

Abstract
During the resolution phase of inflammation, the ‘corpses’ of apoptotic leukocytes are gradually cleared by macrophages. Here we report that during the resolution of peritonitis, the CCR5 chemokine receptor ligands CCL3 and CCL5 persisted in CCR5-deficient mice. CCR5 expression on apoptotic neutrophils and activated apoptotic T cells sequestered and effectively cleared CCL3 and CCL5 from sites of inflammation. CCR5 expression on late apoptotic human polymorphonuclear cells was downregulated by proinflammatory stimuli, including tumor necrosis factor, and was upregulated by ‘proresolution’ lipid mediators, including lipoxin A4, resolvin E1 and protectin D1. Our results suggest that CCR5+ apoptotic leukocytes act as ‘terminators’ of chemokine signaling during the resolution of inflammation.
The resolution of inflammation requires the elimination of local chemical mediators1,2. Resolution is an active rather than a passive process3,4. Therefore, massive apoptosis of the leukocytes that elicit this response is one of the hallmarks of resolution5–7. Those apoptotic cells are gradually cleared by macrophages and other phagocytic cells8–10. Apoptotic cell engulfment by phagocytes is mediated by the interaction between molecules on the surfaces of apoptotic cells and their corresponding receptors on the surfaces of phagocytes, including thrombospondin and CD36 (ref. 11), milk fat globule–epidermal growth factor 8 and αvβ3 integrin9, and others6. Apoptotic cells also serve as ‘resolution cues’ for macrophages, as their engulfment is not accompanied by the release of proinflammatory mediators characteristic of macrophage activation12. Instead, apoptotic cells induce the production of transforming growth factor-β (TGF-β) and interleukin 10 (IL-10)10,13, cytokines that can promote resolution and wound repair. Failure to clear those apoptotic leukocytes can lead to exposure of their nuclear contents, resulting in the development of an autoimmune lupus-like syndrome and accelerated atherosclerosis14,15. Lupus-prone mice have impaired clearance of apoptotic polymorphonuclear cells (PMNs) after thioglycolate injection16. Hence, defects in clearance mechanisms and failure to resolve tissue inflammation in a timely way can underlie disease phenotypes.
During the course of acute inflammation, a temporal switch in the profile of inflammatory lipid-derived mediators occurs; although proinflammatory prostaglandins and leukotrienes dominate in the initial phase, they give way to the ‘proresolution’ lipoxins in the resolution phase3,4,17. In addition to lipoxins, resolvins and protectins are families of lipid mediators derived from ω-3-polyunsaturated fatty acids that are generated during resolution and exert potent antiinflammatory actions4,18,19. TGF-β is another mediator produced in response to apoptotic cells and lipoxin A4 during resolution20 and suppresses inflammation and autoimmune disorders in animal models21.
Although chemokines were initially considered only as chemoattractive cytokines that direct leukocytes to inflammation sites, their considerable involvement in angiogenesis, hematopoiesis, embryonic development, and metastasis is now appreciated22,23. Chemokines exert their actions through G protein–coupled receptors, of which CCR5 is a key representative24. CCR5 binds the chemokines CCL3, CCL4 and CCL5 and is involved in inflammation and autoimmunity22. Along with CXCR4, CCR5 also is pivotal in the pathogenesis of AIDS. These G protein–coupled receptors, along with CD4, seem to be the main coreceptors for human immunodeficiency virus in vivo25.
The inverse correlation between leukocyte apoptosis and proinflammatory chemokine concentrations during the resolution of inflammation suggests the existence of active mechanisms to reduce chemokines in the local resolving milieu. Here we report that CCL3 and CCL5 were increased in peritoneal exudates of Ccr5−/− mice during the resolution of zymosan A–induced peritonitis. Transfer of apoptotic PMNs resulted in CCR5-dependent scavenging of CCL3, CCL4 and CCL5. Moreover, human apoptotic leukocytes expressed more functional CCR5 on their surface. CCR5 surface expression on apoptotic PMNs was reduced by proinflammatory cytokines and was increased by proresolution lipid mediators and cytokines.
RESULTS
Increased chemokines in Ccr5−/− mice
To determine whether CCR5 is involved in chemokine clearance during the resolution of mouse peritonitis, we collected peritoneal exudates from wild-type and Ccr5−/− mice and measured relevant chemokines and cytokines in the exudates. At 12 h after the initiation of peritonitis, Ccr5−/− exudates contained significantly more CCL3 and CCL5 but not CCL4 than did wild-type exudates (Fig. 1a–c). There was also more CCL3 in Ccr5−/− exudates at 4 h after the initiation of peritonitis. In contrast, the rapid reduction in CCL2 between 4 and 12 h after the initiation of peritonitis was similar in wild-type and Ccr5−/− mice (Fig. 1d), and there were no detectable significant differences in the quantity of CXCL12 in wild-type and Ccr5−/− mice (data not shown). There was also significantly more tumor necrosis factor (TNF; Fig. 1e) and IL-1β (Fig. 1f) at 4 h and 12 h, respectively, in Ccr5−/− mice than in wild-type mice. These results suggested that CCR5 is involved in the clearance of CCL3 and CCL5 during the resolution of peritonitis. At 4 h after the initiation of peritonitis, Ccr5−/− peritoneal exudates contained fewer PMNs than did their wild-type counterparts (Fig. 2a). At 12 h after the onset of peritonitis, Ccr5−/− mice also had fewer monocytes and macrophages (Fig. 2b).
Figure 1.
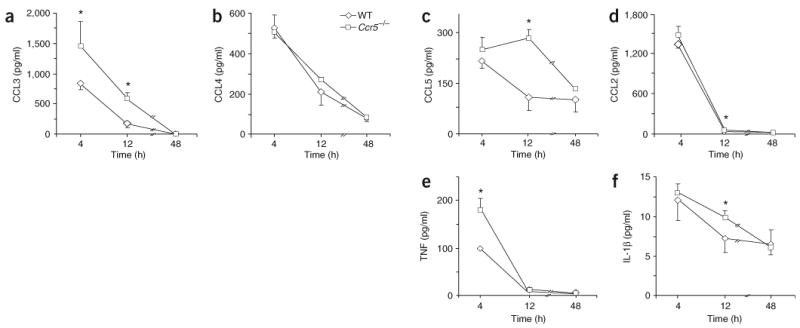
Increased CCL3 and CCL5 in peritoneal exudates from Ccr5−/− mice during the resolution of peritonitis. Peritoneal exudates from Ccr5−/− or wild-type (WT) mice were collected 4, 12 and 48 h after the initiation of peritonitis and CCL3 (a), CCL4 (b), CCL5 (c), CCL2 (d), TNF (e) and IL-1β (f) were measured by Luminex methodology. *, P < 0.05, wild-type versus Ccr5−/−. Results are the mean ± s.e.m. of three experiments.
Figure 2.
