

Abstract
Clinical studies of replicating adenoviruses for the treatment of cancer have demonstrated their safety but have yielded disappointing results, indicating the need for new strategies to improve their efficacy. We hypothesized that the efficacy of a replicating adenovirus could be improved by expression of tissue inhibitor of metalloproteinases-2 (TIMP-2), a 21-kDa unglycosylated secretory protein. TIMP-2 specifically inhibits the active forms of a number of matrix metalloproteinases (MMPs) that play a role in the degradation of basement membranes and the extracellular matrix and are therefore involved in the control of the growth, invasion and metastasis of tumor cells, as well as angiogenesis. In addition, TIMP-2 can abrogate tumor growth and angiogenesis by a variety of mechanisms independent of MMP inhibition. In this study, we demonstrate that expression of TIMP-2 enhanced the antitumor efficacy of a replicating adenovirus in vivo, by reducing both tumor growth and angiogenesis.
Keywords: Angiogenesis, oncolysis, replicating adenovirus, TIMP-2, tumor growth
ABBREVIATIONS: MMP matrix metalloproteinase, TIMP-2 tissue inhibitor of metalloproteinases-2
INTRODUCTION
Oncolytic viruses tailored to replicate selectively within tumor cells are novel anticancer agents with great therapeutic potential.1 The selective replication of the viruses in cancer cells amplifies the initial viral inoculum, leading to destruction of the infected cells by virus-mediated lysis. The viral progeny are thereby released and can spread through the tumor mass to infect neighboring cancer cells, resulting in self-perpetuating cycles of infection, replication and oncolysis. Replication-selective viruses therefore overcome a major limitation of replication-defective vectors for cancer gene therapy, which are unable to infect all, or even most, cells within a solid three-dimensional tumor mass. The safety of oncolytic viruses derives from the restriction of their replication to tumor cells, while sparing normal cells.
While several oncolytic viruses have been identified to date, replication-selective adenoviruses possess a number of advantages.2 Human serotype 5 adenoviruses are associated with relatively mild diseases, their biology is relatively well characterized and their genomes can be manipulated with relative ease. Moreover, adenoviruses can be purified to high titer and are stable in the bloodstream, two features which afford the prospect of intravenous administration to treat disseminated cancer cells. Strategies to restrict the replication of adenoviruses to tumor cells have either involved placing the expression of viral genes, most commonly the E1A gene, under the control of tumor- or tissue-specific promoters, or have been based on the complete or partial deletion of viral genes that are required for replication in normal cells, but not in tumor cells.2 In recognition of their potential, replication-selective adenoviruses have been rapidly translated into human clinical trials in patients with advanced cancer, where the safety of these agents has been demonstrated.3 However, clinical studies of replicating adenoviruses have yielded disappointing results, indicating the need for new strategies to improve their efficacy.
The growth, invasion and metastasis of tumor cells, as well as angiogenesis, involve the degradation of basement membranes and the extracellular matrix.4 This process is controlled by a variety of proteolytic enzymes secreted by both tumor and stromal cells, including matrix metalloproteinases (MMPs), members of a family of zinc-dependent endopeptidases.5 MMPs are upregulated in many forms of cancer and their expression is associated with a poor prognosis.6, 7 The MMPs are synthesized as inactive zymogens and activated by proteinase cleavage. Their activity is tightly regulated by a group of endogenous inhibitors, including tissue inhibitors of metalloproteinases (TIMPs). The four mammalian TIMPs identified to date have many basic similarities, but differ in their structural features, biochemical properties and expression patterns.8, 9
TIMP-2 is a 21-kDa unglycosylated protein that is secreted in a soluble form by endothelial cells and fibroblasts.10 TIMP-2 binds in a 1:1 stoichiometric ratio to the active forms of a number of MMPs, including membrane type 1 MMP (MT1-MMP), MMP-2 and MMP-9, thereby specifically abrogating the MMP activity associated with tumor growth and angiogenesis.8 In addition to its antitumor activity as an inhibitor of MMPs, TIMP-2 can also inhibit tumor growth and angiogenesis by a variety of mechanisms independent of MMP-inhibition.11–13 TIMP-2 is unique among the members of the TIMP family or synthetic MMP inhibitors in being able to directly inhibit the proliferation of endothelial cells in response to angiogenic stimuli such as fibroblast growth factor 2 or vascular endothelial growth factor A.11, 14 Seo et al. have shown that the growth-inhibitory activity of TIMP-2 for human microvascular endothelial cells is mediated through binding to α3β1 integrin and induction of protein tyrosine phosphatase activity.11 Oh et al. have recently demonstrated that TIMP-2 inhibits endothelial cell migration through an indirect MMP-inhibitor effect that requires transcription, synthesis, and cell surface localization of the RECK gene product.13 Furthermore, Feldman et al. have reported that upregulation of mitogen-activated protein kinase phosphatase 1 in tumors overexpressing TIMP-2 results in dephosphorylation of p38 mitogen-activated protein kinase, leading to inhibition of tumor growth and angiogenesis.12
A number of early studies using TIMP-2 delivered as a recombinant protein or via plasmid-mediated expression demonstrated the feasibility of employing TIMP-2 for anticancer therapy. To this end, TIMP-2 was shown to block both tumor growth and local invasion through extracellular matrices. Subsequent studies demonstrated that delivery of TIMP-2 by replication-defective adenoviral vectors results in the reduction of tumor growth, angiogenesis and metastasis.15, 16 Moreover, in addition to having a direct effect on cancer growth, adenovirus-mediated delivery has demonstrated the potential of TIMP-2 in protection of normal organs against metastatic cancer cells.15
Based on this biology, we hypothesized that the efficacy of a replicating adenovirus could be enhanced by expression of TIMP-2. In this strategy, the replicating adenovirus would kill cancer cells directly from within by oncolysis, while secretion of TIMP-2 by the infected cells would complement this therapeutic effect by restricting tumor growth and angiogenesis via both MMP-dependent and –independent mechanisms.
MATERIALS AND METHODS
Viruses
Adwt300, a wild-type human adenovirus serotype 5, was obtained from the American Type Culture Collection (Manassas, VA). Ad-TIMP-2, an E1-, E3-deleted replication-deficient Ad5 vector which expresses TIMP-2 under the control of the CMV promoter, has been described previously.17 AdLacZ is an E1-, E3-deleted replication-deficient Ad5 vector which expresses β-galactosidase under the control of the CMV promoter. The wild-type adenovirus and the vectors were propagated in the permissive 293 cell line and purified by two rounds of cesium chloride density centrifugation. To determine the viral particle concentration, the virus was diluted in 10 mM Tris (pH 8.0)-1 mM EDTA-0.1% SDS, incubated at 56ºC for 10 min, and the absorbance at 260 nm was measured. Under these conditions, an absorbance of 1 corresponds to 1.1 x 1012 particles/ml.18
Cell Lines
MDA-MB-231 human breast cancer cells and LNCaP human prostate cancer cells were acquired from the American Type Culture Collection. SKOV3.ip1 human ovarian cancer cells were obtained from Janet Price (Baylor University, Houston, TX). 293 cells were purchased from Microbix (Toronto, Ontario, Canada). MDA-MB-231, SKOV3.ip1 and 293 cells were maintained in DMEM/Ham’s F-12 medium supplemented with 10 % fetal calf serum (FCS), penicillin (100 units/ml) and streptomycin (100 μg/ml). LNCaP cells were maintained in RPMI 1640 medium containing 10 % FCS, penicillin (100 units/ml), streptomycin (100 μg/ml), sodium pyruvate (1 μM) and sodium bicarbonate (1.5 g/l). All cells were propagated at 37ºC in a humidified 5% CO2 atmosphere.
