

Summary
Long-chain acyl-CoA synthetases (ACSL) activate fatty acids (FA) and provide substrates for virtually every metabolic pathway that catabolizes FA or synthesizes complex lipids. We have hypothesized that each of the five cloned ACSL isoforms partitions FA towards specific downstream pathways. Adult heart expresses all five cloned ACSL isoforms, but their independent functional roles have not been elucidated. Studies implicate ACSL1 in both oxidative and lipid synthetic pathways. To clarify the functional role of ACSL1 and the other ACSL isoforms (3–6), we examined ACS specific activity and Acsl mRNA expression in the developing mouse heart which increases FA oxidative pathways for energy production after birth. Compared to the embryonic heart, ACS specific activity was 14-fold higher on post-natal day 1 (P1). On P1, as compared to the fetus, only Acsl1 mRNA increased, whereas transcripts for the other Acsl isoforms remained the same, suggesting that ACSL1 is the major isoform responsible for activating long-chain FA for myocardial oxidation after birth. In contrast, the mRNA abundance of Acsl3 was highest on E16, and decreased dramatically by P7, suggesting that ACSL3 may play a critical role during the development of the fetal heart. Our data support the hypothesis that each ACSL has a specific role in the channeling of FA towards distinct metabolic fates.
Keywords: fatty acid activation, heart ontogeny, heart fatty acid oxidation, heart lipid synthesis, heart phospholipid composition
Abbreviations: ACO, acyl-CoA oxidase; ACS, acyl-CoA synthetase; ACSL and Acsl, long chain acyl-CoA synthetase protein and mRNA, respectively; CPT-1, carnitine palmitoyl transferase-1; DTT, dithiothreitol; E16 embryonic day 16; FA, fatty acid; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; LCAD, long-chain acyl dehydrogenase; MCAD, medium-chain acyl dehydrogenase; P1, postnatal day 1; PPAR, peroxisome proliferator-activated receptor
Introduction
For the mouse fetus the primary energy source is maternally –supplied glucose, but after birth the energy source shifts abruptly from carbohydrate to lipid when 70 percent of the calories ingested by the suckling pup come from milk triacylglycerol [1]. During the third week of life the pup begins to nibble increasing amounts of chow, and after weaning the mouse again eats a diet that is about 60% carbohydrate and less than 14% fat. Fuel use by the developing rodent heart depends on lactate and glucose in the fetus, and on oxidative metabolism of glucose and glycogen immediately after birth [2]. By 1 week of age, however, the heart derives more than 70% of its energy from the oxidation of fatty acids [2–4] and this preferred use of fatty acids for energy persists throughout life [5].
The postnatal dependence of the heart on fatty acid as its primary energy source is aided by rapid postnatal increases in the activities of enzymes required for β-oxidation, including MCAD, LCAD, M-CPT1, and ACO [6–10]. These increases in activity are controlled by up-regulation of gene transcription [10, 11].
The use of fatty acids for oxidation depends on their initial activation by long chain acyl-CoA synthetases (ACSLs) which catalyze the conversion of long chain fatty acids to long chain fatty acyl-CoAs. ACSL activity is thus an integral initial step for virtually every metabolic pathway that degrades fatty acids or synthesizes complex lipids [11–13]. The formation of acyl-CoAs occurs in a two step process. The first step converts free fatty acid to an acyl-AMP intermediate through the pyrophosphorolysis of ATP. In the second step the thiol group of CoA displaces AMP to form acyl-CoA [14]. Acyl-CoAs then enter metabolic pathways, including β-oxidation, fatty acid retroconversion, desaturation, and elongation, glycerolipid synthesis, phospholipid reacylation, cholesterol ester synthesis, and wax ester formation. Acyl-CoAs also activate numerous cell processes and act as signaling molecules [12, 13, 15–18].
Five ACSL isoforms have been cloned in humans and rodents. ACSL1, 5, and 6 share over 60% amino acid identity [19]. ACSL3 and 4 have 68% identity with each other and approximately 30 percent homology with ACSL1 [20]. The ACSL isoforms differ in their tissue distribution, organelle location, relative expression, enzyme kinetics, regulation, and substrate preferences [12, 13, 21, 22]. Most ACSL isoforms are intrinsic membrane proteins, but several have secondary start sites that cleave the transmembrane domain and render the enzyme membrane associated [23].
Most tissues express more than one ACSL isoform [24], and we have hypothesized that the individual ACSL isoforms each channel fatty acids toward distinct metabolic fates. The developmental expression and role of individual ACSL isoforms in the heart is not known. All five ACSL isoforms are expressed in mouse and rat heart, with Acsl1 having the most abundant transcripts [24, 25]. Expression of Acsl1 is low in liver and heart from mice lacking PPARα [26, 27], a transcription factor which directs the program of fatty acid oxidation [28]. Myocardial contraction is poor in PPARα−/− mice both under basal conditions and after beta1 adrenergic stimulation, and the heart becomes fibrotic [29]. In addition, oleate and PPARα agonists increase Acsl1 expression in cultured rat cardiomyocytes [25]. These data suggest that ACSL1 might provide FA destined for oxidation. In contrast, fasting or a high-fat diet does not increase Acsl1 expression [25]. However, heart specific overexpression of ACSL1 results in excess myocardial TAG accumulation [30], suggesting that ACSL1 channels FA towards esterification.
In order to clarify the normal functional role of ACSL1 and the ACSL isoforms 3–6 in heart, we examined mRNA expression and activities in developing mouse heart. Because the fuel sources and heart requirements for energy substrates change dramatically from embryo to adult, we hypothesized that characterizing ACSL expression and activity during heart development would allow us to better understand the function of the individual ACSL isoforms.
Materials and Methods
Animals
C57BL/6J mice were obtained from Jackson Laboratories. Mice were housed on a 12-h/12-h light/dark cycle with free access to water and Prolab RMH 3000 SP76 rodent diet. Pregnancies were timed by single night mating and visualization of a vaginal plug. Litter sizes were maintained at 5–6 pups per litter. Pups were weaned on postnatal day 21 and then housed in groups of 4. All experiments were conducted in accordance with protocols approved by the University of North Carolina Institutional Animal Care and Use Committee.
Tissue Extraction
Mice were anesthetized by halothane inhalation. Hearts and brains were excised on embryonic day 16 (E16), and on postnatal days 1, 7, 18, and 25. Adult mice were sacrificed at 2 months. Mouse hearts were removed and immediately placed in RNALater (Qiagen) or snap frozen in liquid nitrogen.
Total Membrane Isolation
On ice, hearts were resuspended in 1 ml of Medium I (10 mM Tris HCl, pH 7.4, 0.25 M sucrose) plus 1 mM dithiothreitol (DTT) and protease inhibitors (33.3 μM 4-(2-Aminoethyl)-bezenesulfonylfluoride (AEBSF), 1.033 mM EDTA, 4.3 μM bestain, 0.5 μM E-64, 33.3 μM leupeptin, 33.3 μM aprotinin) (Protease Inhibitor Cocktail for general use, Sigma). Tissue was homogenized on ice with a motor-driven Teflon-glass homogenizer for 15 up-and-down strokes. The homogenate was centrifuged at 1000 x g for 10 min at 5ºC and the supernatant was placed in a new ultracentrifuge tube. The pellet was resuspended in 1 ml of Medium I plus 1 mM DTT and recentrifuged at 1000 x g for 10 min. The resulting supernatant was added to the previous supernatant, and a total membrane fraction was collected by centrifugation at 100,000 x g for 3 h. The membrane pellet was resuspended in 200 μl of Medium I plus 1 mM DTT and homogenized on ice. Protein concentration was determined using the Bicinchonic Acid method (Pierce) using bovine serum albumin (BSA) as the protein standard. The membrane fraction was stored in aliquots at −80°C.