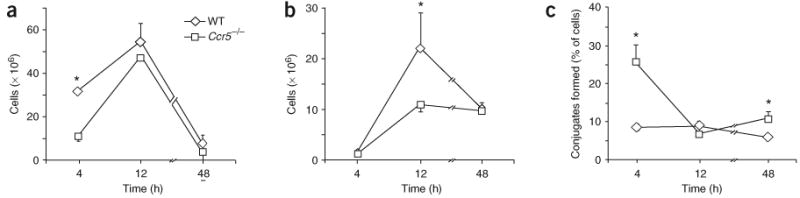
Ccr5−/− macrophages engulf apoptotic PMNs more efficiently than do wild-type macrophages. Flow cytometry of PMNs (a), monocytes and macrophages (b) and PMN-macrophage conjugates (c) from peritoneal exudates of Ccr5−/− or wild-type mice, stained with anti-Ly6G and anti-F4/80. *, P < 0.05, wild-type versus Ccr5−/−. Results are the mean ± s.e.m. of three experiments.
The initial step in the engulfment of apoptotic cells by phagocytes involves the formation of conjugates of both cells, called ‘tethering’26. We identified PMN-macrophage conjugates in peritoneal exudates 48 h after peritonitis induction, based on staining for the Ly6G and F4/80 surface markers and based on measurements of forward versus side scatter (Supplementary Fig. 1 online). The percentage of PMN-macrophage conjugates at 4 and 48 h after peritonitis induction was significantly greater in Ccr5−/− peritoneal exudates than in their wild-type counterparts (Fig. 2c). In addition, F4/80 expression on the surface of single macrophages and on PMN-macrophage conjugates in Ccr5−/− peritoneal exudates was greater than in wild-type mice at 48 h after the onset of peritonitis (Fig. 3). Thus, macrophages from Ccr5−/− mice show early maturation and enhanced attachment to apoptotic PMNs.
Supplementary Figure 1. Ly-6G+F4/80+ in peritoneal exudates are PMN-macrophage conjugates.
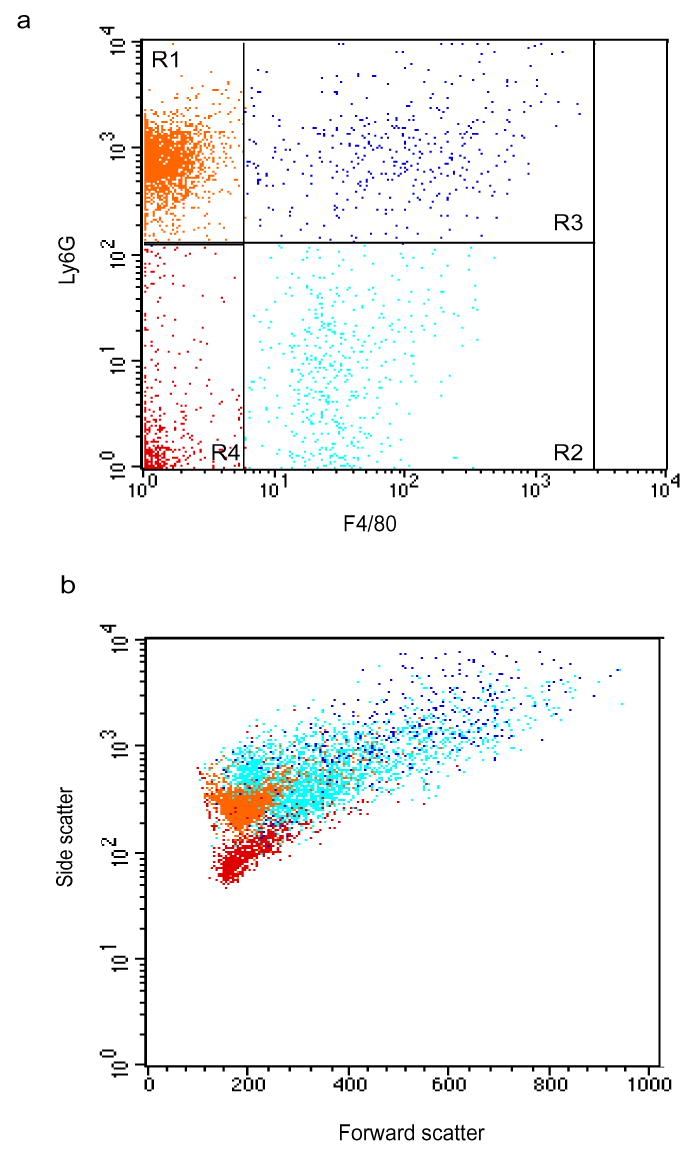
Leukocytes recovered from peritoneal exudates 48 h after peritonitis initiation were stained with anti-Ly-6G and anti-F4/80 and analyzed by flow cytometry. Dot plots represent Ly-6G and F4/80 staining (a) and forward versus side scatter (b). PMNs (R1, orange cells), macrophages (R2, cyan cells) and PMN-macrophage conjugates (R3, blue cells) are indicated. Results are respresentative of six experiments.
Figure 3.
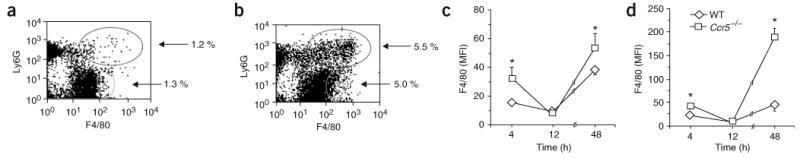
Ccr5−/− macrophages are more mature than wild-type macrophages. (a,b) Flow cytometry of cells from the peritoneal exudates of wild-type mice (a) and Ccr5−/− mice (b), stained with anti-Ly6G and anti-F4/80 and analyzed 48 h after initiation of peritonitis. Numbers for outlined areas indicate percent Ly6G+F4/80+ cells (top) or Ly6G−F4/80+ cells (bottom). (c,d) Average mean fluorescence intensity (MFI) of F4/80 staining on monocytes and macrophages (c) and PMN-macrophage conjugates (d) at various time points (horizontal axes). *, P < 0.05, wild-type versus Ccr5−/−. Results are representative of three experiments (a,b) and the mean ± s.e.m. of three experiments (c,d).
CCR5-dependent chemokine scavenging by apoptotic PMNs
To determine whether CCR5 on apoptotic PMNs is involved in clearing CCL3 and CCL5, we transferred sorted apoptotic PMNs from wild-type and Ccr5−/− mice into the peritonea of Ccr5−/− mice 12 h after injecting the recipient mice with zymosan A. At 1 h after PMN transfer, we collected peritoneal exudates and measured proinflammatory cytokines and chemokines. Recipients of Ccr5−/− PMNs had significantly more CCL3–CCL5 in their exudates than did mice that received wild-type PMNs (Fig. 4a). Recipients of Ccr5−/− PMNs had slightly more TNF and IL-1β, whereas the amount of CCL2 was not statistically different for recipients of wild-type versus Ccr5−/− PMNs.
Figure 4.

