor neurons and preganglionic sympathetic neurons, our data indicate that the loss of preganglionic neurons does not lower heart rate, and the absence of endogenous CT-1 does not significantly alter cardiac function or infarct size. Further studies will be required to understand fully the compensatory mechanisms that promote myocyte survival and development in the absence of CT-1.
Figure 2.

Dobutamine challenge. Heart rate and cardiac function were measured in anesthetized wild type (squares, n=4) and CT-1 null mice (circles, n=5) following ganglionic blockade. No significant differences were detected in basal or stimulated heart rate (A), dP/dtMAX (C), or dP/dtMIN (D). Basal left ventricular pressure (LVP) did not differ between genotypes, but average LVP following dobutamine challenge was significantly lower in CT-1 −/− mice (* p<0.05, unpaired t-test). The data shown are Mean ± SE. (E, F) Representative LVP trace from a single WT (E) or CT1−/− (F) mouse during dobutamine administration. Black arrowheads mark the beginning of dobutamine infusion, and gray arrowheads mark the end.
Acknowledgments
The authors thank Emily Pratt and for assistance quantifying risk and infarct size. This work was supported by HL68231 (BAH).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pennica D, King KL, Shaw KJ, Luis E, Rullamas J, Luoh SM, et al. Expression cloning of cardiotrophin 1, a cytokine that induces cardiac myocyte hypertrophy. Proc Natl Acad Sci U S A. 1995;92:1142–6. doi: 10.1073/pnas.92.4.1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wollert KC, Taga T, Saito M, Narazaki M, Kishimoto T, Glembotski CC, et al. Cardiotrophin-1 activates a distinct form of cardiac muscle cell hypertrophy. Assembly of sarcomeric units in series via gp130/leukemia inhibitory factor receptor-dependent pathways. J Biol Chem. 1996;271:9535–45. doi: 10.1074/jbc.271.16.9535. [DOI] [PubMed] [Google Scholar]
- 3.Taga T, Kishimoto T. Gp130 and the interleukin-6 family of cytokines. Annu Rev Immunol. 1997;15:797–819. doi: 10.1146/annurev.immunol.15.1.797. [DOI] [PubMed] [Google Scholar]
- 4.Heinrich PC, Behrmann I, Müller-Newen G, Schaper F, Graeve L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem J. 1998;334:297–314. doi: 10.1042/bj3340297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heinrich PC, Behrmann I, Haan S, Hermanns HM, Muller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. 2003;374:1–20. doi: 10.1042/BJ20030407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sheng Z, Pennica D, Wood WI, Chien KR. Cardiotrophin-1 displays early expression in the murine heart tube and promotes cardiac myocyte survival. Development. 1996;122:419–28. doi: 10.1242/dev.122.2.419. [DOI] [PubMed] [Google Scholar]
- 7.Sheng Z, Knowlton K, Chen J, Hoshijima M, Brown JH, Chien KR. Cardiotrophin 1(CT-1) Inhibition of Cardiac Myocyte Apoptosis via a Mitogen-activated Protein Kinase-dependent Pathway. Divergence from Downstream CT-1 Signals for Myocardial Cell Hypertrophy. J Biol Chem. 1997;272:5783–91. doi: 10.1074/jbc.272.9.5783. [DOI] [PubMed] [Google Scholar]
- 8.Brar BK, Stephanou A, Liao Z, O'Leary RM, Pennica D, Yellon DM, et al. Cardiotrophin-1 can protect cardiac myocytes from injury when added both prior to simulated ischaemia and at reoxygenation. Cardiovasc Res. 2001;51:265–74. doi: 10.1016/s0008-6363(01)00294-2. [DOI] [PubMed] [Google Scholar]