FCS was purchased from Gibco-BRL, Grand Island, NY and media and supplements were from Mediatech, Herndon, VA.
Quantification of Adenoviral DNA Replication
Monolayers of MDA-MB-231, SKOV3.ip1 or LNCaP cells in 6-well plates were coinfected with Adwt300 at a multiplicity of infection (MOI) of 1 viral particle per cell and Ad-TIMP-2 at MOIs of 0, 0.1, 1 or 10 viral particles per cell. Ten days post-infection, attached and detached cells were harvested and DNA extracted using the DNeasy Tissue Kit (Qiagen, Valencia, CA). The DNA was subjected to quantitative real-time PCR using primers and a probe specific for the adenoviral E1A region (forward primer: 5’-AACCAGTTGCCGTGAGAGTTG-3’; reverse primer: 5’-CTCGTTAAGCAAGTCCTCGATACA-3’; probe: 5’-CACAGCCTGGCGACGCCCA-3’). Human beta-actin DNA was also amplified to allow normalization of the data (forward primer: 5’-TAAGTAGGCGCACAGTAGGTCTGA-3’; reverse primer: 5’-AAAGTGCAAAGAACACGGCTAAG-3’; probe: 5’-CAGACTCCCCATCCCAAGACCCCA-3’).
Adenovirus Yield Assay
Monolayers of MDA-MB-231, SKOV3.ip1 or LNCaP cells in 6-well plates were coinfected with Adwt300 at an MOI of 1 viral particle per cell and Ad-TIMP-2 at MOIs of 0, 0.1, 1 and 10 viral particles per cell. Ten days post-infection, the cells and media were harvested and the number of infectious particles determined by titering on 293 cells.19 Forty hours post-infection, 293 cells were fixed with methanol and infected cells were identified in an immunoassay using polyclonal rabbit anti-Ad5 antiserum (Cocalico, Reamstown, PA) as the primary antibody with a horseradish peroxidase-conjugated goat anti-rabbit secondary antibody (Jackson ImmunoResearch Laboratories, West Grove, PA). DAB (Sigma, St Louis, MO) was employed as the chromogenic substrate and brown cells were counted using a light microscope.
CPE Assay
Monolayers of MDA-MB-231, SKOV3.ip1 or LNCaP cells in 24-well plates were coinfected with Adwt300 at an MOI of 1 viral particle per cell and Ad-TIMP-2 at MOIs of 0, 0.1, 1 and 10 viral particles per cell. Ten days post-infection, the cells were fixed and stained with crystal violet.
In Vitro Cytotoxicity Assay
Monolayers of MDA-MB-231, SKOV3.ip1 or LNCaP cells in 96-well plates were coinfected with Adwt300 at an MOI of 1 viral particle per cell and Ad-TIMP-2 at MOIs of 0, 0.1, 1, 10 and 100 viral particles per cell. Ten days post-infection, a commercial cell proliferation assay (Promega, Madison, WI) was used to measure cell survival according to the manufacturer’s instructions.
Animal Experiments
Animal experiments were performed in accordance with federal and institutional guidelines for animal care. MDA-MB-231 tumor xenografts were established by subcutaneous injection of 4 x 106 cells into the flank of 8–10 week-old female NCr athymic nude mice (nu/nu; Taconic, Germantown, NY). On reaching 80–100mm3, the tumor nodules were injected with 50 μl PBS or with a single dose of the following virus treatments in 50 μl PBS: 106 particles of Adwt300 plus 106 particles of Ad-TIMP-2; 106 particles of Ad-TIMP-2 alone; 106 particles of Adwt300 plus 106 particles of AdLacZ, as a control (8 mice per group). Bidimensional tumor measurements were taken twice a week with calipers and the tumor volume was calculated using the simplified formula for a rotational ellipsoid: 0.5 x length x width2 (ref. 20). Animals were followed for 28 days, until the tumor burden in some of the groups became excessive, whereupon the mice were sacrificed and tumors excised.
Immunohistochemistry
Tumors excised from the treated mice were snap frozen in liquid nitrogen and cut into 50 μm sections. The sections were fixed with acetone and endogenous peroxidase activity was quenched by incubation in 0.3% (v/v) H2O2 in methanol for 30 min. Blood vessels were then visualized via a three-step staining procedure using a rat anti-mouse CD31 monoclonal antibody (BD Biosciences, San Jose, CA), followed by a biotin-conjugated goat anti-rat Ig-specific polyclonal antibody (BD Biosciences) and then an avidin: biotinylated HRP complex (Vectastain ABC kit; Vector Laboratories, Burlingame, CA). DAB was employed as the chromogenic substrate. The number of brown-stained blood vessels was counted in three microscopic fields with magnification x10 in tumor sections from three mice per group.
Statistical Analysis
Differences between groups were analyzed using the Student-Fisher t-test. P < 0.05 was considered statistically significant.
RESULTS
In this proof-of-concept study, we employed a two-component model system to investigate the hypothesis that the efficacy of a replicating adenovirus could be enhanced by arming it with TIMP-2. In this regard, coinfection of cells with a wild-type adenovirus and a replication-defective E1-deleted adenoviral vector expressing TIMP-2 allows replication of the vector as a result of trans-complementation by the E1 proteins expressed by the wild-type virus.
Expression of TIMP-2 Does Not Inhibit the Oncolytic Potency of a Replicating Adenovirus in Vitro.
Monolayers of MDA-MB-231 human breast cancer cells, SKOV3.ip1 human ovarian cancer cells and LNCaP human prostate cancer cells (which express MMP-2 and MMP-9 (refs 21–24) were coinfected with a replicating human serotype 5 adenovirus, Adwt300, at a multiplicity of infection (MOI) of 1 viral particle per cell, and a previously described replication-defective Ad5 vector expressing TIMP-2, Ad-TIMP-2 (ref 17), at MOIs of 0.1, 1 and 10 viral particles per cell. As a control, the cells were infected with Adwt300 alone. Ten days post-infection, the cells were harvested and DNA was extracted and subjected to quantitative real-time PCR analysis to determine the number of copies of the adenoviral E1 region, which is indicative of the synthesis of viral DNA by the replicating adenovirus. As shown in Fig. 1, the number of copies of the E1 gene produced by Adwt300 was not significantly affected by coinfection with Ad-TIMP-2 at MOIs of 0.1, 1 or 10 viral particles per cell (P < 0.04 for all cell lines and MOIs tested).
Fig. 1.