CCR5 mediates chemokine scavenging by apoptotic PMNs.(a) Chemokines and cytokines in peritoneal exudates of recipients of PMNs sorted from exudates from Ccr5−/− or wild-type mice 4 h after peritonitis initiation and cultured overnight, then transferred into the peritonea of Ccr5−/− mice 12 h after the initiation of peritonitis; peritoneal exudates of recipient mice were collected 1 h later for analysis. *, P < 0.05, wild-type versus Ccr5−/− PMN recipients. (b) Chemokines and cytokines in peritoneal exudates of recipients of PMN-enriched exudate cells collected 12 h after peritonitis induction, treated with either CCR5 antagonist or negative control and then transferred into inflamed wild-type peritonea 4 h after injection of recipient mice with zymosan A; peritoneal exudates of recipient mice were collected 1 h later for analysis. *, P < 0.05, control versus CCR5 antagonist treatment. Results are mean ± s.e.m. of four experiments.
To further establish the function of apoptotic PMNs in CCR5-mediated chemokine clearance, we isolated peritoneal exudate cell samples enriched in PMNs from wild-type mice 12 h after the induction of peritonitis, treated the cells with a CCR5-specific antagonist or control and transferred the treated cells into the peritonea of wild-type mice 4 h after injecting the recipient mice with zymosan A. PMNs treated with CCR5 antagonist increased the quantity of CCL3–CCL5 in peritoneal exudates relative to control-treated PMNs (Fig. 4b). Recipients of PMNs treated with CCR5 antagonist or control-treated PMNs had similar amounts of TNF and IL-1β, whereas recipients of PMNs treated with CCR5 antagonist had less CCL2. Thus, CCR5+ apoptotic PMNs assist in clearing CCL3–CCL5.
Next we directly determined whether apoptotic human neutrophils express surface CCR5 in vitro and whether the amount of surface CCR5 was affected by modulators of apoptosis. We cultured freshly isolated human PMNs from healthy donors for 22 h at 37 °C in media containing 10% fetal bovine serum without any additional survival factors to induce spontaneous apoptosis, which was reflected by a reduction in forward scatter. There was substantially more CCR5 staining exclusively on late apoptotic PMNs (propidium iodide positive; data not shown). Freshly isolated human PMNs had little Ccr5 mRNA but substantial intracellular CCR5 protein, as demonstrated by intense intracellular staining with monoclonal antibodies to the N terminus or the second extracellular loop of CCR5 (data not shown). These data suggested that little Ccr5 transcription occurs in human PMNs and that CCR5 protein has a long half-life and is externalized during apoptosis. CCR5 expression on the surfaces of late apoptotic PMNs was inhibited by zVAD-fmk, a general caspase inhibitor, in a concentration-dependent way (Fig. 5a). Exposure to TNF, a proinflammatory cytokine that regulates PMN function27, reduced CCR5 surface expression to an extent similar to the reduction after exposure to 10 μM zVAD-fmk. Both zVAD-fmk and TNF also inhibited PMN apoptosis (Fig. 5b). These results suggested that TNF reduces caspase-dependent expression of CCR5 on late apoptotic human neutrophils.
Figure 5.

Regulation of CCR5 expression on late apoptotic human PMNs. (a,b) Flow cytometry of CCR5 surface expression (a) and apoptotic phenotype (b) of human PMNs incubated for 22 h with vehicle, zVAD-fmk or TNF. (c) Flow cytometry of CCR5 surface expression and apoptotic phenotype of human PMNs exposed for 22 h to vehicle, resolution-phase lipid mediators or TGF-β. RvE1-Me, resolvin E1–methyl ester; ATLa, aspirin-triggered lipoxin A4 analog; PD1-Me, PD1–methyl ester. *, P < 0.05, vehicle versus experimental treatment. Results are representative of three experiments (a,b) and the mean ± s.e.m. of three experiments (c).
Specific families of lipid mediators are generated temporally during the progression of inflammation. Several such lipids suppress inflammation and promote immune response resolution4,7. These include the protectins (such as PD1) generated from docosahexaenoic acid, resolvins generated from eicosapentaenoic acid, and lipoxins generated from arachidonic acid. We exposed PMNs to PD1–methyl ester (a metabolically stable analog of aspirin-triggered lipoxin A4 (ATLa)), aspirin-triggered lipoxin A4 analog, resolvin E1–methyl ester, and TGF-β (a proresolution cytokine21), which are mediators generated during immune response resolution20. Exposure of PMNs to as little as 10 nM of each mediator increased CCR5 expression on the surfaces of late apoptotic PMNs (Fig. 5c). These proresolution mediators did not modulate apoptosis of PMNs and therefore did not increase the percentage of CCR5+ cells. Thus, proresolution lipid mediators induce an increase in CCR5 expression on the surfaces of apoptotic PMNs.
To determine whether CCR5 expression conferred chemotactic properties on apoptotic PMN, we did migration experiments using the CCR5 ligand CCL4. A small and statistically insignificant percentage of freshly isolated PMNs migrated toward CCL4 (8.9% ± 1.9% and 10.9% ± 0.1% of cells for vehicle and CCL4, respectively). In comparison, there was no migration of apoptotic PMNs (8.8 ± 1.1% and 8.3 ± 0.9% of cells for vehicle and CCL4, respectively). Therefore, CCR5 expression on apoptotic PMNs does not induce chemotactic responsiveness to CCL4.
CCR5 on late apoptotic T cells
Apoptotic T cells are also cleared by macrophages28. Therefore, to determine whether expression of CCR5 on the surface of T cells is regulated during apoptosis, we isolated peripheral blood T cells from healthy human donors and activated the cells with CD3-specific antibodies. As expected, stimulation with antibody to CD3 (anti-CD3) significantly increased the percentage of dead cells in the late apoptotic T cell population (Fig. 6a). Anti-CD3 stimulation also significantly increased the percentage of CCR5+ cells in the late apoptotic T cell population, but it decreased the percentage of CCR5+ cells in the live and early apoptotic T cell population. In contrast, anti-CD3-induced modulation of Fas (CD95) surface expression resulted in the opposite trend (reduced on late apoptotic cells and increased on live and early apoptotic cells). Incubation of anti-CD3-stimulated T cells with staurosporine, a pharmacological inducer of apoptosis, promoted an increase in the percentage of CCR5+ cells in the late apoptotic T cell population, with the maximum effect at 24 h after treatment (Fig. 6b). As expected, staurosporine treatment resulted in a time-dependent increase in apoptosis among those late apoptotic T cells (Fig. 6c). Exposure of resting peripheral blood T cells to staurosporine alone reduced CCR5 expression (data not shown). Thus, late apoptotic activated peripheral blood T cells have high expression of CCR5 on their surfaces.
Figure 6.

Apoptotic activated peripheral blood T cells have high expression of CCR5. (a) Flow cytometry of the surface phenotype of peripheral blood T cells activated for 3 d by anti-CD3 (5 μg/ml). Gray bar, percent death of all cells. (b,c) Flow cytometry of CCR5 surface expression (b) and apoptotic surface phenotype (c) of T cells activated as described in a and treated for 4–48 h with staurosporine (STS). *, P < 0.05, versus late apoptotic. Results are representative of three experiments.