Assay for ACSL activity
The acyl-CoA synthetase assay contained 50 μM [14C]palmitate in 0.5 mM Triton X-100, 1 μM EDTA, 5 mM ATP, 250 μM CoA, 175 mM Tris HCl, pH 7.4, 8 mM MgCl2, and 5 mM DTT in a total volume of 200 μl. In some assays, varying concentrations of unlabeled arachidonate were added to the assay as a competitor. Triacsin C (10 μM in DMSO) or troglitazone (50 μM in DMSO) was added directly to the enzyme reaction mixture during inhibition assays. DMSO alone does not inhibit ACSL activity [31]. For heart samples from postnatal days 1, 7, 18, and 25, the assay was initiated with 2 to 6 μg protein and ACS specific activity was measured after a 5 min incubation with substrates at 37 C. Assays of ACS activity in adult heart contained 1 to 3 μg protein. Because ACS activity was very low at E16, 2 to 6 μg protein was incubated with substrates for 10 min. The reaction was stopped with 2-propanol:heptane:1 M sulfuric acid (80:20:1) and the acyl-CoA products were extracted from heptane with water. Specific activities were proportional with time for 15 min and with protein to 10 μg, allowing measurement of initial rates.
Quantitative real-time –PCR
The abundance of the ACSL mRNA transcripts was determined by quantitative real-time (qRT) PCR. DNA-free total RNA was isolated from tissue with RNeasy Fibrous Tissue Mini Kit (Qiagen). RNA was checked for purity by determining that the A260:A280 ratio was approximately 1.85. 20 ng of RNA and 20 μl of reaction buffer (0.05 μg forward primer, 0.05 μg reverse primer, 20 μM family probe, 5 units reverse transcriptase (Invitrogen), 15 μl Universal PCR MasterMix (Applied Biosystems). Quadruplicate reactions were run for all samples. qRT-PCR was run on an ABI Prism 7700 Sequence Detection System using a TaqMan® PCR protocol (48 ºC for 30 min, 95 ºC 10 min, 95 ºC 15 sec, 60 ºC 1 min for 40 cycles). Primers and probes for QRT-PCR are listed in Table 1. All probes were dual labeled with FAM (6-carboxyfluorescein) as reporter and TAMRA (6-carboxytetramethylrhodamine) as quencher. The ACSL primers were designed to amplify all splice variants [24]. PCR efficiency was calculated from amplification plots [32] and expression of transcripts is presented as a ratio to the level of GAPDH (endogenous control) and relative expression to E16 was determined by REST-MCS [33, 34]. ACSL abundance as a percent of total ACSL transcripts was determined by taking the inverse of the PCR efficiency raised to the delta Ct for each isoform and time point, adding the total for each time point and calculating % of total.
Table I.
Primer and probe sequences used for qRT-PCR.
| position | accession # | |||
|---|---|---|---|---|
| Acsl1 | forward | GCAAGAACAGCTGAAGCCC | 1573a | BC056644 |
| reverse | AGGTGCCATTTGGCAGCCA | 1651a | ||
| probe | CCAATGTCCCCCGTGTGTAACCA | 1606a | ||
| Acsl 3 | forward | CAATTACAGAAGTGTGGGACT | 1454a | NM_028817.2 |
| reverse | CACCTTCCTCCCAGTTCTTT | 1543a | ||
| probe | TACCGGCAGAGTGGGAGCACCA | 1479a | ||
| Acsl 4 | forward | TATGGGCTGACAGAATCATG | 1396a | NM_207625 |
| reverse | CAACTCTTCCAGTAGTGTAG | 1465a | ||
| probe | TAACTTCAGTAACTGTTCCAGCACC | 1417a | ||
| Acsl 5 | forward | GGCCAAACAGAATGCACAG | 1330a | NM_027976 |
| reverse | GGAGTCCCAACATGACCTG | 1403a | ||
| probe | TGTCCAGTCCCCAGGTGATGTAA | 1361a | ||
| Acsl 6 | forward | TGAATGCACAGCTGGGTGTA | 1383a | AY786363 |
| reverse | ATGTGGTTGCAGGGCAGAG | 1466a | ||
| probe | TGTCCAGTCCCCTGGCGTTGTGA | 1406a | ||
| Aco | forward | CAAGGAAGTGGCGTGGAACT | 1533a | BC054727 |
| reverse | CTGCAAAGACCTTAACGGTC | 1602a | ||
| probe | ACTTCTGTCGACCTTGTTCGCGCAA | 1555a | ||
| Mcad | forward | GAGCAGGTTTCAAGATCGCA | 800a | BC013498 |
| reverse | ACTTCGTGGCTTCGTCTAGA | 904a | ||
| probe | CTAGCCCGACAGCGCCAGCT | 855a | ||
| Gapdh | forward | ATGGGTGTGAACCACGAGAA | 271a | DQ403054.1 |
| reverse | GGCATGGACTGTGGTCATGA | 414a | ||
| probe | TGCATCCTGCACCACCAACTGCTTAG | 321a |
Position relative to translation start for indicated accession number
Extraction and measurement of fatty acids
Hearts were homogenized in water and lipids were extracted by the 2-step Bligh-Dyer method [35]. The lower (chloroform) phase was transferred to a clean tube and evaporated to dryness under nitrogen. The residual lipids were saponified and the fatty acids trans-methylated by sequential 1 ml additions of 4.25% NaOH in CHCl3:MeOH (2:1, v/v) and 1N HCl in saline [36]. Samples were mixed vigorously and then centrifuged at 1500 x g for 5 min. The lower phase containing the fatty acid methyl esters was transferred to a clean, dry tube and evaporated to dryness under nitrogen. Fatty acid methyl esters were then resuspended in 50 μl undecane, and analyzed using capillary gas chromatography (GC). Fatty acid methyl esters were analyzed by Fast GC on a Perkin Elmer AutoSystem XL Gas Chromatograph (Shelton, CT), split injection, with helium as the carrier gas. The methyl esters were separated on a capillary column coated with 70% cyanopropyl polysilphenylene – siloxane (10 m x 0.1 mm ID- BPX70 0.2 μm; SGE, Austin, TX); injector 240ºC and detector 280ºC. Data were analyzed with the Perkin Elmer Totalchrom Chromatography Software, version 6.2. Heptadecanoic acid (17:0) was added to the samples as an internal standard to correct for recovery and quantitation. Individual fatty acids were identified by comparing their retention with authentic standards (Nu Chek Prep, Elysian, MN). The fatty acid analysis was performed by the Biochemistry Core of the University of North Carolina Clinical Nutrition Research Unit.
Results
Heart ACS specific activity increases 14-fold after birth
The fetal heart utilizes glucose primarily, whereas fatty acids derived from milk triacylglycerol become the major fuel after birth. Thus, we expected heart ACSL activity to increase immediately after birth. As reported in rat heart, [37], ACSL specific activity measured with [14C]palmitate as substrate was very low on E16 (1.3 nmol/min/mg). In contrast, on P1, mouse heart ACSL specific activity increased 14-fold to19 nmol/min/mg (Fig. 1). These results are consistent with postnatal up-regulation of a program of fatty acid oxidation. ACSL specific activity remained unchanged through postnatal day 25, although with heart growth, total heart ACSL activity increased 6.3-fold. In the adult mouse heart, ACSL specific activity was 6.6-fold higher (125 nmol/min/mg) than that measured in the neonate and 90-fold higher than present at E16.