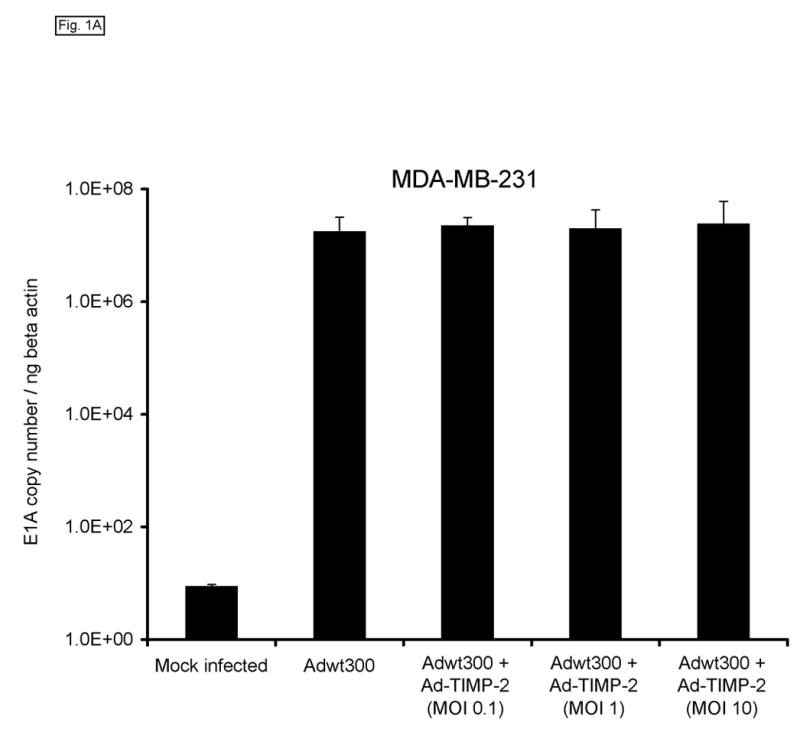
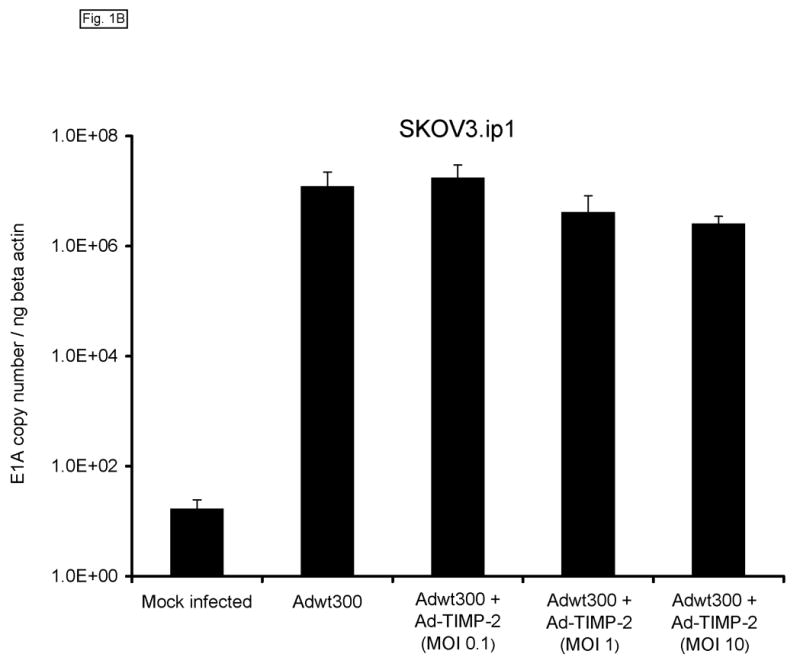
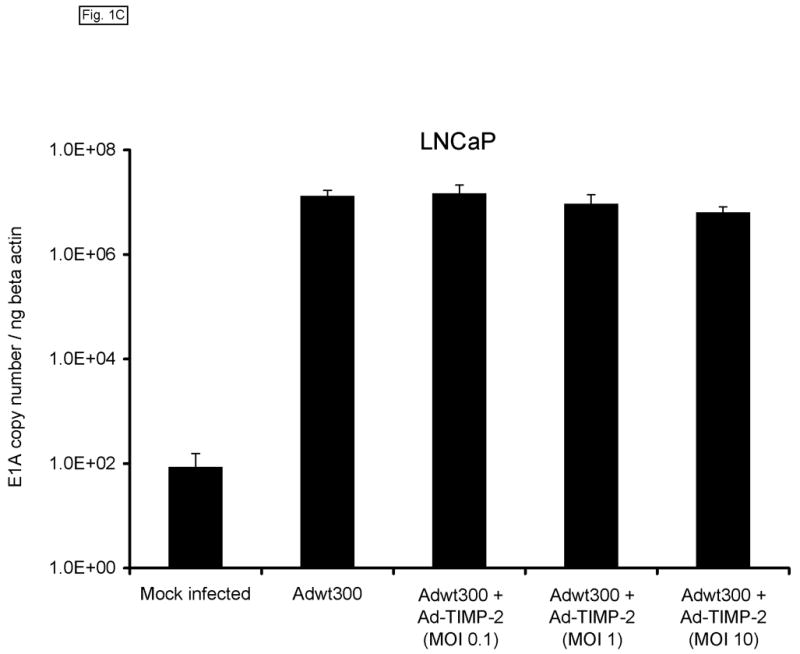
Expression of TIMP-2 does not inhibit adenoviral DNA replication. Monolayers of MDA-MB-231 (A), SKOV3.ip1 (B) and LNCaP (C) cells were coinfected with Adwt300 at an MOI of 1 viral particle per cell and Ad-TIMP-2 at MOIs of 0, 0.1, 1 or 10 viral particles per cell. Ten days post-infection, cells were harvested and DNA extracted and subjected to quantitative real-time PCR to detect the adenoviral E1A region. Human beta-actin DNA was also amplified to allow normalization of the data. Representative results are shown.
We next sought to confirm that expression of TIMP-2 did not interfere with the ability of the replicating adenovirus to produce infectious progeny. To this end, monolayers of MDA-MB-231, SKOV3.ip1 and LNCaP cells were coinfected with a replicating adenovirus, Adwt300, at an MOI of 1 viral particle per cell, and Ad-TIMP-2 at MOIs of 0.1, 1 and 10 viral particles per cell. As a control, cells were infected with Adwt300 alone. Ten days post-infection, the cells and media were harvested and the number of infectious particles was determined by titering on 293 cells.19 As shown in Fig. 2, the number of viral progeny was not significantly affected by coinfection of Adwt300 with Ad-TIMP-2 at MOIs of 0.1 or 10 viral particles per cell (P ≤ 0.05 for all cell lines and MOIs of 0.1 and 10; P = 0.06, 0.08 and 0.07 for MDA-MB-231, SKOV3.ip1 and LNCaP cells, respectively, at an MOI of 1).
Fig. 2.