To determine the specificity and kinetics of the mechanism leading to CCR5 expression on late apoptotic human leukocytes, we treated human CD4+ Jurkat T cells with staurosporine or Fas ligand, a physiological inducer of apoptosis in T cells (Fig. 7). We assessed surface CCR5 expression and apoptotic phenotype after 24 and 48 h, respectively. We classified apoptotic phenotypes into three categories: early apoptotic cells with reduced volume (detected by forward scatter) but not stained by propidium iodide; membrane-permeable cells highly stained by propidium iodide; and cells with degraded DNA that stained weakly with propidium iodide. We defined the last two categories as ‘late apoptotic cells’. We identified live cells by their lack of volume reduction and propidium iodide staining. Live T cells exposed to vehicle alone had little surface expression of CCR5, but small late apoptotic T cells subjected to the same treatment expressed much more CCR5 on their surfaces (Fig. 7a,b). We monitored substantial significant increases in the percentage of CCR5+ cells after the induction of apoptosis by staurosporine (Fig. 7d,e) or Fas ligand (Fig. 7g,h) in late apoptotic T cells (including those containing degraded DNA). In the early apoptotic T cells, an increase in IgG staining but not in CCR5 staining was observed. Hence, it appears that CCR5 expression on early apoptotic T cells is reduced. For the purpose of direct comparison, we found no statistically significant differences in CXCR4 surface expression on live versus late apoptotic cells (Fig. 7c,f). Thus, CCR5 expression was upregulated substantially on late apoptotic cells after treatment with either staurosporine or Fas ligand.
Figure 7.

Late apoptotic T cells have high expression of CCR5. Flow cytometry of CCR5 surface expression and apoptotic phenotypes of Jurkat CD4+ T cells incubated with vehicle (0.2% DMSO for 48 h; a–c), staurosporine (2 μM for 24 h; d–f) or Fas ligand (5 ng/ml for 48 h; g,h) and then stained with propidium iodide (a–g) and mouse IgG (a,d,g), anti-CCR5 (b,e,h) or anti-CXCR4 (c,f). Positively stained late apoptotic cells are outlined. Results are representative of ten experiments.
Characteristics of CCR5 expressed on apoptotic cells
To determine whether CCR5 expressed on late apoptotic T cells is functional, we assessed the binding of CCL4 (a chemokine that binds exclusively to CCR5) along with propidium iodide staining after the induction of apoptosis by staurosporine or Fas ligand. The percentage of late apoptotic T cells that bound CCL4 increased in a concentration-dependent way after incubation with staurosporine or Fas ligand (Supplementary Figure 2 online). In contrast, the percentage of live and early apoptotic T cells that bound CCL4 decreased after treatment with staurosporine or Fas ligand (Supplementary Figure 2). Staurosporine and Fas ligand had similar effects on apoptosis (Supplementary Fig. 2) and CCR5 expression on the whole cell population (data not shown). Therefore, CCL4 binding capacity is modulated in parallel with CCR5 expression during apoptosis of T cells.
Supplementary Figure 2. Increased CCL4 binding to late apoptotic T cells.

(a–c) Late apoptotic (a) or live and early apoptotic (b) Tc ells were incubated with STS, indicated concentrations of Fas ligand, or vehicle for 24 h. Cells were then incubated with biotinylated CCL4 or soybean trypsin inhibitor (STI) as a nonspecific biotinylated probe, followed by FITC-avidin. After washing, CCL4 binding (a,b) and apoptotic phenotype (c) was assessed by flow cytometry. *P < 0.05 versus vehicle alone. Results are representative of six experiments.
To determine whether the CCR5 expressed during apoptosis has different properties than CCR5 expressed in live cells, we compared the binding of CCL4 to staurosporine-treated late apoptotic cells and live cells. CCL4 had more relative binding to late apoptotic T cells than to live cells (Fig. 8a). These results suggested that CCR5 on late apoptotic T cells may have higher affinity for CCL4 than does CCR5 expressed on live cells.
Figure 8.

CCR5 on apoptotic cells has characteristics different from those of CCR5 on live cells. (a) Flow cytometry of CCL4 binding and apoptotic phenotype of T cells incubated for 48 h with staurosporine or vehicle, then incubated with increasing concentrations of biotinylated CCL4 followed by fluorescein isothiocyanate–avidin. *, P < 0.05, versus live cells. (b) Light microscopy (left) and fluorescent microscopy (middle and right) of live cells (top) or apoptotic cells (bottom) from a that were fixed after staining and applied to coverslips. Arrowheads indicate CCR5 clustering. PI, propidium iodide. Original magnification, ×1,000. Results represent mean ± s.e.m. of three experiments (a) or are representative of three experiments (b).
Another mechanism regulating ligand-receptor interactions is avidity modulation. To determine whether there are differences in the spatial distribution of CCR5 on the surfaces of late apoptotic T cells and live cells, we immobilized and fixed CCR5-stained cells and assessed surface CCR5 distribution by fluorescent microscopy. By light microscopy and propidium iodide staining, we noted that late apoptotic T cells had features characteristic of apoptosis, such as reduced volume, ruffled membranes and a condensed nucleus (Fig. 8b). Late apoptotic T cells also bound more CCL4, and CCL4 binding was localized in patches, suggesting the formation of clusters (Fig. 8b). Therefore, CCR5 expressed on late apoptotic T cells is localized in clusters on the cell surface, and such clustering may increase its avidity for ligands.
One of the main intracellular signaling pathways regulating apoptosis involves the activation of caspases, a family of serine proteases that cleave and activate key proteins of the apoptotic cascade29. To determine whether caspases are involved in the regulation of CCR5 expression and CCL4 binding during T cell apoptosis, we incubated T cells with the caspase inhibitor zVAD-fmk before inducing apoptosis by treating cells with staurosporine or Fas ligand. Incubation with zVAD-fmk partially or completely inhibited the staurosporine- or Fas ligand–induced binding of CCL4 to late apoptotic T cells (Supplementary Figure 3 online). Notably, zVAD-fmk did not prevent the reduction in binding of CCL4 to early apoptotic cells that was induced by treatment with staurosporine or Fas ligand (data not shown).
Supplementary Figure 3. Caspase inhibition abrogates apoptosis-induced modulation of CCL4 binding to late apoptotic T cells.

T cells were incubated with vehicle (a–d) or Z-VAD-fmk (e,f) for 20 min and then treated with vehicle (a,b), STS (24 h; c,e) or Fas ligand (48 h; d,f). Cells were incubated with biotinylated CCL4 (open histograms) or STI (filled histograms) and analyzed by flow cytometry. (g) Relative inhibiton of CCL4 binding and apoptosis induced by Z-VAD-fmk. *P < 0.05 versus CCL4 binding. Results are representative of (a–f) and present the mean ± s.e.m. of (g) three experiments.
We also compared the effect of zVAD-fmk on apoptosis and CCL4 binding (Supplementary Figure 3). Incubation with zVAD-fmk completely inhibited both CCL4 binding and apoptosis induced by Fas ligand. Notably, zVAD-fmk strongly inhibited staurosporine-induced binding of CCL4 but weakly inhibited staurosporine-induced apoptosis. These results suggested that caspase-independent apoptotic pathways do not efficiently induce CCR5 expression. Our findings collectively suggest substantial involvement of CCR5-expressing leukocytes in the sequestration of chemokines during the resolution of inflammation (Supplementary Figure 4 online).