Figure 1. Heart ACSL activity increased after birth.
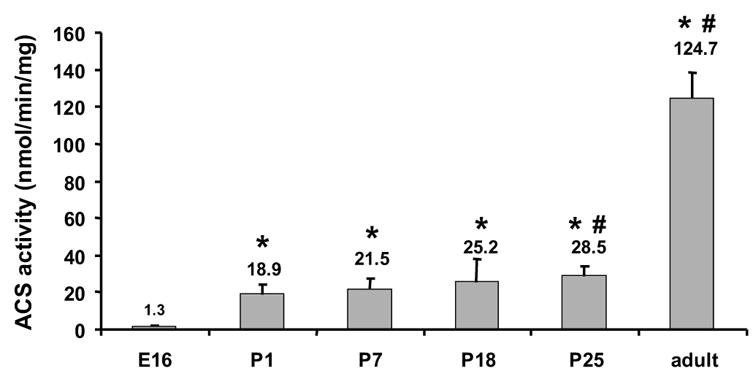
Total membrane protein from hearts at embryonic day 16 (E16), postnatal days 1, 7, 18, 25, and adult were assayed for ACSL activity with [14C]palmitate as substrate as described in Materials and Methods. n = 3 for each time point. * indicates P ≤ 0.0002 vs. E16. # indicates P ≤ 0.02 vs. P1.
Heart ACSL1 mRNA abundance increases after birth
To determine which ACSL isoforms are present in the fetus and which contribute to the postnatal increase in ACS activity, we analyzed the abundance of each of the five Acsl mRNA transcripts. Only the transcript levels for Acsl1 increased after birth (Fig. 2A). The ratio of Acsl1 to Gapdh was 4-fold higher on P1 than on E16 and reached a maximum on P18 at 6-fold higher than E16. The abundance of Acsl1 transcripts was similar to the observed developmental increase in mRNA level of the fatty acid oxidative enzymes MCAD and ACO (Fig. 2C, D), suggesting that Acsl1 message is up-regulated postnatally with the fatty acid oxidation program. As a percent of total Acsl transcripts, the abundance of Acsl1 message rose from 7.5% of total on E16 to 21% on P1 and to 50% on P18 through adult (Fig. 3). The ontological pattern observed for Acsl1 message from E16 to P25 mirrors the developmental increase observed with ACSL activity shown in Fig. 1. Taken together, these data suggest that ACSL1 is the primary isoform in the heart that activates long-chain fatty acids for oxidation.
Figure 2. Heart Acsl1 mRNA increased after birth.
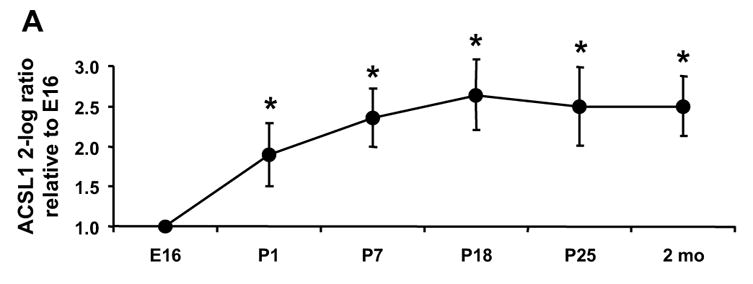
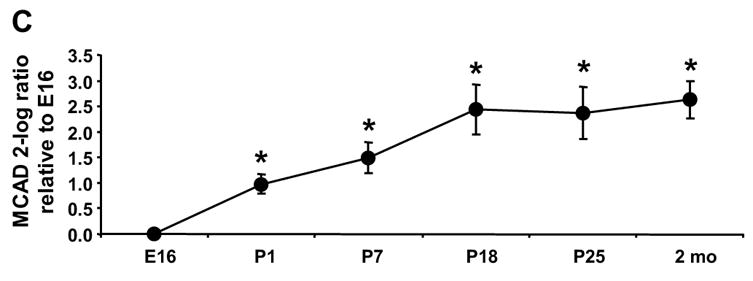
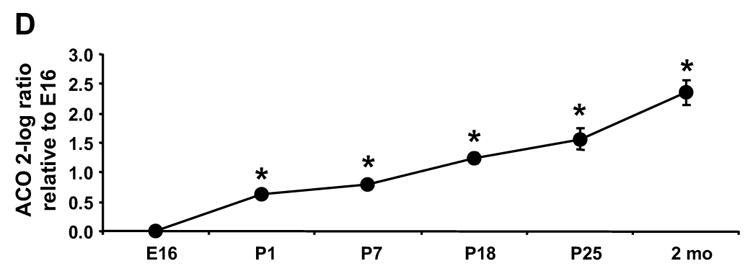
The level of the transcript for each Acsl isoform was determined by qRT-PCR as described in Materials and Methods. The expression of each isoform is presented as the 2-log ratio to Gapdh relative to E16. (A) Acsl1, * indicates P ≤ 0.007 vs. E16 (B) Acsl3, * indicates P ≤ 0.03 vs. E16; (C) Mcad, * indicates P ≤ 0.01 vs. E16; (D) Aco, * indicates P ≤ 0.009 vs. E16; n = 4 for each time point and each gene. Some error bars for standard deviation are within in the symbol.
Figure 3. Acsl3 and 4 transcripts are most abundant in the fetal heart, while Acsl1 and 4 are most abundant in the adult.
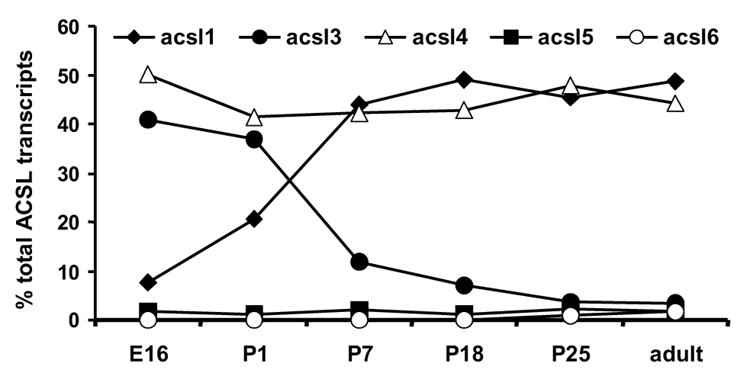
The percent of Acsl1, 3, 4, 5, and 6 mRNAs as a percent of total Acsl transcripts was determined as described in Materials and Methods.
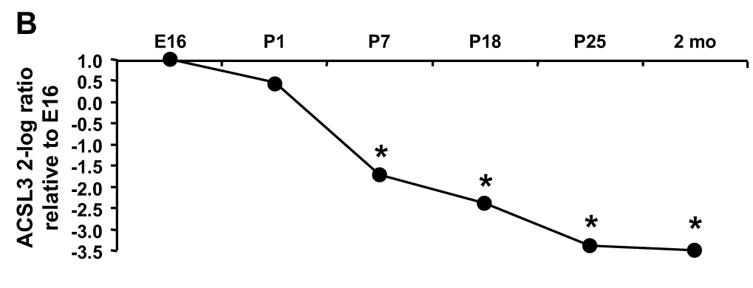
The change in abundance of Acsl3 was the inverse of that observed for Acsl1. The ratio of Acsl3 to Gapdh was highest on E16 and P1 and decreased 70% by P7 (Fig. 2 B). Acsl3 mRNA abundance continued to diminish throughout the neonatal period and was lowest in the adult. Acsl3 message comprised 40% of total Acsl transcripts on E16 and P1, dropped to 12% of total on P7, and only represented 3.5% of transcripts in the adult (Fig. 3). Unlike Acsl1 and 3, the abundance of Acsl4 and 5 mRNA did not change from embryo to neonate to adult (data not shown). Acsl6 mRNA was virtually undetectable until P25 when the percent of Acsl6 rose to 1% of total transcripts (Fig. 3). Although Acsl4 message did not change developmentally, Acsl4 mRNA was abundant, representing 50% of total Acsl transcripts at all time points (Fig. 3). Message for Acsl5 and 6 together comprised less than 5% of total Acsl transcripts (Fig. 3).