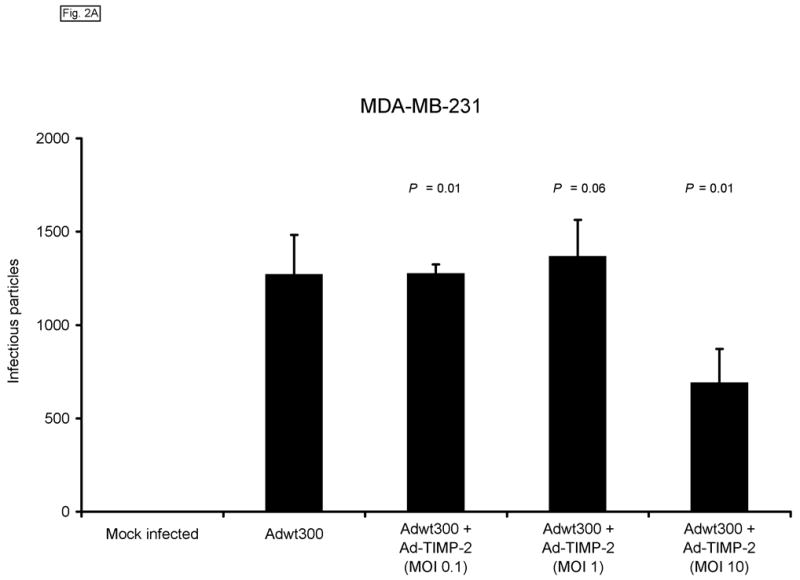
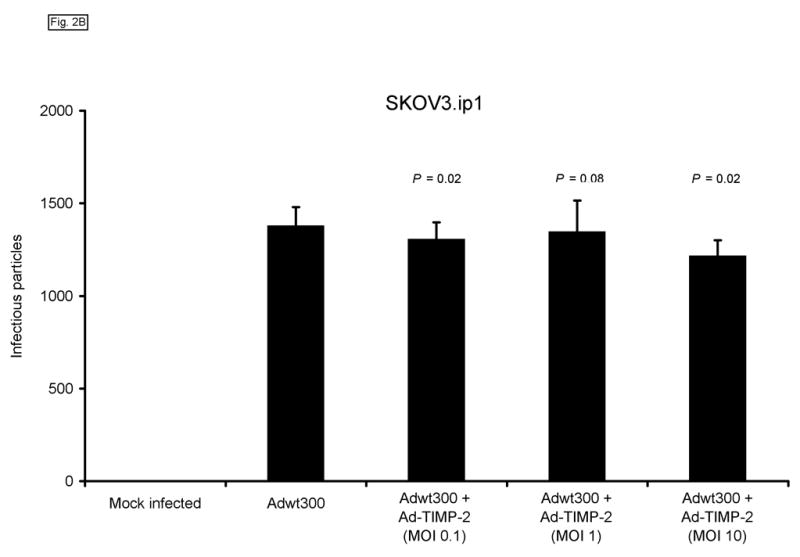
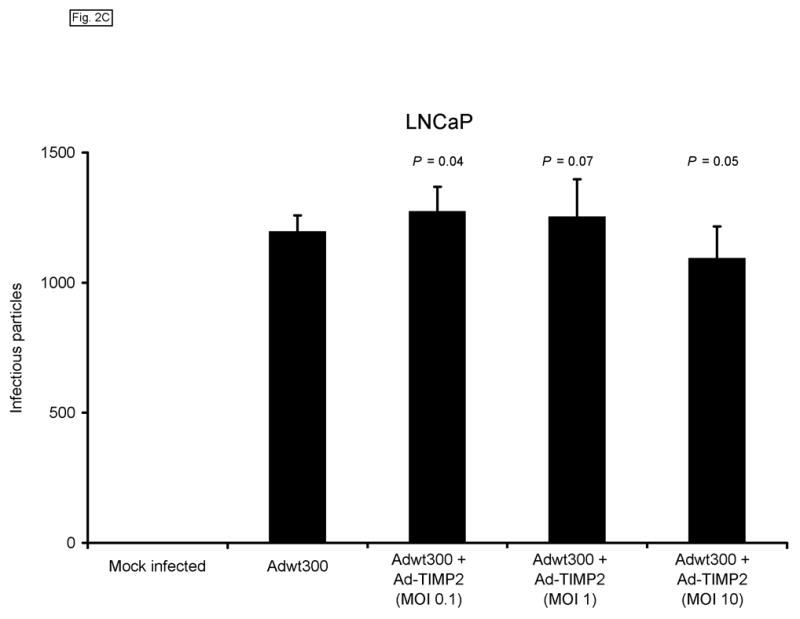
Expression of TIMP-2 does not interfere with the ability of the replicating adenovirus to produce infectious progeny. Monolayers of MDA-MB-231 (A), SKOV3.ip1 (B) and LNCaP (C) cells were coinfected with Adwt300 at an MOI of 1 viral particle per cell and Ad-TIMP-2 at MOIs of 0, 0.1, 1 or 10 viral particles per cell. Ten days post-infection, the cells and media were harvested and the number of infectious particles was determined by titering on 293 cells. Representative results are shown.
We then examined whether expression of TIMP-2 would impair the oncolytic potency of the replicating adenovirus. Monolayers of MDA-MB-231, SKOV3.ip1 and LNCaP cells were coinfected with a replicating adenovirus, Adwt300, at an MOI of 1 viral particle per cell, and Ad-TIMP-2 at MOIs of 0.1, 1 and 10 viral particles per cell. As a control, cells were infected with Adwt300 alone. Ten days post-infection, the cytopathic effect was monitored by staining proteins in the viable cells with crystal violet. As shown in Fig. 3A, expression of TIMP-2 did not inhibit oncolysis of the cancer cells by the replicating adenovirus. This finding was confirmed by a quantitative assay in which viable MDA-MB-231 cells were counted (Fig. 3B; P ≤ 0.05 for all MOIs tested).). Hence, the expression of TIMP-2 did not inhibit the oncolytic potency of the replicating adenovirus in vitro.
Fig. 3.
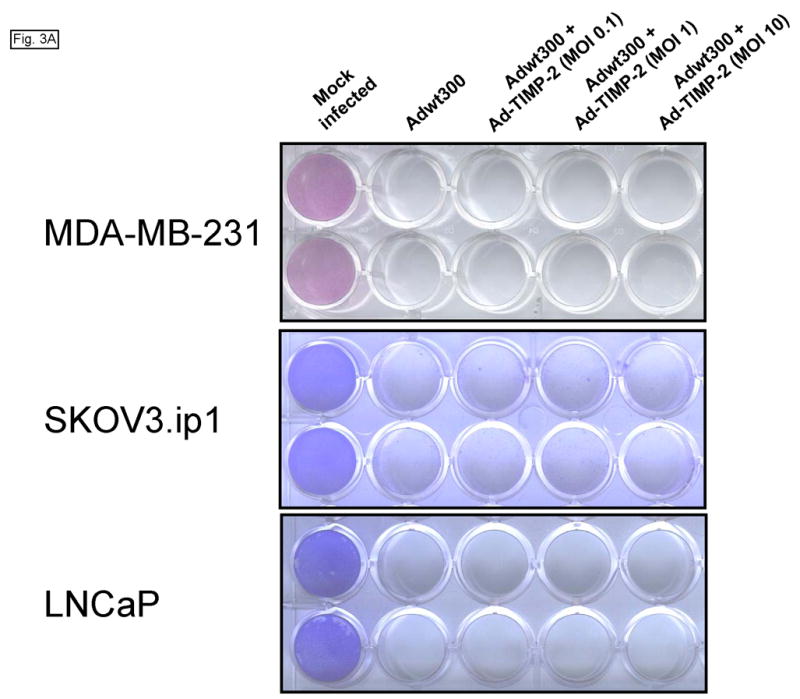
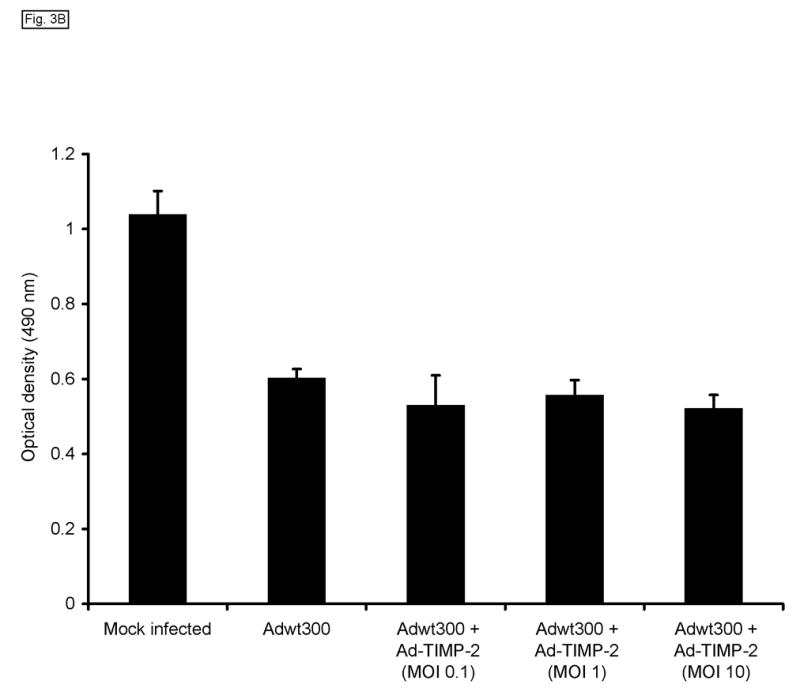
Expression of TIMP-2 does not inhibit the oncolytic potency of the replicating adenovirus. Monolayers of MDA-MB-231, SKOV3.ip1 and LNCaP cells were coinfected with Adwt300, at an MOI of 1 viral particle per cell, and Ad-TIMP-2 at MOIs of 0, 0.1, 1 and 10 viral particles per cell. (A) Ten days post-infection, the cytopathic effect was monitored by staining proteins in the viable cells with crystal violet. (B) Ten days post-infection, a cell proliferation assay was performed to count viable cells. Representative results with MDA-MB-231 cells are shown.