Supplementary Figure 4.

Scheme of the role of apoptotic leukocytes and pro-resolving lipid mediators in the resolution of inflammation.
DISCUSSION
Here we have reported that apoptotic mouse PMNs and T cells scavenged CCR5 ligands during the resolution of zymosan A–initiated peritonitis. Expression of CCR5 on the surfaces of apoptotic human PMNs and T cells was dependent on caspase activity. CCR5 on the surfaces of apoptotic PMNs was downregulated by TNF and was upregulated by mediators that promote the resolution of inflammation, such as PD1, resolvin E1, aspirin-triggered lipoxin A4 analog and TGF-β. In addition, CCR5 on late apoptotic T cells had higher avidity toward its ligands than did CCR5 on live T cells. Those findings identify a function for apoptotic PMNs and T cells during the resolution of inflammation and suggest that apoptotic PMNs and T cells are capable of reducing local quantities of soluble bioactive mediators in the inflammatory milieu.
For a return to homeostasis and limiting the infiltration of immune cells into inflamed sites1, an active counter-regulation or ‘shutdown’ of proinflammatory mediators, including chemokines, is needed4. At present, chemokine proteolysis by means of proteases, including elastase, cathepsin G, matrix metalloproteases, CD26 and others, is believed to be the main mechanism responsible for neutralizing or reducing chemokine activity30. However, chemokines processed by such proteases do not necessarily show reduced bioactivity, and increased bioactivity has been noted in some cases30. CCL3, CCL4 and CCL5 are all processed by CD26, which removes the first two N-terminal amino acids. That alteration, however, does not reduce their action through CCR5 (refs. 31–33).
CCR5 ligands as well as other chemokines can be scavenged by two members of the G protein–coupled receptor family: D6 and Duffy antigen receptor for chemokines. Those ‘silent’ chemokine receptors bind to multiple chemokines without transmitting detectable intracellular signals30. D6-deficient mice have defective clearance of β-chemokines after inflammation, which leads to an inflammatory pathology resembling that of human psoriasis34. Another chemokine deactivation mechanism involves the induction of ‘uncoupled’ CCR1, CCR2 and CCR5 on dendritic cells or monocytes treated with IL-10 (ref. 35). Those receptors fail to transmit signals and instead act as ‘decoys’ that ‘soak up’ and sequester chemokines.
CCR5 and its ligands are paramount in the pathogenesis of inflammation and autoimmune disorders22. The effect of CCR5 ligands could be abolished by their timely removal during the resolution of inflammation. Here we have identified a mechanism that could serve to dampen chemokine activity, stop leukocyte infiltration and clear inflammatory sites. Apoptotic PMNs and T cells had high expression of CCR5, which had more avidity toward its ligands, and functioned as ‘chemokine-scavenging devices’. Our in vivo results have demonstrated that apoptotic PMNs scavenged chemokines during the resolution of acute inflammation. Those findings, combined with published evidence indicating that apoptotic cells are engulfed and processed by phagocytes6,36, complete the sequence of CCR5 ligand clearance. Hence, during resolution, the PMNs and T cells that were attracted to sites of inflammation by CCR5 ligands undergo apoptosis, which results in increased CCR5 surface expression, which facilitates scavenging of CCR5 ligands. CCR5 ‘scavengers’ are ultimately engulfed and cleared by macrophages. The removal of proinflammatory chemokines from the resolving milieu would limit further leukocyte recruitment, prevent excessive tissue damage and promote a return to catabasis. EP1, a receptor for PGE2 involved in the resolution of inflammation3, was not upregulated on apoptotic leukocytes, whereas the ‘silent’ chemokine receptor D6 was highly upregulated on the surface of apoptotic PMNs and T cells (n = 3; data not shown). Those findings emphasize the function of apoptotic leukocytes as selective scavengers of proinflammatory chemokines but apparently not of proresolution mediators.
Although CCL3 and CCL5 persisted during early resolution (4–12 h) in Ccr5−/− mice, CCL3 and CCL5 decreased during the later phase of resolution (48 h). That suggested the existence of additional mechanisms for removing those chemokines. Ccr5−/− mice have delayed clearance of influenza virus and exacerbated lung inflammation37, but those characteristics have been attributed to the lack of prevention of macrophage apoptosis through CCL5-CCR5 signaling rather than to persistent expression of CCR5 ligands.
Chemokine scavenging by ‘silent’ or ‘decoy’ receptors is accompanied by deficiencies in intracellular signaling induced by G protein–coupled receptors, which prevent unwanted migration and activation of scavenging cells. Apoptotic leukocytes are the ultimate scavenging entity, as they are essentially inert38, having lost their membrane integrity and being in the process of losing their cytoskeletal building blocks. As might be expected, we found that apoptotic human PMNs did not migrate toward CCL4. That finding was consistent with the idea that CCR5 expressed on their surface does not confer migratory functions.
Our findings have indicated that macrophages infiltrating the inflamed peritonea of Ccr5−/− mice had a more mature phenotype (manifested by their high expression of F4/80) as well as an enhanced ability to engulf apoptotic PMNs. CCL5 exacerbates septic peritonitis through binding to CCR1 (ref. 39) but also promotes macrophage survival during viral infection via CCR5 (ref. 37). Therefore, it remains to be determined whether the changes in Ccr5−/− macrophage maturation were a result of excessive exposure to CCL3 and CCL5 or of a lack of inhibitory signals generated by CCR5. Regardless of which is true, our results have indicated that CCR5 has a key and previously unappreciated function in controlling the inflammation-resolution axis during mouse peritonitis.
We found that induction of apoptosis in Jurkat cells or unactivated T cells triggered downregulation of CCR5 expression. That is consistent with published findings showing that CCR5 is downregulated on lymphocytes and monocytes treated with rapamycin40, which induces cell cycle arrest. CCR5 expression was substantially upregulated by treatment of late apoptotic Jurkat cells and activated T cells with Fas ligand and staurosporine or with staurosporine alone, respectively. Therefore, it seems that an activated state, such as that induced by exposure to antigen, which would be encountered in inflammatory scenarios in vivo, is required for chemokine scavenging by apoptotic T cells mediated by CCR5.
TNF is a pluripotent cytokine that acts as a ‘double-edged sword’ during the onset and progression of inflammation and autoimmunity41. In contrast, lipoxin A4, resolvin E1 and PD1 are antiinflammatory mediators that promote resolution4,7,8. Lipoxin A4, resolvin E1 and PD1 stop PMN recruitment and promote resolution4,20,42. TGF-β inhibits the apoptosis of PMNs but promotes the apoptosis of T cells43,44. TGF-β is also released by apoptotic T cells45 and by macrophages that have engulfed apoptotic leukocytes during the resolution of inflammation13,28. TNF reduced, whereas lipoxin A4, resolvin E1, PD1 and TGF-β enhanced, CCR5 expression on apoptotic PMNs. That dichotomy is in agreement with the opposing functions of TNF and proresolution mediators during inflammation and its resolution. In addition, it is noteworthy that unlike proinflammatory lipid mediators such as LTB4 (ref. 46), proresolution lipid mediators did not enhance PMN apoptosis. Therefore, those mediators seem to promote expression of CCR5 exclusively on PMNs that have already initiated apoptosis.