- 9.Liao Z, Brar BK, Cai Q, Stephanou A, O'Leary RM, Pennica D, et al. Cardiotrophin-1 (CT-1) can protect the adult heart from injury when added both prior to ischaemia and at reperfusion. Cardiovasc Res. 2002;53:902–10. doi: 10.1016/s0008-6363(01)00531-4. [DOI] [PubMed] [Google Scholar]
- 10.Yoshida K, Taga T, Saito M, Suematsu S, Kumanogoh A, Tanaka T, et al. Targeted disruption of gp130, a common signal transducer for the interleukin 6 family of cytokines, leads to myocardial and hematological disorders. Proc Natl Acad Sci U S A. 1996;93:407–11. doi: 10.1073/pnas.93.1.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Betz UAK, Bloch W, van den Broek M, Yoshida K, Taga T, Kishimoto T, et al. Postnatally Induced Inactivation of gp130 in Mice Results in Neurological, Cardiac, Hematopoietic, Immunological, Hepatic, and Pulmonary Defects. The Journal of Experimental Medicine. 1998;188:1955–65. doi: 10.1084/jem.188.10.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oppenheim RW, Wiese S, Prevette D, Armanini M, Wang S, Houenou LJ, et al. Cardiotrophin-1, a muscle-derived cytokine, is required for the survival of subpopulations of developing motoneurons. J Neurosci. 2001;21:1283–91. doi: 10.1523/JNEUROSCI.21-04-01283.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ruixing Y, Dezhai Y, Jiaquan L. Effects of cardiotrophin-1 on hemodynamics and cardiomyocyte apoptosis in rats with acute myocardial infarction. J Med Invest. 2004;51:29–37. doi: 10.2152/jmi.51.29. [DOI] [PubMed] [Google Scholar]
- 14.Ghosh S, Ng LL, Talwar S, Squire IB, Galinanes M. Cardiotrophin-1 protects the human myocardium from ischemic injury. Comparison with the first and second window of protection by ischemic preconditioning. Cardiovasc Res. 2000;48:440–7. doi: 10.1016/s0008-6363(00)00186-3. [DOI] [PubMed] [Google Scholar]
- 15.Aoyama T, Takimoto Y, Pennica D, Inoue R, Shinoda E, Hattori R, et al. Augmented expression of cardiotrophin-1 and its receptor component, gp130, in both left and right ventricles after myocardial infarction in the rat. J Mol Cell Cardiol. 2000;32:1821–30. doi: 10.1006/jmcc.2000.1218. [DOI] [PubMed] [Google Scholar]
- 16.Freed DH, Moon MC, Borowiec AM, Jones SC, Zahradka P, Dixon IM. Cardiotrophin-1: expression in experimental myocardial infarction and potential role in post-MI wound healing. Mol Cell Biochem. 2003;254:247–56. doi: 10.1023/a:1027332504861. [DOI] [PubMed] [Google Scholar]
- 17.Holtmann B, Wiese S, Samsam M, Grohmann K, Pennica D, Martini R, et al. Triple Knock-Out of CNTF, LIF, and CT-1 Defines Cooperative and Distinct Roles of these Neurotrophic Factors for Motoneuron Maintenance and Function. J Neurosci. 2005;25:1778–87. doi: 10.1523/JNEUROSCI.4249-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oberle S, Schober A, Meyer V, Holtmann B, Henderson C, Sendtner M, et al. Loss of leukemia inhibitory factor receptor beta or cardiotrophin-1 causes similar deficits in preganglionic sympathetic neurons and adrenal medulla. J Neurosci. 2006;26:1823–32. doi: 10.1523/JNEUROSCI.4127-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wolff RA, Chien GL, Van Winkle DM. Propidium Iodide Compares Favorably with Histology and Triphenyl Tetrazolium Chloride in the Assessment of Experimentally-induced Infarct Size. J Mol Cell Cardiol. 2000;32:225–32. doi: 10.1006/jmcc.1999.1074. [DOI] [PubMed] [Google Scholar]