A Replicating Adenovirus Expresses a Greater Level of TIMP-2 Than a Replication-Defective Adenoviral Vector
We hypothesized that the level of expression of TIMP-2 that could be achieved by coinfection of the replicating adenovirus and Ad-TIMP-2 would be significantly greater than could be achieved by the replication-defective adenoviral vector alone. To investigate this, monolayers of MDA-MB-231, SKOV3.ip1 and LNCaP cells were coinfected with a replicating adenovirus, Adwt300, at an MOI of 1 viral particle per cell, and Ad-TIMP-2 at MOIs of 1 and 10 viral particles per cell. As a control, cells were infected with Ad-TIMP-2 alone at MOIs of 1 and 10 viral particles per cell. Expression and secretion of TIMP-2 were assayed by harvesting the culture medium at 3, 6, 9 and 12 days post-infection and subjecting it to immunoblot analysis using an anti-TIMP-2 monoclonal antibody. As shown in Fig. 4A, at each MOI tested, higher levels of TIMP-2 were secreted by cells coinfected with Ad-TIMP-2 and the replicating adenovirus, than cells infected with the replication-defective Ad-TIMP-2 alone. This observation was confirmed by an enzyme-linked immunosorbent assay in which the levels of TIMP-2 in the culture medium of infected MDA-MB-231 cells were quantified at various time-points post-infection (Fig. 4B).
Fig. 4.
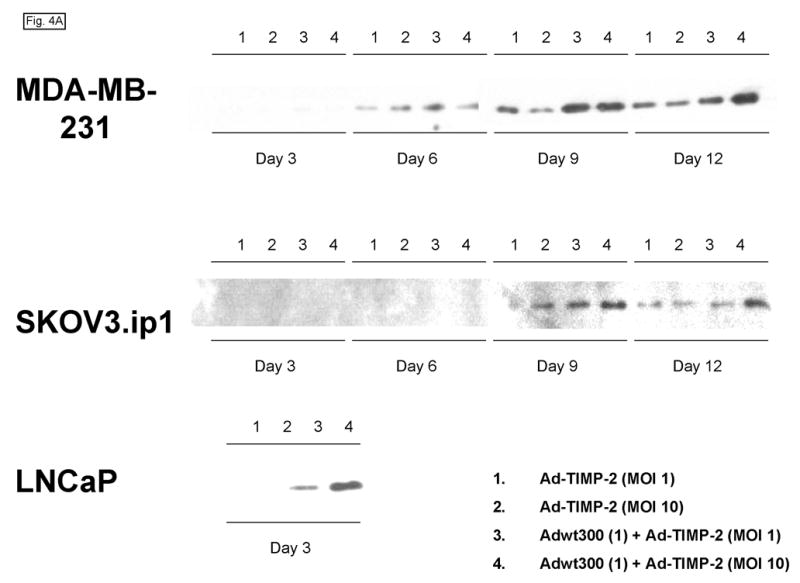
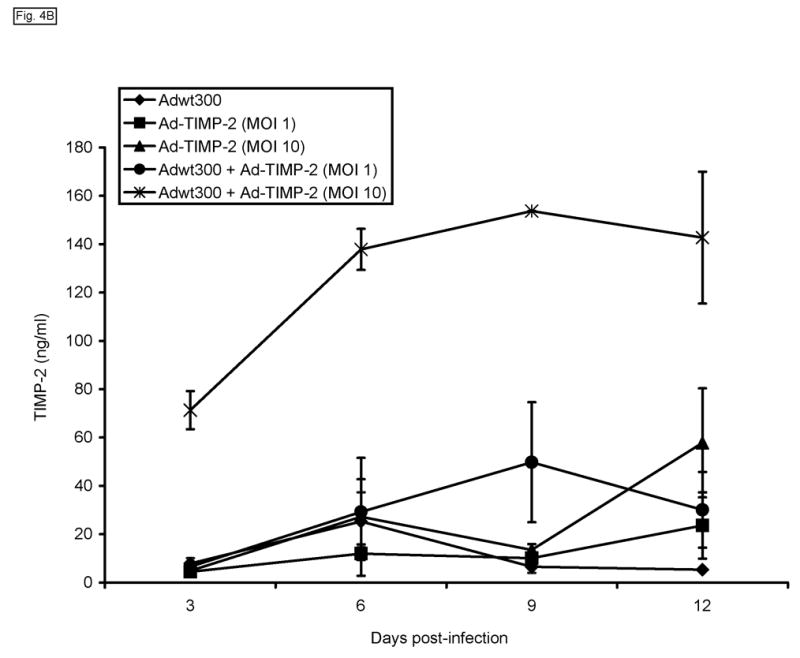
A replicating adenovirus expresses a greater level of TIMP-2 than a replication-defective adenoviral vector. Monolayers of MDA-MB-231, SKOV3.ip1 and LNCaP cells in 6-well plates were coinfected with Adwt300 at an MOI of 1 viral particle per cell and Ad-TIMP-2 at MOIs of 1 and 10 viral particles per cell. As a control, cells were infected with Ad-TIMP-2 alone at MOIs of 1 and 10 viral particles per cell. (A) Expression and secretion of TIMP-2 were assayed by harvesting the culture medium at 3, 6, 9 and 12 days post-infection and subjecting it to immunoblot analysis using an anti-TIMP-2 monoclonal antibody. (B) The levels of TIMP-2 in the culture medium of MDA-MB-231 cells at various time-points post-infection were quantified in an ELISA (Fig. 4b). Hence, a greater level of TIMP-2 was expressed by a replicating adenovirus than by a replication-defective adenoviral vector.