Our findings have shown that late apoptotic leukocytes have high, caspase-dependent expression of surface CCR5. Surface CCR5 on apoptotic leukocytes was differentially regulated by proinflammatory and proresolution mediators. Thus, apoptotic leukocytes can actively participate in terminating chemokine signaling by reducing the quantities of those mediators in the local milieu during the resolution of inflammation.
METHODS
Reagents
BSA, Histopaque-1077, poly-L-lysine and propidium iodide were from Sigma; RPMI 1640 medium, FBS, glutamine, penicillin and streptomycin were from Bio Whittaker; staurosporine was from MP Biomedicals; Fas ligand was from Upstate Biotechnology; and recombinant human TNF and TGF-β were from Pepro Tech. Aspirin-triggered lipoxin A4 analog (15-epi-16-(para-fluoro)-phenoxy-LXA4), resolvin E1–methyl ester (5S,12R,18R-trihydroxy-6Z,8E,10E,14Z,16E-EPA) and PD1–methyl ester (10R,17S-dihydroxydocosa-4Z,7Z,11E,13E,15Z,19Z-hexaenoic acid) were prepared by total organic synthesis and were characterized, including nuclear magnetic resonance spectroscopy, according to published criteria18,46,47.
Mouse peritonitis
Peritonitis was produced as described20 in 6- to 10-week-old FVB or C57BL/6 male and female mice (Charles River Laboratories) fed laboratory Rodent Diet 5001 (Purina Mills), in accordance with the Harvard Medical Area Standing Committee on Animals (02570; 10.17.05).
Chemokine clearance in vivo.
Ccr5−/− mice48,49 were backcrossed for ten generations to C57BL/6 mice. Peritonitis was initiated in age- and sex-matched wild-type and Ccr5−/− mice and exudates were collected after 4, 12 and 48 h. CCL2–CCL5, TNF and IL-1β in cell-free exudates were measured by Luminex methodology as described below. In some experiments, exudates were collected from wild-type and Ccr5−/− mice after 4 h and PMNs (more than 97% Ly6G+F4/80−) were isolated with a FACSAria (BD Biosciences). PMNs were cultured overnight to promote apoptosis, and cells were transferred into Ccr5−/− mice (2.5 × 106 cells per mouse) 12 h after the initiation of peritonitis. Exudates were collected 1 h after cell transfer, and chemokines and cytokines were measured. The percent change was calculated as follows: (amount of mediator in recipients of Ccr5−/− PMNs)/(amount of mediator in recipients of wild-type PMNs) × 100. In other experiments, peritoneal exudates were collected from FVB mice 12 h after injection of zymosan A and samples were enriched for PMNs by plastic adherence for 1 h in culture media. Nonadherent cells were collected and washed and were incubated for 20 min at 37 °C with the CCR5-specific antagonist SMM chemokine50 or its reduced and unfolded control counterpart (2 μg/ml; Raylight). Cells were then transferred into recipient wild-type mice (16 × 106 cells per mouse) 4 h after the initiation of peritonitis; exudates were collected 1 h later and chemokines and cytokines were measured. The percentage of change was calculated as follows: (amount of mediator in recipients of CCR5 antagonist–loaded PMNs)/(amount of mediator in recipients of control-loaded PMNs) ×100. TNF, IL-1β and CCL2-5 were measured by ‘multiplexed’ sandwich immunoassay (Biosource) with a Luminex xMAP 100 (MiraiBio). CCL5 measurements were confirmed by standard enzyme-linked immunosorbent assay (Biosource).
Human cells
Peripheral blood T cells and PMNs were separated from the venous blood of healthy volunteers as described19,27 and in accordance with the Brigham and Women’s Hospital Human Research Committee (88-02642; 12/21/05). Human PMN isolate preparations contained 96% ± 3% PMNs, as determined by light microscopy and Wright-Giemsa staining.
Apoptosis induction
Spontaneous apoptosis of PMNs was detected after 22 h of incubation in culture media. In some experiments, zVAD-fmk (10–50 μM), TNF (40 ng/ml), resolvin E1–methyl ester, aspirin-triggered lipoxin A4 analog, PD1–methyl ester (10 nM) or TGF-β (10 ng/ml) was added. Vehicle treatment was 0.05% (volume/volume) ethanol. Peripheral blood T cells were activated by incubation for 3 d in 24-well plates coated with anti-CD3 (5 μg/ml; R&D Systems). Jurkat cells or activated peripheral blood T cells were incubated for 4–48 h with staurosporine (1–2 μM) or Fas ligand (0.05–5 ng/ml), after which cells were collected and used for flow cytometry or binding assays. In some experiments, zVAD-fmk (10–50 μM; R&D Systems) was added to cells 20 min before the addition of apoptosis-inducing agents. Vehicle treatment was 0.2% (volume/volume) dimethyl sulfoxide.
CCR5 surface expression
After the induction of apoptosis, T cells or PMNs (0.5 × 106 cells per sample) were stained with mouse immunoglobulin G (IgG; BD Pharmingen), mouse anti-CCR5 or mouse anti-CXCR4 (1 μg per sample in flow cytometry buffer (PBS, 1% FBS and 0.01% sodium azide, wt/vol); R&D Systems), followed by fluorescein isothiocyanate–conjugated goat anti-mouse IgG (1:100 dilution). Cells were resuspended in flow cytometry buffer containing 2 μg/ml of propidium iodide and were analyzed on a FACSort (BD Biosciences). Values obtained for IgG staining were subtracted from values obtained for anti-CCR5 staining.
Statistical analysis
In vitro and in vivo experiments were analyzed by the two-tailed Student’s t-test, with P values less than 0.05 considered statistically significant. Further methods are described in Supplementary Methods online.
Supplemental Methods
Chemokine binding assays
Apoptosis was induced in Jurkat cells as described above, and cells (0.5×106/sample) were bound by CCL4 or STI using the Fluorokine™ kit (R&D Systems) according to the manufacturer’s instructions. Next, cells were washed and resuspended in binding buffer with 2 μg/ml PI and analyzed on a FACSort flow cytometer (BD Biosciences). Values obtained for STI binding were subtracted from values obtained for CCL4 binding. In some experiments, bound cells were fixed with 3.7% paraformaldehyde, immobilized on poly-L-lysine-coated cover slips, and visualized with a light or fluorescent microscope (Eclipse E600, Nikon).
Chemotaxis assays
Human PMNs were labeled with CFSE (10 μM) for 30 min, washed, and either cultured for 22 h to initiate apoptosis or used immediately. Chemotaxis was measured by adding CCL4 (25 nM) or vehicle to the lower well of a chemotaxis chamber, and 2×105 PMNs to the upper well. After 3 h the fluorescence intensity of the cells in the lower well was determined using Cytofluor™ 2300 (PerSeptive Biosystems), and the percentage of cells that migrated towards CCL4 was calculated.