- 20.Aizawa-Abe M, Ogawa Y, Masuzaki H, Ebihara K, Satoh N, Iwai H, et al. Pathophysiological role of leptin in obesity-related hypertension. J Clin Invest. 2000;105:1243–52. doi: 10.1172/JCI8341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Derouet D, Rousseau F, Alfonsi F, Froger J, Hermann J, Barbier F, et al. Neuropoietin, a new IL-6-related cytokine signaling through the ciliary neurotrophic factor receptor. Proc Natl Acad Sci U S A. 2004;101:4827–32. doi: 10.1073/pnas.0306178101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chandrasekar B, Mitchell DH, Colston JT, Freeman GL. Regulation of CCAAT/Enhancer binding protein, interleukin-6, interleukin-6 receptor, and gp130 expression during myocardial ischemia/reperfusion. Circulation. 1999;99:427–33. doi: 10.1161/01.cir.99.3.427. [DOI] [PubMed] [Google Scholar]
- 23.Gwechenberger M, Mendoza LH, Youker KA, Frangogiannis NG, Smith CW, Michael LH, et al. Cardiac myocytes produce interleukin-6 in culture and in viable border zone of reperfused infarctions. Circulation. 1999;99:546–51. doi: 10.1161/01.cir.99.4.546. [DOI] [PubMed] [Google Scholar]
- 24.Kucia M, Dawn B, Hunt G, Guo Y, Wysoczynski M, Majka M, et al. Cells Expressing Early Cardiac Markers Reside in the Bone Marrow and Are Mobilized Into the Peripheral Blood After Myocardial Infarction. Circ Res. 2004;95:1191–9. doi: 10.1161/01.RES.0000150856.47324.5b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zou Y, Takano H, Mizukami M, Akazawa H, Qin Y, Toko H, et al. Leukemia Inhibitory Factor Enhances Survival of Cardiomyocytes and Induces Regeneration of Myocardium After Myocardial Infarction. Circulation. 2003;108:748–53. doi: 10.1161/01.CIR.0000081773.76337.44. [DOI] [PubMed] [Google Scholar]
- 26.Ono K, Matsumori A, Shioi T, Furukawa Y, Sasayama S. Cytokine gene expression after myocardial infarction in rat hearts: possible implication in left ventricular remodeling. Circulation. 1998;98:149–56. doi: 10.1161/01.cir.98.2.149. [DOI] [PubMed] [Google Scholar]
- 27.Wollert KC, Chien KR. Cardiotrophin-1 and the role of gp130-dependent signaling pathways in cardiac growth and development. J Mol Med. 1997;75:492–501. doi: 10.1007/s001090050134. [DOI] [PubMed] [Google Scholar]
- 28.Escary JL, Perreau J, Dumenil D, Ezine S, Brulet P. Leukaemia inhibitory factor is necessary for maintenance of haematopoietic stem cells and thymocyte stimulation. Nature. 1993;363:361–4. doi: 10.1038/363361a0. [DOI] [PubMed] [Google Scholar]
- 29.Stewart CL, Kaspar P, Brunet LJ, Bhatt H, Gadi I, Kontgen F, et al. Blastocyst implantation depends on maternal expression of leukaemia inhibitory factor. Nature. 1992;359:76–9. doi: 10.1038/359076a0. [DOI] [PubMed] [Google Scholar]
- 30.Ware CB, Horowitz MC, Renshaw BR, Hunt JS, Liggitt D, Koblar SA, et al. Targeted disruption of the low-affinity leukemia inhibitory factor receptor gene causes placental, skeletal, neural and metabolic defects and results in perinatal death. Development. 1995;121:1283–99. doi: 10.1242/dev.121.5.1283. [DOI] [PubMed] [Google Scholar]
- 31.Hirota H, Yoshida K, Kishimoto T, Taga T. Continuous activation of gp130, a signal-transducing receptor component for interleukin 6-related cytokines, causes myocardial hypertrophy in mice. Proc Natl Acad Sci U S A. 1995;92:4862–6. doi: 10.1073/pnas.92.11.4862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hamanaka I, Saito Y, Nishikimi T, Magaribuchi T, Kamitani S, Kuwahara K, et al. Effects of cardiotrophin-1 on hemodynamics and endocrine function of the heart. Am J Physiol Heart Circ Physiol. 2000;279:H388–H396. doi: 10.1152/ajpheart.2000.279.1.H388. [DOI] [PubMed] [Google Scholar]
- 33.Jin H, Yang R, Ko A, Pennica D, Wood WI, Paoni NF. Effects of cardiotrophin-1 on haemodynamics and cardiac function in conscious rats. Cytokine. 1998;10:19–25. doi: 10.1006/cyto.1997.0241. [DOI] [PubMed] [Google Scholar]