Expression of TIMP-2 Enhances Antitumor Efficacy of a Replicating Adenovirus in Vivo
We next wished to determine whether expression of TIMP-2 would enhance the antitumor efficacy of the replicating adenovirus in vivo. Female athymic nude mice bearing subcutaneous MDA-MB-231 xenografts on the flank were given a single intratumoral injection of one of the following treatments in 50 μl PBS: 106 particles of Adwt300 plus 106 particles of Ad-TIMP-2; 106 particles of Ad-TIMP-2 alone; 106 particles of Adwt300 plus 106 particles of AdLacZ, a control replication-defective adenoviral vector expressing β-galactosidase; or PBS alone (8 mice per group). Bidimensional tumor measurements were taken twice a week with calipers, and the tumor volume was calculated using the simplified formula for a rotational ellipsoid: 0.5 x length x width2 (ref. 20). Tumor growth kinetics are shown in Fig. 5. Twenty-eight days post-injection of the virus, tumors treated with the replicating adenovirus plus Ad-TIMP-2 were significantly smaller than tumors injected with the replicating adenovirus plus the irrelevant vector, AdLacZ (P = 0.04), with Ad-TIMP-2 alone (P = 0.03), or with PBS (P = 0.006). Hence, expression of TIMP-2 by a replicating adenovirus enhanced the inhibition of tumor growth in vivo.
Fig. 5.
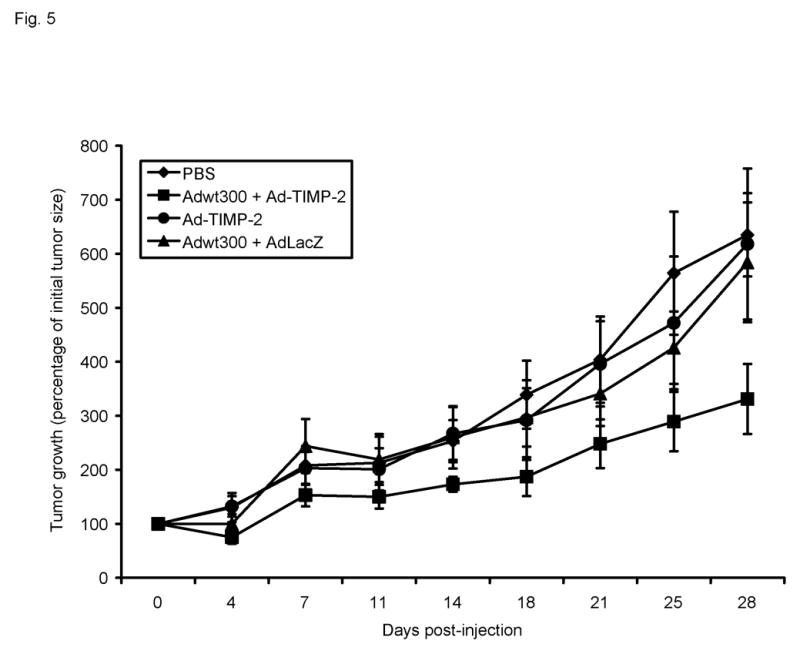
Growth kinetics of subcutaneous MDA-MB-231 tumors in athymic nude mice. Tumor nodules were injected with 50 μl PBS or with a single dose of the following virus treatments in 50 μl PBS: 106 particles of Adwt300 alone; 106 particles of Adwt300 plus 106 particles of Ad-TIMP-2; 106 particles of Ad-TIMP-2 alone; 106 particles of Adwt300 plus 106 particles of AdLacZ. Data points represent the mean ± SE of the tumor size in each group at the indicated time points (n = 8).
In addition to playing a role in tumor growth, MMP-dependent matrix proteolysis is also involved in the process of angiogenesis. To analyze angiogenesis, tumor sections from these mice were subjected to immunohistochemical staining using a primary antibody against mouse CD31 (also known as PECAM-1, platelet endothelial cell adhesion molecule) to visualize endothelial cells. As shown in Figs. 6A and B, significantly fewer blood vessels were observed in sections of tumors treated with the replicating adenovirus plus Ad-TIMP-2 compared to tumors injected with the replicating adenovirus plus the irrelevant vector, AdLacZ, with Ad-TIMP-2 alone, or with PBS (P < 0.01 for all groups tested). Hence, expression of TIMP-2 by a replicating adenovirus reduced angiogenesis in vivo.
Fig. 6.
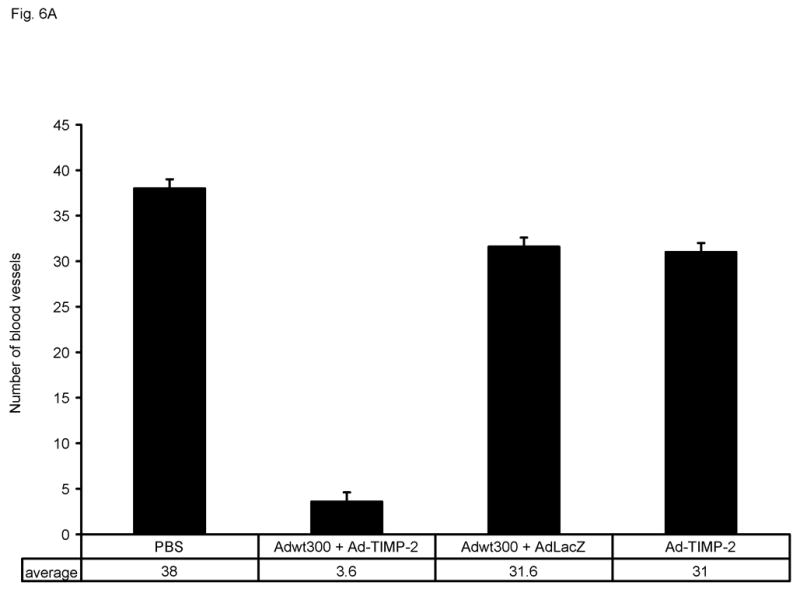
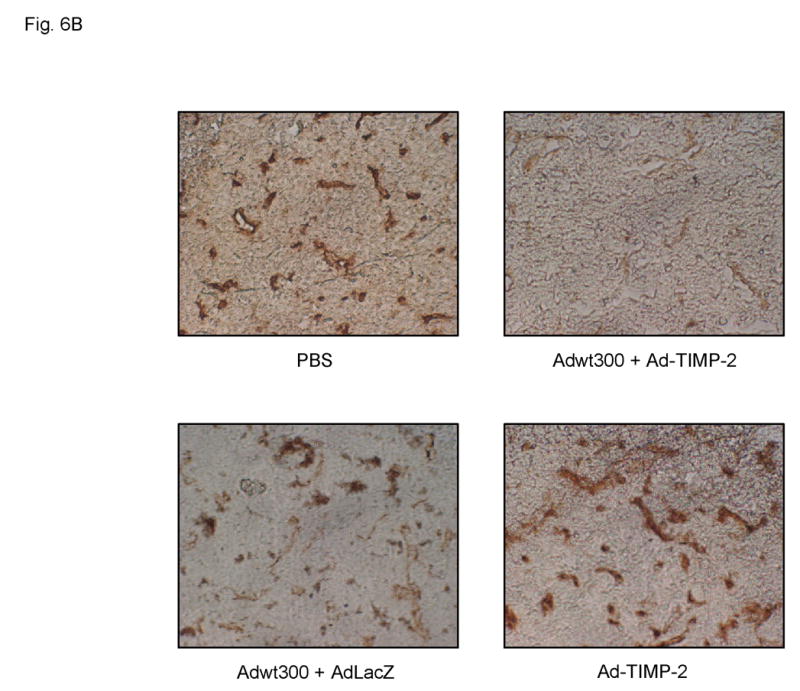
Immunohistochemical staining of blood vessels in MDA-MB-231 tumors. Tumors were excised from athymic nude mice 28 days after treatment. Tumor sections were subjected to immunohistochemistry using a rat anti-mouse CD31 monoclonal antibody as the primary antibody, followed by a biotin-conjugated goat anti-rat Ig-specific polyclonal antibody and then an avidin: biotinylated HRP complex with an HRP-conjugated goat anti-rabbit secondary antibody. DAB was employed as the chromogenic substrate. (A) Number of blood vessels per microcopic field. Data represent the mean ± SE of number of blood vessels in tumor sections from three mice per group. (B) Representative tumor sections in which the endothelia of blood vessels are stained brown.