Heart ACS activity is inhibited by triacsin C
To determine the contribution of the separate ACSL isoforms to the ACSL specific activity measured, we used the inhibitors triacsin C and troglitazone. Triacsin C inhibits only ACSL 1, 3 and 4 [31, 38] and thiazolidinediones inhibit only ACSL4 [31, 38]. Incubating heart total membranes with 10 μM triacsin C inhibited ACS activity measured with [14C]palmitate as substrate 87 to 97% at all developmental time points (Fig. 4), suggesting that ACSL5 and ACSL6, which are not inhibited by triacsin C, contribute minimally to heart ACS specific activity. This result is consistent with our qRT-PCR analysis which showed that Acsl5 and 6 message levels were very low (Fig. 3). Contrary to the abundance of Acsl4 transcript, treatment of heart membranes with 50 μM troglitazone inhibited ACSL activity less than 5%, indicating that ACSL4 contributed very little to total heart ACSL activity (Fig. 4). It is unclear why troglitazone appeared to increase ACSL specific activity 10–20% in the P18, P25 and adult samples. Taken together, the mRNA expression data and inhibition studies strongly suggest that ACSL3 may provide the majority of fetal and immediate postnatal ACSL activity and that ACSL1 is the major contributor to postnatal and adult activity.
Figure 4. Triacsin C inhibited ACSL activity at all developmental time points.
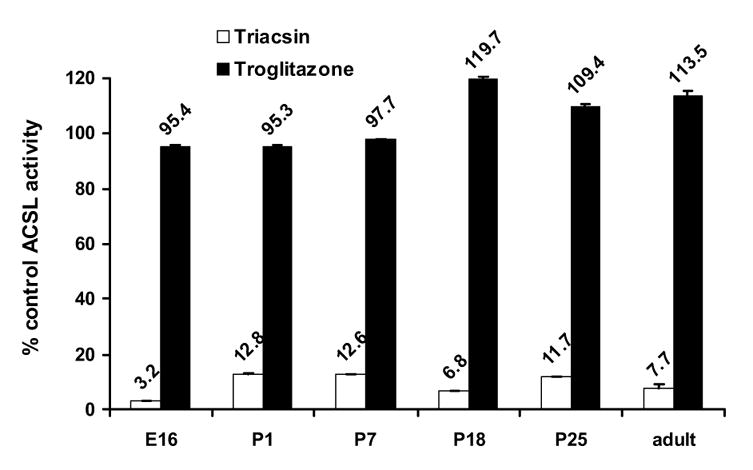
Total membrane protein from hearts at embryonic day 16 (E16) and postnatal days 1, 7, 18, 25, and adult were assayed for ACSL activity in the presence of 10 μM triacsin C (❏) or 50 μM troglitazone (■) as described in Materials and Methods. Data are presented as a percent of control ACSL activity assayed in the presence of DMSO. n = 3 for each time point.
Arachidonate (20:4) inhibits the synthesis of palmitoyl-CoA in heart membranes from E16
Because a significant number of Acsl3 transcripts were most apparent at E16 and P1, we assessed the relative contribution of the ACSL3 isoform to total ACSL specific activity. When assayed with [14C]palmitate, purified ACSL4 is inhibited 50% by < 1 μM 20:4, whereas arachidonate is a poor substrate for ACSL3 and 50 μM arachidonate is required to inhibit ACSL3 50% [38]. Thus, one would expect competitive inhibition at low concentrations of 20:4 in membranes that contained relatively more ACSL4 activity, however, arachidonate had no effect on palmitoyl-CoA synthetase activity in heart membranes from adult mice (Fig. 5). In P1 samples, arachidonate inhibited ACSL activity a modest 13%. In E16 heart membranes, arachidonate inhibited palmitoyl-CoA synthesis as much as 44%, but inhibition required high concentrations of 20:4. Further, because troglitazone had little effect on ACSL activity (Fig. 4), the data suggest that ACSL3 is the major activity competed for by arachidonate on E16 and that ACSL3 contributes considerably to the activation of long-chain fatty acids in the embryonic heart.
Figure 5. Arachidonic acid inhibited palmitoyl-synthase activity at embryonic day 16.
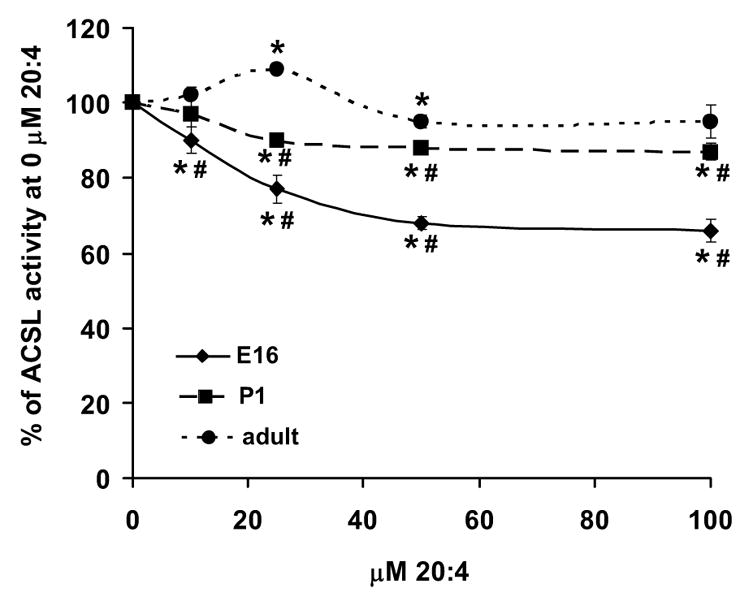
Total membrane protein from hearts at embryonic day 16 (♦), postnatal day 1 (■), and adult (●) were assayed for [14C]palmitoyl synthase activity in the presence of increasing concentrations of arachidonic acid (20:4) as described in Materials and Methods. Data are presented as the percent of ACS activity at 0 μM 20:4. n = 3 for each point. Some error bars for standard deviation are within the symbol. * indicates P ≤ 0.05 vs. 0 μM 20:4. # indicates P ≤ 0.04 vs. adult.
Fetal heart phospholipids are enriched in myristate (14:0) and arachidonate (20:4)
Although each of the ACSL isoforms can use fatty acids of 12–22 carbons, the isoforms differ in their preferred fatty acid substrates. Because ACSL3 displays the highest activity with laurate (12:0), myristate (14:0), arachidonate (20:4), and eicosapentaenoic acid (20:5) [39], we determined the FA composition of heart phospholipids to confirm that ACSL3 contributes functionally to heart FA metabolism.