Acknowledgments
We thank M.H. Small for assistance in manuscript preparation, and J. Deady for technical assistance. Supported by the US National Institutes of Health (GM38765, DK-074448 and P50-DE016191 to C.N.S., and DK-074449 to A.D.L.) and the Arthritis Foundation (A.A.).
Footnotes
Note: Supplementary information is available on the Nature Immunology website.
AUTHOR CONTRIBUTIONS
All authors were involved in experimental planning and data analysis and contributed to manuscript preparation. A.A. carried out FACS analysis, binding, chemotaxis, human cell isolation and cell culture; G.F. carried out experiments, FACS analysis, human cell isolation and culture; Y.-P.S. carried out peritonitis experiments and related analyses; A.K. and T.E.V.D. carried out Luminex analyses and related experimental design; A.D.L. carried out experiments with CCR5-deficient mice.
COMPETING INTERESTS STATEMENT
The authors declare competing financial interests (see the Nature Immunology website for details).
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions/
References
- 1.Majno G, Joris I. Cells, Tissues, and Disease: Principles of General Pathology. Oxford University Press: New York; 2004. [Google Scholar]
- 2.Serhan CN. Lipoxins and novel aspirin-triggered 15-epi-lipoxins (ATL): a jungle of cell-cell interactions or a therapeutic opportunity? Prostaglandins. 1997;53:107–137. doi: 10.1016/s0090-6980(97)00001-4. [DOI] [PubMed] [Google Scholar]
- 3.Levy BD, Clish CB, Schmidt B, Gronert K, Serhan CN. Lipid mediator class switching during acute inflammation: signals in resolution. Nat Immunol. 2001;2:612–619. doi: 10.1038/89759. [DOI] [PubMed] [Google Scholar]
- 4.Serhan CN. A search for endogenous mechanisms of anti-inflammation uncovers novel chemical mediators: missing links to resolution. Histochem Cell Biol. 2004;122:305–321. doi: 10.1007/s00418-004-0695-8. [DOI] [PubMed] [Google Scholar]
- 5.Rossi AG, Haslett C. In: Proinflammatory and Antiinflammatory Peptides. Said SI, editor. Marcel Dekker; New York: 1998. pp. 9–24. [Google Scholar]
- 6.Savill J, Dransfield I, Gregory C, Haslett C. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat Rev Immunol. 2002;2:965–975. doi: 10.1038/nri957. [DOI] [PubMed] [Google Scholar]
- 7.Gilroy DW, Perretti M. Aspirin and steroids: new mechanistic findings and avenues for drug discovery. Curr Opin Pharmacol. 2005;5:405–411. doi: 10.1016/j.coph.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 8.Godson C, et al. Cutting edge: lipoxins rapidly stimulate nonphlogistic phagocytosis of apoptotic neutrophils by monocyte-derived macrophages. J Immunol. 2000;164:1663–1667. doi: 10.4049/jimmunol.164.4.1663. [DOI] [PubMed] [Google Scholar]
- 9.Hanayama R, et al. Identification of a factor that links apoptotic cells to phagocytes. Nature. 2002;417:182–187. doi: 10.1038/417182a. [DOI] [PubMed] [Google Scholar]
- 10.Byrne A, Reen DJ. Lipopolysaccharide induces rapid production of IL-10 by monocytes in the presence of apoptotic neutrophils. J Immunol. 2002;168:1968–1977. doi: 10.4049/jimmunol.168.4.1968. [DOI] [PubMed] [Google Scholar]
- 11.Savill J, Hogg N, Ren Y, Haslett C. Thrombospondin cooperates with CD36 and the vitronectin receptor in macrophage recognition of neutrophils undergoing apoptosis. J Clin Invest. 1992;90:1513–1522. doi: 10.1172/JCI116019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim S, Elkon KB, Ma X. Transcriptional suppression of interleukin-12 gene expression following phagocytosis of apoptotic cells. Immunity. 2004;21:643–653. doi: 10.1016/j.immuni.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 13.Mitchell S, et al. Lipoxins, aspirin-triggered epi-lipoxins, lipoxin stable analogues, and the resolution of inflammation: stimulation of macrophage phagocytosis of apoptotic neutrophils in vivo. J Am Soc Nephrol. 2002;13:2497–2507. doi: 10.1097/01.asn.0000032417.73640.72. [DOI] [PubMed] [Google Scholar]
- 14.Hanayama R, et al. Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science. 2004;304:1147–1150. doi: 10.1126/science.1094359. [DOI] [PubMed] [Google Scholar]
- 15.Aprahamian T, et al. Impaired clearance of apoptotic cells promotes synergy between atherogenesis and autoimmune disease. J Exp Med. 2004;199:1121–1131. doi: 10.1084/jem.20031557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Potter PK, Cortes-Hernandez J, Quartier P, Botto M, Walport MJ. Lupus-prone mice have an abnormal response to thioglycolate and an impaired clearance of apoptotic cells. J Immunol. 2003;170:3223–3232. doi: 10.4049/jimmunol.170.6.3223. [DOI] [PubMed] [Google Scholar]
- 17.Bandeira-Melo C, et al. Cyclooxygenase-2-derived prostaglandin E2 and lipoxin A4 accelerate resolution of allergic edema in Angiostrongylus costaricensis-infected rats: relationship with concurrent eosinophilia. J Immunol. 2000;164:1029–1036. doi: 10.4049/jimmunol.164.2.1029. [DOI] [PubMed] [Google Scholar]
- 18.Arita M, et al. Stereochemical assignment, antiinflammatory properties, and receptor for the ω-3 lipid mediator resolvin E1. J Exp Med. 2005;201:713–722. doi: 10.1084/jem.20042031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ariel A, et al. The docosatriene protectin D1 Is produced by TH2 skewing and promotes human T cell apoptosis via lipid raft clustering. J Biol Chem. 2005;280:43079–43086. doi: 10.1074/jbc.M509796200. [DOI] [PubMed] [Google Scholar]
- 20.Bannenberg GL, et al. Molecular circuits of resolution: formation and actions of resolvins and protectins. J Immunol. 2005;174:4345–4355. doi: 10.4049/jimmunol.174.7.4345. [DOI] [PubMed] [Google Scholar]
- 21.Wahl SM, Swisher J, McCartney-Francis N, Chen W. TGF-β: the perpetrator of immune suppression by regulatory T cells and suicidal T cells. J Leukoc Biol. 2004;76:15–24. doi: 10.1189/jlb.1103539. [DOI] [PubMed] [Google Scholar]
- 22.Luster AD, Alon R, von Andrian UH. Immune cell migration in inflammation: present and future therapeutic targets. Nat Immunol. 2005;6:1182–1190. doi: 10.1038/ni1275. [DOI] [PubMed] [Google Scholar]