DISCUSSION
In recognition of their potential, replication-selective adenoviruses have been rapidly translated into human clinical trials in patients with advanced cancer, where the safety of these agents has been demonstrated.3 However, clinical studies of replicating adenoviruses have yielded disappointing results, indicating the need for new strategies to improve their efficacy. In one approach to improve the efficacy of replication-selective adenoviruses, they have been engineered to deliver therapeutic transgenes.25 In most cases, such “armed” oncolytic adenoviruses have been designed to carry therapeutic genes, such as the suicide genes cytosine deaminase and herpes simplex virus thymidine kinase,26–28 that will augment the virus-mediated eradication of the infected tumor cells. However, rather than arm a replicating adenovirus with a protein which would merely act by killing the infected cancer cells, we hypothesized that it would be rational to arm the replicating adenovirus with a secreted protein with a distinct mechanism of action within the microenvironment of the cancer cells.
To this end, we propose to enhance the efficacy of a replicating adenovirus by arming it with TIMP-2. TIMP-2 possesses a number of attractive features that favor its use in this therapeutic strategy. It is a relatively small (21 kDa) unglycosylated protein that is naturally secreted in a soluble form.8 It has been recognized for some time that TIMP-2 binds in a 1:1 stoichiometric ratio to the active forms of a number of MMPs, including MT1-MMP, MMP-2 and MMP-9, thereby specifically inhibiting the MMP activity associated with tumor growth and angiogenesis.29 Indeed, in recent studies MT-MMPs have been shown to be the key mediators of angiogenesis30, 31 providing the rationale for the overexpression of TIMP-2 to directly block tumor cell growth locally. A distinct advantage of TIMP-2 over other TIMPs is that it has recently been shown to inhibit tumor growth and angiogenesis by a variety of novel mechanisms independent of MMP-inhibition.11–13 TIMP-2 is unique among the members of the TIMP family or synthetic MMP inhibitors in being able to directly inhibit the proliferation of endothelial cells in response to angiogenic stimuli such as fibroblast growth factor 2 or vascular endothelial growth factor A.11, 14 Seo et al. have shown that the growth-inhibitory activity of TIMP-2 for human microvascular endothelial cells is mediated through binding to 3ß1 integrin and induction of protein tyrosine phosphatase activity.11 Oh et al. have recently demonstrated that TIMP-2 inhibits endothelial cell migration through an indirect MMP-inhibitor effect that requires transcription, synthesis, and cell surface localization of the RECK gene product.13 Furthermore, Feldman et al. have reported that upregulation of mitogen-activated protein kinase phosphatase 1 in tumors overexpressing TIMP-2 results in dephosphorylation of p38 mitogen-activated protein kinase, leading to inhibition of tumor growth and angiogenesis.12 Thus, there is a clear rationale for a therapeutic strategy exploiting the localized overexpression of TIMP-2 in the tumor microenvironment.
We hypothesized that the replicating adenovirus would kill cancer cells directly from within by oncolysis, while secretion of TIMP-2 by the infected cells would complement this therapeutic effect by restricting tumor growth and angiogenesis via both MMP-dependent and -independent mechanisms. In the proof-of-concept study described in this manuscript, we employed a two-component model system to investigate this hypothesis. In this regard, coinfection of cells with a wild-type adenovirus and a replication-defective E1-deleted adenoviral vector expressing TIMP-2 allows replication of the vector as a result of trans-complementation by the E1 proteins expressed by the wild-type virus.
For such a dual-action, armed replicating adenovirus to be of utility, it is important that the expression of TIMP-2 should not impair its oncolytic potency. Accordingly, we demonstrated that the oncolytic potency of a replicating adenovirus in MMP-2- and MMP-9-positive MDA-MB-231 human breast cancer cells was not inhibited by expression of TIMP-2. Moreover, we showed that a greater level of TIMP-2 was expressed by a replicating adenovirus than by a replication-defective adenoviral vector. Having performed in vitro studies to establish these two key indicators of efficacy, we evaluated the expression of TIMP-2 by a replicating adenovirus in the treatment of solid tumors in vivo. Expression of TIMP-2 by the replicating adenovirus was shown both to enhance the inhibition of tumor growth and to reduce angiogenesis in vivo. We have therefore shown that the therapeutic efficacy of a replicating adenovirus can be enhanced expression of TIMP-2.
TIMP-2 plays a central role in many physiological processes by limiting MMP activity and through other non-MMP inhibitory functions.8 As such it will be important to limit TIMP-2 transgene expression locally within the tumor. Now that we have proof-of-concept for usage of TIMP-2 in the oncolytic virus setting, we can develop a single-agent armed replicating adenovirus using both transcriptional and transductional targeting approaches to limit transgene expression to the tumor without compromising efficacy.
Acknowledgments
This work was supported by National Institutes of Health grant R01 CA108585 and DOD grant W81XWH-04-1-0800.