The most abundant fatty acids in adult heart phospholipids were 22:6, 18:0, 18:2, 16:0, 18:1, 20:4, and 22:5 (presented in descending order of abundance, Table 2), consistent with previous reports [40]. In P1 hearts, however, the most prevalent fatty acids were 18:1>16:0>18:0, 18:2>20:4>22:6>22:5. In hearts from day 16 embryos, 16:0 was most abundant followed by 18:0, 20:4, 18:1, 18:2, 22:6, and 22:5. These differences in the distribution of fatty acids between heart lipids isolated from the embryo and adult are consistent with the hypothesis that different ACSL isoforms activate fatty acids for phospholipid synthesis during each of these times, but may also reflect, in part, the availability of the different fatty acid species.
Table II.
Fatty acid composition of heart phospholipids
| Fatty Acid | E16 | P1 | Adult |
|---|---|---|---|
| 14:0 | 0.91 | 0.59 | 0.27± 0.08 |
| 14:1 | 0.00 | 0.04 | 0.02 ± 0.02 |
| 16:0 | 23.12 | 17.96 | 13.90 ± 0.99 |
| 16:1 | 2.07 | 1.43 | 0.46 ± 0.06 |
| 18:0 | 16.10 | 13.11 | 17.94 ± 0.27 |
| 18:1 | 14.93 | 23.54 | 9.68 ± 0.44 |
| 18:2 | 10.91 | 12.17 | 14.94 ± 1.24 |
| 20:0 | 0.46 | 0.43 | 0.27 ± 0.17 |
| 18:3 | 0.25 | 0.20 | 0.12 ± 0.01 |
| 20:1 | 0.19 | 0.2 6 | 0.36 ± 0.01 |
| 20:2 | 1.04 | 1.09 | 0.60 ± 0.06 |
| 20:3 | 1.15 | 0.99 | 0.67 ± 0.11 |
| 20:4 | 15.84 | 10.07 | 6.70 ± 0.19 |
| 20:3 | 0.63 | 0.47 | 0.08 ± 0.14 |
| 22:0 | 0.48 | 0.64 | 0.03 ± 0.05 |
| 22:1 | 0.58 | 0.35 | 0.25 ± 0.04 |
| 20:5 | 0.15 | 0.08 | 0.30 ± 0.07 |
| 24:0 | 0.15 | 0.14 | 0.00 ± 0.01 |
| 24:1 | 0.09 | 0.04 | 0.07 ± 0.01 |
| 22:5 | 1.99 | 2.77 | 1.71 ± 0.14 |
| 22:6 | 8.95 | 9.02 | 31.63 ± 3.29 |
Heart lipids were extracted and fatty acid composition was determined as described in Materials and Methods. Data are presented as percent of total fatty acids recovered. For E16 and P1, the values presented are an average from a pool of 8–10 hearts. For the adult, n=3.
Of the major fatty acids, docosahexaenoic acid (22:6) was 3-fold higher in adult mouse heart than in E16 and P1 (Table 2). High levels of 22:6 have been previously reported in rodent heart and muscle phospholipids [40, 41]. In other tissues, such as liver and adipose, 22:6 is a minor constituent of membrane phospholipids [41, 42]. Our data do not support a role for an ACSL isoform in 22:6 metabolism because only the expression of Acsl1 transcripts is increased in the adult, and ACSL1 does not thioesterify 22:6 efficiently [43]. Fatty acid transport proteins-1 and -6 are present in adult heart [29] and may contribute to increased 22:6 presence.
In contrast, arachidonic acid (20:4) was 2- to 2.4-fold higher in P1 and E16 heart lipids, respectively, compared to adult (Fig. 6). Although all ACSL isoforms can activate 20:4, it is a preferred fatty acid for ACSL3, 4, and 6 [20, 38, 39, 43, 44]. Unique to ACSL3 is its high activity with 12- and 14-carbon fatty acid substrates [39]. To differentiate between contributions from ACSL3, 4, and 6, we examined the prevalence of myristate (14:0) and 20:4 in E16, P1, and adult hearts. Interestingly, we observed that both 14:0 and 20:4 were 3-fold higher in E16 heart phospholipids than in those from adult hearts (Fig. 6). These data are consistent with the high expression levels of Acsl3 mRNA in the day 16 embryo, and suggest that the ACSL3 isoform contributes to the unique fatty acid profile of embryonic heart lipids.
Figure 6. Phospholipids were enriched in myristate and arachidonate at embryonic day 16.
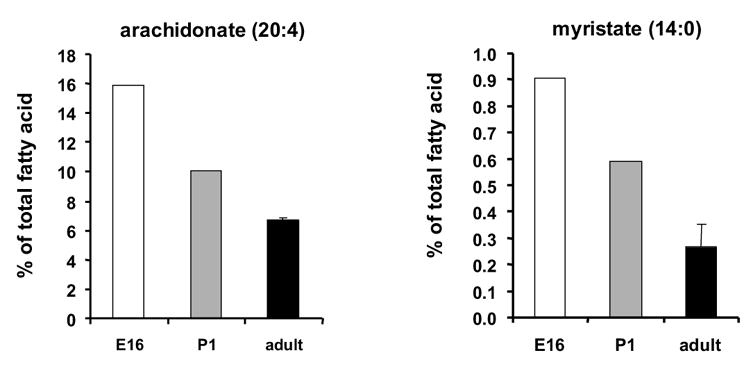
The fatty acid composition of total phospholipids from hearts at embryonic day 16 (E16 ❏), postnatal day 1 ( ), and adult (■) was determined as described in Materials and Methods. For E16 and P1, the value presented is an average of a pool of 8–10 hearts. n = 3 for the adult.
Discussion
Long-chain acyl-CoA synthetases are required to activate fatty acids before their entry into all degradative or synthetic pathways with the exception of eicosanoid synthesis. Studies of the five cloned ACSL isoforms in fibroblasts, liver cell lines, and neuronal cell lines, have suggested that each ACSL isoform channels fatty acids toward separate metabolic fates. Studies using triacsin C, a fungal derived competitive inhibitor of ACSL1, 3, and 4 [31, 38], showed that 5 μM triacsin C inhibited de novo synthesis of triacylglycerol 93% and phospholipids 83% in human fibroblasts, but did not impair phospholipid reacylation [45]. In primary rat hepatocytes triacsin C inhibited de novo TAG synthesis 73%, but inhibited β-oxidation only 33% [46]. Similarly, troglitazone, an inhibitor of ACSL4 activity, decreased the incorporation of oleate into triacylglycerol and oxidative products 50% and 20%, respectively, but did not affect the incorporation of oleate into phospholipid [46, 47]. Also in hepatocytes, overexpression of ACSL1 increased oleate incorporation into diacylglycerol and phospholipids, and decreased incorporation into cholesterol esters and secreted triacylglycerol [48]. In the rat hepatoma cell line McArdle –RH 7777, overexpression of ACSL5 increased de novo synthesis of TAG, but did not alter fatty acid oxidation [49]. In addition, ACSL1 overexpression in PC12 neuronal cells increased the internalization of oleate by 55%, and increased arachidonate and docosahexanoic acid uptake by 25% [50]. In contrast, overexpression of ACSL6 increased internalization of oleate by 90%, arachidonate by 115%, and docosahexanoic acid by 70% [50]. Taken together these data support the hypothesis that ACSL isoforms are functionally distinct. Although all five ACSL isoforms are expressed in the heart, little is known about their individual functional roles, so we examined Acsl mRNA expression and activity in the developing mouse heart in order to determine the isoforms most critical for FA oxidation.