- 34.Yao L, Kohno M, Noma T, Murakami K, Tsuji T, Yu Y, et al. Acute effect of human cardiotrophin-1 on hemodynamic parameters in spontaneously hypertensive rats and Wistar Kyoto rats. Hypertens Res. 2001;24:717–21. doi: 10.1291/hypres.24.717. [DOI] [PubMed] [Google Scholar]
- 35.Yamaguchi N, de Champlain J, Nadeau R. Correlation between the response of the heart to sympathetic stimulation and the release of endogenous catecholamines into the coronary sinus of the dog. Circ Res. 1975;36:662–8. doi: 10.1161/01.res.36.5.662. [DOI] [PubMed] [Google Scholar]
- 36.Cao WH, Morrison SF. Disinhibition of rostral raphe pallidus neurons increases cardiac sympathetic nerve activity and heart rate. Brain Res. 2003;980:1–10. doi: 10.1016/s0006-8993(03)02981-0. [DOI] [PubMed] [Google Scholar]
- 37.Esler M, Lambert G, Brunner-La Rocca HP, Vaddadi G, Kaye D. Sympathetic nerve activity and neurotransmitter release in humans: translation from pathophysiology into clinical practice. Acta Physiol Scand. 2003;177:275–84. doi: 10.1046/j.1365-201X.2003.01089.x. [DOI] [PubMed] [Google Scholar]
- 38.Stephanou A, Brar B, Heads R, Knight RD, Marber MS, Pennica D, et al. Cardiotrophin-1 induces heat shock protein accumulation in cultured cardiac cells and protects them from stressful stimuli. J Mol Cell Cardiol. 1998;30:849–55. doi: 10.1006/jmcc.1998.0651. [DOI] [PubMed] [Google Scholar]
- 39.Talwar S, Squire IB, O'brien RJ, Downie PF, Davies JE, Ng LL. Plasma cardiotrophin-1 following acute myocardial infarction: relationship with left ventricular systolic dysfunction. Clin Sci (Lond) 2002;102:9–14. [PubMed] [Google Scholar]
- 40.Brar BK, Stephanou A, Pennica D, Latchman DS. CT-1 mediated cardioprotection against ischaemic re-oxygenation injury is mediated by PI3 kinase, Akt and MEK1/2 pathways. Cytokine. 2001;16:93–6. doi: 10.1006/cyto.2001.0951. [DOI] [PubMed] [Google Scholar]
- 41.Kunisada K, Tone E, Fujio Y, Matsui H, Yamauchi-Takihara K, Kishimoto T. Activation of gp130 transduces hypertrophic signals via STAT3 in cardiac myocytes. Circulation. 1998;98:346–52. doi: 10.1161/01.cir.98.4.346. [DOI] [PubMed] [Google Scholar]
- 42.Fuchs M, Hilfiker A, Kaminski K, Hilfiker-Kleiner D, Guener Z, Klein G, et al. Role of interleukin-6 for LV remodeling and survival after experimental myocardial infarction. FASEB J. 2003;17:2118–20. doi: 10.1096/fj.03-0331fje. [DOI] [PubMed] [Google Scholar]
- 43.Kunisada K, Hirota H, Fujio Y, Matsui H, Tani Y, Yamauchi-Takihara K, et al. Activation of JAK-STAT and MAP Kinases by Leukemia Inhibitory Factor Through gp130 in Cardiac Myocytes. Circulation. 1996;94:2626–32. doi: 10.1161/01.cir.94.10.2626. [DOI] [PubMed] [Google Scholar]
- 44.Yasukawa H, Hoshijima M, Gu Y, Nakamura T, Pradervand S, Hanada T, et al. Suppressor of cytokine signaling-3 is a biomechanical stress-inducible gene that suppresses gp130-mediated cardiac myocyte hypertrophy and survival pathways. J Clin Invest. 2001;108:1459–67. doi: 10.1172/JCI13939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Masu Y, Wolf E, Holtmann B, Sendtner M, Brem G, Thoenen H. Disruption of the CNTF gene results in motor neuron degeneration. Nature. 1993;365:27–32. doi: 10.1038/365027a0. [DOI] [PubMed] [Google Scholar]
- 46.DeChiara TM, Vejsada R, Poueymirou WT, Acheson A, Suri C, Conover JC, et al. Mice lacking the CNTF receptor, unlike mice lacking CNTF, exhibit profound motor neuron deficits at birth. Cell. 1995;83:313–22. doi: 10.1016/0092-8674(95)90172-8. [DOI] [PubMed] [Google Scholar]