- 23.Strieter RM, Belperio JA, Phillips RJ, Keane MP. CXC chemokines in angiogenesis of cancer. Semin Cancer Biol. 2004;14:195–200. doi: 10.1016/j.semcancer.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 24.Oppermann M. Chemokine receptor CCR5: insights into structure, function, and regulation. Cell Signal. 2004;16:1201–1210. doi: 10.1016/j.cellsig.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 25.Alkhatib G, et al. CC CKR5: a RANTES, MIP-1α, MIP-1β receptor as a fusion cofactor for macrophage-tropic HIV-1. Science. 1996;272:1955–1958. doi: 10.1126/science.272.5270.1955. [DOI] [PubMed] [Google Scholar]
- 26.Arur S, et al. Annexin I is an endogenous ligand that mediates apoptotic cell engulfment. Dev Cell. 2003;4:587–598. doi: 10.1016/s1534-5807(03)00090-x. [DOI] [PubMed] [Google Scholar]
- 27.Hachicha M, Pouliot M, Petasis NA, Serhan CN. Lipoxin (LX)A4 and aspirin-triggered 15-epi-LXA4 inhibit tumor necrosis factor 1α-initiated neutrophil responses and trafficking: regulators of a cytokine-chemokine axis. J Exp Med. 1999;189:1923–1930. doi: 10.1084/jem.189.12.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huynh ML, Fadok VA, Henson PM. Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-β1 secretion and the resolution of inflammation. J Clin Invest. 2002;109:41–50. doi: 10.1172/JCI11638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Green D, Kroemer G. The central executioners of apoptosis: caspases or mitochondria? Trends Cell Biol. 1998;8:267–271. doi: 10.1016/s0962-8924(98)01273-2. [DOI] [PubMed] [Google Scholar]
- 30.Rot A, von Andrian UH. Chemokines in innate and adaptive host defense: basic chemokinese grammar for immune cells. Annu Rev Immunol. 2004;22:891–928. doi: 10.1146/annurev.immunol.22.012703.104543. [DOI] [PubMed] [Google Scholar]
- 31.Proost P, et al. Cleavage by CD26/dipeptidyl peptidase IV converts the chemokine LD78β into a most efficient monocyte attractant and CCR1 agonist. Blood. 2000;96:1674–1680. [PubMed] [Google Scholar]
- 32.Guan E, Wang J, Roderiquez G, Norcross MA. Natural truncation of the chemokine MIP-1β/CCL4 affects receptor specificity but not anti-HIV-1 activity. J Biol Chem. 2002;277:32348–32352. doi: 10.1074/jbc.M203077200. [DOI] [PubMed] [Google Scholar]
- 33.Oravecz T, et al. Regulation of the receptor specificity and function of the chemokine RANTES (regulated on activation, normal T cell expressed and secreted) by dipeptidyl peptidase IV (CD26)-mediated cleavage. J Exp Med. 1997;186:1865–1872. doi: 10.1084/jem.186.11.1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jamieson T, et al. The chemokine receptor D6 limits the inflammatory response in vivo. Nat Immunol. 2005;6:403–411. doi: 10.1038/ni1182. [DOI] [PubMed] [Google Scholar]
- 35.D’Amico G, et al. Uncoupling of inflammatory chemokine receptors by IL-10: generation of functional decoys. Nat Immunol. 2000;1:387–391. doi: 10.1038/80819. [DOI] [PubMed] [Google Scholar]
- 36.Chan A, Magnus T, Gold R. Phagocytosis of apoptotic inflammatory cells by microglia and modulation by different cytokines: mechanism for removal of apoptotic cells in the inflamed nervous system. Glia. 2001;33:87–95. doi: 10.1002/1098-1136(20010101)33:1<87::aid-glia1008>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 37.Tyner JW, et al. CCL5–CCR5 interaction provides antiapoptotic signals for macrophage survival during viral infection. Nat Med. 2005;11:1180–1187. doi: 10.1038/nm1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Whyte MK, Meagher LC, MacDermot J, Haslett C. Impairment of function in aging neutrophils is associated with apoptosis. J Immunol. 1993;150:5124–5134. [PubMed] [Google Scholar]
- 39.Ness TL, et al. CCR1 and CC chemokine ligand 5 interactions exacerbate innate immune responses during sepsis. J Immunol. 2004;173:6938–6948. doi: 10.4049/jimmunol.173.11.6938. [DOI] [PubMed] [Google Scholar]
- 40.Heredia A, et al. Rapamycin causes down-regulation of CCR5 and accumulation of anti-HIV β-chemokines: an approach to suppress R5 strains of HIV-1. Proc Natl Acad Sci USA. 2003;100:10411–10416. doi: 10.1073/pnas.1834278100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.O’Shea JJ, Ma A, Lipsky P. Cytokines and autoimmunity. Nat Rev Immunol. 2002;2:37–45. doi: 10.1038/nri702. [DOI] [PubMed] [Google Scholar]
- 42.Serhan CN, et al. Design of lipoxin A4 stable analogs that block transmigration and adhesion of human neutrophils. Biochemistry. 1995;34:14609–14615. doi: 10.1021/bi00044a041. [DOI] [PubMed] [Google Scholar]
- 43.Brunetti M, et al. Polymorphonuclear leukocyte apoptosis is inhibited by platelet-released mediators, role of TGFβ-1. Thromb Haemost. 2000;84:478–483. [PubMed] [Google Scholar]
- 44.Chen W, et al. Requirement for transforming growth factor β1 in controlling T cell apoptosis. J Exp Med. 2001;194:439–453. doi: 10.1084/jem.194.4.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen W, Frank ME, Jin W, Wahl SM. TGF-β released by apoptotic T cells contributes to an immunosuppressive milieu. Immunity. 2001;14:715–725. doi: 10.1016/s1074-7613(01)00147-9. [DOI] [PubMed] [Google Scholar]
- 46.Hebert MJ, Takano T, Holthofer H, Brady HR. Sequential morphologic events during apoptosis of human neutrophils. Modulation by lipoxygenase-derived eicosanoids. J Immunol. 1996;157:3105–3115. [PubMed] [Google Scholar]
- 47.Serhan CN, et al. Anti-inflammatory actions of neuroprotectin D1/protectin D1 and its natural stereoisomers: assignments of dihydroxy-containing docosatrienes. J Immunol. 2006;176:1848–1859. doi: 10.4049/jimmunol.176.3.1848. [DOI] [PubMed] [Google Scholar]
- 48.Wysocki CA, et al. Differential roles for CCR5 expression on donor T cells during graft-versus-host disease based on pretransplant conditioning. J Immunol. 2004;173:845–854. doi: 10.4049/jimmunol.173.2.845. [DOI] [PubMed] [Google Scholar]
- 49.Kuziel WA, et al. CCR5 deficiency is not protective in the early stages of atherogenesis in apoE knockout mice. Atherosclerosis. 2003;167:25–32. doi: 10.1016/s0021-9150(02)00382-9. [DOI] [PubMed] [Google Scholar]
- 50.Kumar S, et al. SMM-chemokines: a class of unnatural synthetic molecules as chemical probes of chemokine receptor biology and leads for therapeutic development. Chem Biol. 2006;13:69–79. doi: 10.1016/j.chembiol.2005.10.012. [DOI] [PubMed] [Google Scholar]