During the development of the embryo to neonate to the 2-month old adult mouse, many ontological changes occur. In heart, the most pronounced change is the switch in substrate for ATP generation. The fetal heart uses lactate and glucose, but immediately after birth glycogen and glucose oxidation provide ATP, and subsequently fatty acids provide the primary fuel [2]. Concomitant with the substrate switch, the pathway of fatty acid oxidation is up-regulated via PPARα. PPARα mRNA increases dramatically in mouse hearts on P1, as compared to embryonic day 16.5 [10]. Transcript levels decrease on day 7, but in the adult equaled the amount observed on day 1. The expression of the fatty acid oxidation enzymes MCAD and CPT-1, followed a similar pattern.
We observed a 14-fold increase in ACSL specific activity between E16 and P1 and a further 5-fold increase in the 2 month old adult (Fig. 1). Of the five ACSL isoforms, only the transcripts for Acsl1 increased after birth (Fig. 2A). Acsl1 mRNA increased 3.7-fold on P1 compared to E16, and reached a maximal 5.5-fold increase on P18. This pattern of Acsl1 mRNA expression matches the developmental pattern of expression observed for transcripts of Mcad and Aco (Fig. 2C, D), genes required for fatty acid oxidation and regulated by PPARα [6–10]. The Acsl1 promoter contains a PPAR response element [51, 52], and Acsl1 transcript abundance is diminished in the absence of PPARα [26, 27]. A study of rat cardiomyocytes demonstrated that fatty acid and PPARα agonists are able to upregulate Acsl1 [25]. Our data are consistent with Acsl1 being a PPARα regulated gene during heart development. Because the expression of the transcripts for the other Acsl isoforms decreased or remained unchanged after birth compared to the fetus (Fig. 2B), it appears that transcription of Acsl3, 4, 5, and 6 is not up-regulated by PPARα.
The increase in ACS specific activity we observed in developing mouse heart mirrored the pattern observed for Acsl1 mRNA expression (Figs. 1 and 2A). However, the magnitude of the increase in ACSL activity was 3.8-fold greater than the increase in Acsl1 mRNA. Discordance between mRNA and protein has been previously reported for ACSL isoforms [24]. The post-natal increase in heart ACSL activity is similar to the increase in post-natal palmitoyl-CoA synthetase activity previously reported in both heart and liver [37]. Our studies with the inhibitors triacsin C and troglitazone were consistent with ACSL1 being the predominant isoform contributing to ACSL activity in postnatal heart (Fig. 4). Taken together, these data lead us to conclude that after birth ACSL1 becomes the major isoform providing activated fatty acids for oxidation.
Our results show that in the adult heart the number of Acsl3, 5, and 6 transcripts was low relative to Acsl1 and 4 (Fig. 3), consistent with previous reports of Acsl expression in adult heart [24, 25]. Acsl3 transcripts were 16-fold more abundant at E16 than in the adult (Fig. 2 B). Although ACSL activity was low on E16, arachidonate inhibited palmitoyl-CoA synthetase activity (Fig. 5), indicating that the acyl-CoA synthetase(s) catalyzing the reaction preferred the polyunsaturated fatty acid compared to 16:0. Both ACSL3 and ACSL4 prefer 20:4 over 16:0 [38], but because 50 μM troglitazone minimally inhibited ACSL activity at all developmental time points (Fig. 4), we concluded that ACSL4 did not contribute significantly to heart ACSL activity.
We propose that ACSL3 plays a significant role in the embryonic heart. Phospholipids from hearts on E16 are enriched in myristate and arachidonate (Fig. 6), fatty acids uniquely preferred by ACSL3 [39], suggesting that ACSL3 contributes functionally to heart fatty acid metabolism in the fetus. Strengthening the argument that ASCL3 is important for fetal heart development are the presence of DNA motifs for the cardiogenic transcription factors, GATA and Nkx2.5, in the 5′ flanking region of Acsl3 [53, 54]. Although transactivation assays with the ACSL3 5′ flanking region demonstrated that the region containing the GATA and Nkx2.5 motifs contains a negative promoter element [53], the cell line used did not express the cardiac GATA-4 and Nkx2.5 transcription factors. Further studies on GATA-4 and Nkx2.5 regulation of ACSL3 transcription in rat heart will be needed to determine whether these transcription factors contribute to high ACSL3 transcript levels in the embryo.
Although ACSL4 transcript is abundant in mouse heart (Fig. 3), we do not believe that ACSL4 contributes significantly to total ACSL activity because troglitazone, which inhibits only ACSL4 [31, 38], did not inhibit ACS specific activity at any developmental time point (Fig. 4). ACSL4 may play a role in heart lipoprotein synthesis as has been proposed for ACSL4 in liver [12, 13, 21, 22]. Heart lipoprotein synthesis is increased under conditions of diabetes [55]. If ACSL4 is important for lipoprotein assembly, we would expect ACSL4 to be up-regulated under the same conditions.
In summary, our data support the hypothesis that each ACSL isoform has a specific role in the channeling of fatty acids toward distinct metabolic fates. In addition, some of these roles may be further modulated by subcellular location and/or tissue specificity. For example, we found that the expression of ACSL1 mRNA and activity is increased concomitant with the upregulation of the FA oxidation program, suggesting that ACSL1 is the major isoform responsible for activating long-chain fatty acids for myocardial β-oxidation after birth. This result is in contrast to the function of ACSL1 in liver, where overexpression of ACSL1 in hepatocytes does not alter oleate incorporation in β-oxidation products, but does increase oleate incorporation into diacylglycerol and phospholipids [48]. Further studies of the function of ACSL isoforms in different tissues will be needed to complete our understanding of the functional role of ACSL in various fatty acid utilizing pathways.