References
- 1.Chiocca EA. Oncolytic viruses. Nat Rev Cancer. 2002;2:938–50. doi: 10.1038/nrc948. [DOI] [PubMed] [Google Scholar]
- 2.Alemany R, Balague C, Curiel DT. Replicative adenoviruses for cancer therapy. Nat Biotechnol. 2000;18:723–27. doi: 10.1038/77283. [DOI] [PubMed] [Google Scholar]
- 3.Bell JC, Lichty B, Stojdl D. Getting oncolytic virus therapies off the ground. Cancer Cell. 2003;4:7–11. doi: 10.1016/s1535-6108(03)00170-3. [DOI] [PubMed] [Google Scholar]
- 4.Liotta LA, Kohn EC. The microenvironment of the tumour-host interface. Nature. 2001;411:375–9. doi: 10.1038/35077241. [DOI] [PubMed] [Google Scholar]
- 5.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161–74. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 6.Kanayama H, Yokota K, Kurokawa Y, Murakami Y, Nishitani M, Kagawa S. Prognostic values of matrix metalloproteinase-2 and tissue inhibitor of metalloproteinase-2 expression in bladder cancer. Cancer. 1998;82:1359–66. [PubMed] [Google Scholar]
- 7.Thomas P, Khokha R, Shepherd FA, Feld R, Tsao MS. Differential expression of matrix metalloproteinases and their inhibitors in non-small cell lung cancer. J Pathol. 2000;190:150–6. doi: 10.1002/(SICI)1096-9896(200002)190:2<150::AID-PATH510>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 8.Baker AH, Edwards DR, Murphy G. Metalloproteinase inhibitors: biological actions and therapeutic opportunities. J Cell Sci. 2002;115:3719–27. doi: 10.1242/jcs.00063. [DOI] [PubMed] [Google Scholar]
- 9.Visse R, Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ Res. 2003;92:827–39. doi: 10.1161/01.RES.0000070112.80711.3D. [DOI] [PubMed] [Google Scholar]
- 10.Boone TC, Johnson MJ, De Clerck YA, Langley KE. cDNA cloning and expression of a metalloproteinase inhibitor related to tissue inhibitor of metalloproteinases. Proc Natl Acad Sci U S A. 1990;87:2800–4. doi: 10.1073/pnas.87.7.2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seo DW, Li H, Guedez L, Wingfield PT, Diaz T, Salloum R, Wei BY, Stetler-Stevenson WG. TIMP-2 mediated inhibition of angiogenesis: an MMP-independent mechanism. Cell. 2003;114:171–80. doi: 10.1016/s0092-8674(03)00551-8. [DOI] [PubMed] [Google Scholar]
- 12.Feldman AL, Stetler-Stevenson WG, Costouros NG, Knezevic V, Baibakov G, Alexander HR, Jr, Lorang D, Hewitt SM, Seo DW, Miller MS, O'Connor S, Libutti SK. Modulation of tumor-host interactions, angiogenesis, and tumor growth by tissue inhibitor of metalloproteinase 2 via a novel mechanism. Cancer Res. 2004;64:4481–6. doi: 10.1158/0008-5472.CAN-03-2929. [DOI] [PubMed] [Google Scholar]
- 13.Oh J, Seo DW, Diaz T, Wei B, Ward Y, Ray JM, Morioka Y, Shi S, Kitayama H, Takahashi C, Noda M, Stetler-Stevenson WG. Tissue inhibitors of metalloproteinase 2 inhibits endothelial cell migration through increased expression of RECK. Cancer Res. 2004;64:9062–9. doi: 10.1158/0008-5472.CAN-04-1981. [DOI] [PubMed] [Google Scholar]
- 14.Fernandez CA, Butterfield C, Jackson G, Moses MA. Structural and functional uncoupling of the enzymatic and angiogenic inhibitory activities of tissue inhibitor of metalloproteinase-2 (TIMP-2): loop 6 is a novel angiogenesis inhibitor. J Biol Chem. 2003;278:40989–95. doi: 10.1074/jbc.M306176200. [DOI] [PubMed] [Google Scholar]
- 15.Brand K, Baker AH, Perez-Canto A, Possling A, Sacharjat M, Geheeb M, Arnold W. Treatment of colorectal liver metastases by adenoviral transfer of tissue inhibitor of metalloproteinases-2 into the liver tissue. Cancer Res. 2000;60:5723–30. [PubMed] [Google Scholar]
- 16.Li H, Lindenmeyer F, Grenet C, Opolon P, Menashi S, Soria C, Yeh P, Perricaudet M, Lu H. AdTIMP-2 inhibits tumor growth, angiogenesis, and metastasis, and prolongs survival in mice. Hum Gene Ther. 2001;12:515–26. doi: 10.1089/104303401300042429. [DOI] [PubMed] [Google Scholar]
- 17.Baker AH, Wilkinson GW, Hembry RM, Murphy G, Newby AC. Development of recombinant adenoviruses that drive high level expression of the human metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 and -2 genes: characterization of their infection into rabbit smooth muscle cells and human MCF-7 adenocarcinoma cells. Matrix Biol. 1996;15:383–95. doi: 10.1016/s0945-053x(96)90158-4. [DOI] [PubMed] [Google Scholar]
- 18.Mittereder N, March KL, Trapnell BC. Evaluation of the concentration and bioactivity of adenovirus vectors for gene therapy. J Virol. 1996;70:7498–509. doi: 10.1128/jvi.70.11.7498-7509.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bewig B, Schmidt WE. Accelerated titering of adenoviruses. Biotechniques. 2000;28:870–3. doi: 10.2144/00285bm08. [DOI] [PubMed] [Google Scholar]
- 20.Dethlefsen LA, Prewitt JM, Mendelsohn ML. Analysis of tumor growth curves. J Natl Cancer Inst. 1968;40:389–405. doi: 10.1093/jnci/40.2.389. [DOI] [PubMed] [Google Scholar]
- 21.Yoneda T, Sasaki A, Dunstan C, Williams PJ, Bauss F, De Clerck YA, Mundy GR. Inhibition of osteolytic bone metastasis of breast cancer by combined treatment with the bisphosphonate ibandronate and tissue inhibitor of the matrix metalloproteinase-2. J Clin Invest. 1997;99:2509–17. doi: 10.1172/JCI119435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Afzal S, Lalani EN, Poulsom R, Stubbs A, Rowlinson G, Sato H, Seiki M, Stamp GW. MT1-MMP and MMP-2 mRNA expression in human ovarian tumors: possible implications for the role of desmoplastic fibroblasts. Hum Pathol. 1998;29:155–65. doi: 10.1016/s0046-8177(98)90226-x. [DOI] [PubMed] [Google Scholar]
- 23.Huang S, Van Arsdall M, Tedjarati S, McCarty M, Wu W, Langley R, Fidler IJ. Contributions of stromal metalloproteinase-9 to angiogenesis and growth of human ovarian carcinoma in mice. J Natl Cancer Inst. 2002;94:1134–42. doi: 10.1093/jnci/94.15.1134. [DOI] [PubMed] [Google Scholar]
- 24.Wilson SR, Gallagher S, Warpeha K, Hawthorne SJ. Amplification of MMP-2 and MMP-9 production by prostate cancer cell lines via activation of protease-activated receptors. Prostate. 2004;60:168–74. doi: 10.1002/pros.20047. [DOI] [PubMed] [Google Scholar]
- 25.Hermiston T. Gene delivery from replication-selective viruses: arming guided missiles in the war against cancer. J Clin Invest. 2000;105:1169–72. doi: 10.1172/JCI9973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Freytag SO, Rogulski KR, Paielli DL, Gilbert JD, Kim JH. A novel three-pronged approach to kill cancer cells selectively: concomitant viral, double suicide gene, and radiotherapy. Hum Gene Ther. 1998;9:1323–33. doi: 10.1089/hum.1998.9.9-1323. [DOI] [PubMed] [Google Scholar]
- 27.Wildner O, Morris JC, Vahanian NN, Ford H, Jr, Ramsey WJ, Blaese RM. Adenoviral vectors capable of replication improve the efficacy of HSVtk/GCV suicide gene therapy of cancer. Gene Ther. 1999;6:57–62. doi: 10.1038/sj.gt.3300810. [DOI] [PubMed] [Google Scholar]
- 28.Wildner O, Blaese RM, Morris JC. Therapy of colon cancer with oncolytic adenovirus is enhanced by the addition of herpes simplex virus-thymidine kinase. Cancer Res. 1999;59:410–3. [PubMed] [Google Scholar]
- 29.Coussens LM, Fingleton B, Matrisian LM. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science. 2002;295:2387–92. doi: 10.1126/science.1067100. [DOI] [PubMed] [Google Scholar]
- 30.Chun TH, Sabeh F, Ota I, Murphy H, McDonagh KT, Holmbeck K, Birkedal-Hansen H, Allen ED, Weiss SJ. MT1-MMP-dependent neovessel formation within the confines of the three-dimensional extracellular matrix. J Cell Biol. 2004;167:757–67. doi: 10.1083/jcb.200405001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sabeh F, Ota I, Holmbeck K, Birkedal-Hansen H, Soloway P, Balbin M, Lopez-Otin C, Shapiro S, Inada M, Krane S, Allen E, Chung D, Weiss SJ. Tumor cell traffic through the extracellular matrix is controlled by the membrane-anchored collagenase MT1-MMP. J Cell Biol. 2004;167:769–81. doi: 10.1083/jcb.200408028. [DOI] [PMC free article] [PubMed] [Google Scholar]