Acknowledgments
This work was supported by DK59931 (TML), DK59935 (RAC), and DK56350 to the University of North Carolina Clinical Nutrition Research Unit from the National Institutes of Health, by 230323N from the American Heart Association (TML), and AgriFunGen (ACN).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Holsinger VH. Handbook of Milk Composition. Vol. 9. Academic Press; San Diego: 1995. [Google Scholar]
- 2.Makinde AO, Kantor PF, Lopaschuk GD. Maturation of fatty acid and carbohydrate metabolism in the newborn heart. Mol Cell Biochem. 1998;188:49–56. [PubMed] [Google Scholar]
- 3.Werner JC, Whitman V, Fripp RR, Schuler HG, Musselman J, Sham RL. Fatty acid and glucose utilization in isolated, working fetal pig hearts. Am J Physiol. 1983;245:E19–23. doi: 10.1152/ajpendo.1983.245.1.E19. [DOI] [PubMed] [Google Scholar]
- 4.Fisher DJ, Heymann MA, Rudolph AM. Myocardial oxygen and carbohydrate consumption in fetal lambs in utero and in adult sheep. Am J Physiol. 1980;238:H399–405. doi: 10.1152/ajpheart.1980.238.3.H399. [DOI] [PubMed] [Google Scholar]
- 5.Grynberg A, Demaison L. Fatty acid oxidation in the heart. J Cardiovasc Pharmacol. 1996;28(suppl 1):S11–S17. doi: 10.1097/00005344-199600003-00003. [DOI] [PubMed] [Google Scholar]
- 6.Hainline BE, Kahlenbeck DJ, Grant J, Strauss AW. Tissue specific and developmental expression of rat long-and medium-chain acyl-CoA dehydrogenases. Biochim Biophys Acta. 1993;1216:460–8. doi: 10.1016/0167-4781(93)90015-6. [DOI] [PubMed] [Google Scholar]
- 7.Nagao M, Parimoo B, Tanaka K. Developmental, nutritional, and hormonal regulation of tissue-specific expression of the genes encoding various acyl-CoA dehydrogenases and alpha-subunit of electron transfer flavoprotein in rat. J Biol Chem. 1993;268:24114–24. [PubMed] [Google Scholar]
- 8.Kelly DP, Gordon JI, Alpers R, Strauss AW. The tissue-specific expression and developmental regulation of two nuclear genes encoding rat mitochondrial proteins. Medium chain acyl-CoA dehydrogenase and mitochondrial malate dehydrogenase. J Biol Chem. 1989;264:18921–5. [PubMed] [Google Scholar]
- 9.Disch DL, Rader TA, Cresci S, Leone TC, Barger PM, Vega R, Wood PA, Kelly DP. Transcriptional control of a nuclear gene encoding a mitochondrial fatty acid oxidation enzyme in transgenic mice: role for nuclear receptors in cardiac and brown adipose expression. Mol Cell Biol. 1996;16:4043–51. doi: 10.1128/mcb.16.8.4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lehman JJ, Barger PM, Kovacs A, Saffitz JE, Medeiros DM, Kelly DP. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J Clin Invest. 2000;106:847–56. doi: 10.1172/JCI10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kornberg A, Pricer WEJ. Enzymatic synthesis of the conenzyme A derivatives of long-chain fatty acids. J Biol Chem. 1953;204:329–343. [PubMed] [Google Scholar]
- 12.Coleman RA, Lewin TM, Muoio DM. Physiological and nutritional regulation of enzymes of triacylglycerol synthesis. Ann Rev Nutr. 2000;20:77–103. doi: 10.1146/annurev.nutr.20.1.77. [DOI] [PubMed] [Google Scholar]
- 13.Coleman RA, Lewin TM, Van Horn CG, Gonzalez-Baro MR. Do long-chain acyl-CoA synthetases regulate fatty acid entry into synthetic versus degradative pathways? J Nutr. 2002;132:2123–6. doi: 10.1093/jn/132.8.2123. [DOI] [PubMed] [Google Scholar]
- 14.Groot PH, Scholte HR, Hulsmann WC. Fatty acid activation: specificity, localization, and function. Adv Lipid Res. 1976;14:75–126. doi: 10.1016/b978-0-12-024914-5.50009-7. [DOI] [PubMed] [Google Scholar]
- 15.Faergman NJ, Knudsen J. Role of long-chain fatty acyl-CoA esters in the regulation of metabolism and in cell signalling. Biochem J. 1997;323:1–12. doi: 10.1042/bj3230001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Prentki M, Corkey BE. Are the β-Cell signaling molecules malonyl-CoA and cytosolic long-chain acyl-CoA implicated in multiple tissue defects of obesity and NIDDM. Diabetes. 1996;45:273–283. doi: 10.2337/diab.45.3.273. [DOI] [PubMed] [Google Scholar]
- 17.Schnurr JA, Shockey JM, de Boer GJ, Browse JA. Fatty acid export from the chloroplast. Molecular characterization of a major plastidial acyl-coenzyme A synthetase from Arabidopsis. Plant Physiol. 2002;129:1700–9. doi: 10.1104/pp.003251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stoveken T, Kalscheuer R, Malkus U, Reichelt R, Steinbuchel A. The wax ester synthase/acyl coenzyme A:diacylglycerol acyltransferase from Acinetobacter sp. strain ADP1: characterization of a novel type of acyltransferase. J Bacteriol. 2005;187:1369–76. doi: 10.1128/JB.187.4.1369-1376.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oikawa E, Iijima H, Suzuki T, Sasano H, Sato H, Kamataki A, Nagura H, Kang MJ, Fujino T, Suzuki H, Yamamoto TT. A novel acyl-CoA synthetase, ACS5, expressed in intestinal epithelial cells and proliferating preadipocytes. J Biochem. 1998;124:679–685. doi: 10.1093/oxfordjournals.jbchem.a022165. [DOI] [PubMed] [Google Scholar]
- 20.Kang MJ, Fujino T, Sasano H, Minekura H, Yabuki N, Nagura H, Iijima H, Yamamoto TT. A novel arachidonate-preferring acyl-CoA synthetase is present in steroidogenic cells of the rat adrenal, ovary, and testis. Proc Natl Acad Sci. 1997;94:2880–2884. doi: 10.1073/pnas.94.7.2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lewin TM, Kim JH, Granger DA, Vance JE, Coleman RA. Acyl-CoA synthetase isoforms 1, 4, and 5 are present in different subcellular membranes in rat liver and can be inhibited independently. J Biol Chem. 2001;276:24674–24679. doi: 10.1074/jbc.M102036200. [DOI] [PubMed] [Google Scholar]
- 22.Lewin TM, Van Horn CG, Krisans SK, Coleman RA. Rat liver acyl-CoA synthetase 4 is a peripheral-membrane protein located in two distinct subcellular organelles, peroxisomes, and mitochondrial-associated membrane. Arch Biochem Biophys. 2002;404:263–70. doi: 10.1016/s0003-9861(02)00247-3. [DOI] [PubMed] [Google Scholar]
- 23.Cao Y, Murphy KJ, McIntyre TM, Zimmerman GA, Prescott SM. Expression of fatty acid-CoA ligase 4 during development and in brain. FEBS Lett. 2000;467:263–267. doi: 10.1016/s0014-5793(00)01159-5. [DOI] [PubMed] [Google Scholar]
- 24.Mashek DG, Li LO, Coleman RA. Rat long-chain acyl-CoA synthetase mRNA, protein, and activity vary in tissue distribution and in response to diet. J Lipid Res. 2006;47:2004–10. doi: 10.1194/jlr.M600150-JLR200. [DOI] [PubMed] [Google Scholar]
- 25.Durgan DJ, Smith JK, Hotze MA, Egbejimi O, Cuthbert KD, Zaha VG, Dyck JR, Abel ED, Young ME. Distinct transcriptional regulation of long-chain acyl-CoA synthetase isoforms and cytosolic thioesterase 1 in the rodent heart by fatty acids and insulin. Am J Physiol Heart Circ Physiol. 2006;290:H2480–97. doi: 10.1152/ajpheart.01344.2005. [DOI] [PubMed] [Google Scholar]
- 26.Aoyama T, Peters JM, Iritani N, Nakajima T, Furihata K, Hashimoto T, Gonzalez FJ. Altered constitutive expression of fatty acid-metabolizing enzymes in mice lacking the peroxisome proliferator-activated receptor alpha (PPARalpha) J Biol Chem. 1998;273:5678–5684. doi: 10.1074/jbc.273.10.5678. [DOI] [PubMed] [Google Scholar]
- 27.Djouadi F, Brandt JM, Weinheimer CJ, Leone TC, Gonzalez FJ, Kelly DP. The role of the peroxisome proliferator-activated receptor alpha (PPAR alpha) in the control of cardiac lipid metabolism. Prostaglandins Leukot Essent Fatty Acids. 1999;60:339–43. doi: 10.1016/s0952-3278(99)80009-x. [DOI] [PubMed] [Google Scholar]
- 28.Kliewer SA, Xu HE, Lambert MH, Willson TM. Peroxisome proliferator-activated receptors: from genes to physiology. Recent Prog Horm Res. 2001;56:239–63. doi: 10.1210/rp.56.1.239. [DOI] [PubMed] [Google Scholar]
- 29.Loichot C, Jesel L, Tesse A, Tabernero A, Schoonjans K, Roul G, Carpusca I, Auwerx J, Andriantsitohaina R. Deletion of peroxisime proliferator-activated receptor-{alpha} Iinduces an alteration of cardiac functions. Am J Physiol Heart Circ Physiol. 2006 doi: 10.1152/ajpheart.01065.2004. [DOI] [PubMed] [Google Scholar]
- 30.Chiu HC, Kovacs A, Ford DA, Hsu FF, Garcia R, Herrero P, Saffitz JE, Schaffer JE. A novel mouse model of lipotoxic cardiomyopathy. J Clin Invest. 2001;107:813–822. doi: 10.1172/JCI10947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim JH, Lewin TM, Coleman RA. Selective inhibition of recombinant rat acyl-CoA synthetases 1, 4, and 5 by triacsin C and thiazolidinediones. J Biol Chem. 2001;276:24667–24673. doi: 10.1074/jbc.M010793200. [DOI] [PubMed] [Google Scholar]
- 32.Ramakers C, Ruijter JM, Deprez RH, Moorman AF. Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neurosci Lett. 2003;339:62–6. doi: 10.1016/s0304-3940(02)01423-4. [DOI] [PubMed] [Google Scholar]
- 33.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pfaffl MW, Horgan GW, Dempfle L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002;30:e36. doi: 10.1093/nar/30.9.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 36.Tacconi M, Wurtman RJ. Rat brain phosphatidyl-N,N-dimethylethanolamine is rich in polyunsaturated fatty acids. J Neurochem. 1985;45:805–9. doi: 10.1111/j.1471-4159.1985.tb04064.x. [DOI] [PubMed] [Google Scholar]
- 37.Warshaw JB. Cellular energy metabolism during fetal development. IV. Fatty acid activation, acyl transfer and fatty acid oxidation during development of the chick and rat. Dev Biol. 1972;28:537–44. doi: 10.1016/0012-1606(72)90001-2. [DOI] [PubMed] [Google Scholar]
- 38.Van Horn CG, Caviglia JM, Li LO, Wang S, Granger DA, Coleman RA. Characterization of recombinant long-chain rat acyl-CoA synthetase isoforms 3 and 6: identification of a novel variant of isoform 6. Biochemistry. 2005;44:1635–42. doi: 10.1021/bi047721l. [DOI] [PubMed] [Google Scholar]
- 39.Fujino T, Kang MJ, Suzuki H, Iijima H, Yamamoto T. Molecular characterization and expression of rat acyl-CoA synthetase 3. J Biol Chem. 1996;271:16748–16752. doi: 10.1074/jbc.271.28.16748. [DOI] [PubMed] [Google Scholar]
- 40.Pamplona R, Portero-Otin M, Ruiz C, Gredilla R, Herrero A, Barja G. Double bond content of phospholipids and lipid peroxidation negatively correlate with maximum longevity in the heart of mammals. Mech Ageing and Dev. 1999;112:169–183. doi: 10.1016/s0047-6374(99)00045-7. [DOI] [PubMed] [Google Scholar]
- 41.Owens K, Hughes BP. Lipids of dystrophic and normal mouse muscle: whole tissue and particulate fractions. J Lipid Res. 1970;11:486–95. [PubMed] [Google Scholar]
- 42.Christiansen EN, Lund JS, Rortveit T, Rustan AC. Effect of dietary n-3 and n-6 fatty acids on fatty acid desaturation in rat liver. Biochim Biophys Acta. 1991;1082:57–62. doi: 10.1016/0005-2760(91)90299-w. [DOI] [PubMed] [Google Scholar]
- 43.Iijima H, Fujino T, Minekura H, Suzuki H, Kang MJ, Yamamoto T. Biochemical studies of two rat acyl-CoA synthetases, ACS1 and ACS2. Eur J Biochem. 1996;242:186–190. doi: 10.1111/j.1432-1033.1996.0186r.x. [DOI] [PubMed] [Google Scholar]
- 44.Fujino T, Yamamoto T. Cloning and functional expression of a novel long-chain acyl-CoA synthetase expressed in brain. J Biochem. 1992;111:197–203. doi: 10.1093/oxfordjournals.jbchem.a123737. [DOI] [PubMed] [Google Scholar]
- 45.Igal RA, Wang P, Coleman RA. Triacsin C blocks de novo synthesis of glycerolipids and cholesterol esters but not recycling of fatty acid into phospholipid: evidence for functionally separate pools of acyl-CoA. Biochem J. 1997;324:529–534. doi: 10.1042/bj3240529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Muoio DM, Lewin TM, Weidmar P, Coleman RA. Acyl-CoAs are functionally channeled in liver: Potential role of acyl-CoA synthetase. Am J Physiol. 2000;279:E1366–E1373. doi: 10.1152/ajpendo.2000.279.6.E1366. [DOI] [PubMed] [Google Scholar]
- 47.Fulgencio JP, Kohl C, Girard J, Pegorier JP. Troglitazone inhibits fatty acid oxidation and esterification, and gluconeogenesis in isolated hepatocytes from starved rats. Diabetes. 1996;45:1556–1562. doi: 10.2337/diab.45.11.1556. [DOI] [PubMed] [Google Scholar]
- 48.Li LO, Mashek DG, Jie A, Doughman SD, Newgard CB, Coleman RA. Overexpression of rat longchain acyl-CoA synthetase-1 alters fatty acid metabolism in rat primary hepatocytes. J Biol Chem. 2006 doi: 10.1074/jbc.M604427200. [DOI] [PubMed] [Google Scholar]
- 49.Mashek DG, McKenzie MA, Van Horn CG, Coleman RA. Rat long chain acyl-CoA synthetase 5 increases fatty acid uptake and partitioning to cellular triacylglycerol in McArdle-RH7777 cells. J Biol Chem. 2006;281:945–50. doi: 10.1074/jbc.M507646200. [DOI] [PubMed] [Google Scholar]
- 50.Marszalek JR, Kitidis C, Dararutana A, Lodish HF. Acyl-CoA synthetase 2 overexpression enhances fatty acid internalization and neurite outgrowth. J Biol Chem. 2004;279:23882–91. doi: 10.1074/jbc.M313460200. [DOI] [PubMed] [Google Scholar]
- 51.Suzuki H, Watanabe M, Fujino T, Yamamoto T. Multiple promoters in rat acyl-CoA synthetase gene mediate differential expression of multiple transcripts with 5′-end heterogeneity. J Biol Chem. 1995;270:9676–9682. doi: 10.1074/jbc.270.16.9676. [DOI] [PubMed] [Google Scholar]
- 52.Martin G, Schoonjans K, Lefebvre AM, Staels B, Auwerx J. Coordinate regulation of the expression of the fatty acid transport protein and acyl-CoA synthetase genes by PPARα and PPARγ activators. J Biol Chem. 1997;272:28210–28217. doi: 10.1074/jbc.272.45.28210. [DOI] [PubMed] [Google Scholar]
- 53.Minekura H, Fujino T, Kang MJ, Fujita T, Endo Y, Yamamoto TT. Human acyl-coenzyme A synthetase 3 cDNA and localization of its gene (ACS3) to chromosome band 2q34–q35. Genomics. 1997;42:180–181. doi: 10.1006/geno.1997.4710. [DOI] [PubMed] [Google Scholar]
- 54.Charron F, Nemer M. GATA transcription factors and cardiac development. Semin Cell Dev Biol. 1999;10:85–91. doi: 10.1006/scdb.1998.0281. [DOI] [PubMed] [Google Scholar]
- 55.Nielsen LB, Bartels ED, Bollano E. Overexpression of apolipoprotein B in the heart impedes cardiac triglyceride accumulation and development of cardiac dysfunction in diabetic mice. J Biol Chem. 2002;277:27014–20. doi: 10.1074/jbc.M203458200. [DOI] [PubMed] [Google Scholar]