

Abstract
The protein pirin, which is involved in a variety of biological processes, is conserved from prokaryotic microorganisms, fungi, and plants to mammals. It acts as a transcriptional cofactor or an apoptosis-related protein in mammals and is involved in seed germination and seedling development in plants. In prokaryotes, while pirin is stress induced in cyanobacteria and may act as a quercetinase in Escherichia coli, the functions of pirin orthologs remain mostly uncharacterized. We show that the Serratia marcescens pirin (pirinSm) gene encodes an ortholog of pirin protein. Protein pull-down and bacterial two-hybrid assays followed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and electrospray ionization-tandem mass spectrometry analyses showed the pyruvate dehydrogenase (PDH) E1 subunit as a component interacting with the pirinSm gene. Functional analyses showed that both PDH E1 subunit activity and PDH enzyme complex activity are inhibited by the pirinSm gene in S. marcescens CH-1. The S. marcescens CH-1 pirinSm gene was subsequently mutated by insertion-deletion homologous recombination. Accordingly, the PDH E1 and PDH enzyme complex activities and cellular ATP concentration increased up to 250%, 140%, and 220%, respectively, in the S. marcescens CH-1 pirinSm mutant. Concomitantly, the cellular NADH/NAD+ ratio increased in the pirinSm mutant, indicating increased tricarboxylic acid (TCA) cycle activity. Our results show that the pirinSm gene plays a regulatory role in the process of pyruvate catabolism to acetyl coenzyme A through interaction with the PDH E1 subunit and inhibiting PDH enzyme complex activity in S. marcescens CH-1, and they suggest that pirinSm is an important protein involved in determining the direction of pyruvate metabolism towards either the TCA cycle or the fermentation pathways.
The protein pirin is widely found in mammals, plants, fungi, and also prokaryotic organisms (32). While the cellular functions of pirin show diversity and pirin homologs play important roles in a number of different biological processes, cellular localization of pirin is not restricted to specific compartments. In eukaryotes, pirin was initially isolated through a yeast two-hybrid screen from the HeLa cell cDNA library and is localized within cell nuclei; it acts as an interactor with nuclear factor I/CCAAT box transcription factor (32). In an attempt to identify downstream nuclear targets of Bcl-3 by using yeast two-hybrid screening of the expression cDNA library derived from human activated B cells, it was found that pirin interacts with and increases the DNA-binding activity of Bcl-3-p50 complex (Bcl-3 is a member of the IκB family that inhibits NF-κB activity) (6). A recent report from Orzaez et al. further showed that lepirin, a tomato homolog of human pirin, is involved in programmed cell death (21). On the other hand, in Arabidopsis, a pirin ortholog (AtPirin1) was isolated as a protein interacting with the α subunit of G protein through the yeast two-hybrid screen. AtPirin1 is involved in the regulation of seed germination and early seedling development of Arabidopsis (16). The human pirin crystalline structure was subsequently determined by Pang et al. (22), who showed that pirin comprises two β-barrel domains, with a potential Fe(II) cofactor bound within the cavity of the N-terminal domain. These findings suggest an enzymatic role for pirin, most likely in biological redox reactions involving oxygen (22).
In prokaryotes, pirA, encoding an ortholog of pirin, together with an adjacent gene, pirB, is up-regulated under high salinity and some other stress conditions in the cyanobacterium Synechocystis sp. strain PCC 6803 (13). However, induction of the pirAB genes is not related to programmed cell death, and disruption of pirA did not affect the cellular gene expression profile (13). Adams and Jia (1) determined the crystalline structure of the pirin homolog YhhW from Escherichia coli, in which the YhhW structure shows similarity to human pirin. Through active-site analysis, YhhW was further shown to act as a 2,3-dioxygenase that degrades the antioxidant quercetin to 2-protocatechuoylphloroglucinol, with concomitant release of CO as a by-product (1).
Previous studies on selection for precocious-swarming mutants derived from Serratia marcescens CH-1 by transposon mutagenesis (15, 28) identified a mutant strain in which a pirin gene homolog was inserted by use of a mini-Tn5 transposon (P.-C. Soo and H.-C. Lai, unpublished data). In comparison to human pirin, which is a 32-kDa protein consisting of 290 amino acids, and to E. coli pirin (YhhW), which is a 25.4-kDa protein with 231 amino acids, bioinformatic analyses identified a 312-amino-acid, 35-kDa pirin ortholog (pirinSm) in S. marcescens strain Db11 (Sanger Institute; http://www.sanger.ac.uk/cgi-bin/BLAST/submitblast/s_marcescens). Subsequently, a 5-kb pirinSm gene locus was cloned and sequenced in S. marcescens strain CH-1. In this study, using protein pull-down and bacterial two-hybrid screening assays followed by protein identification by electrospray ionization-tandem mass spectrometry (ESI-MS/MS) analyses, we showed that the pirin ortholog in S. marcescens CH-1 interacts with the E1 subunit of pyruvate dehydrogenase (PDH) complex. PDH E1 is one of the three subunits (E1, pyruvate dehydrogenase; E2, dihydrolipoamide dehydrogenase transacetylase; and E3, lipoamide dehydrogenase) of the PDH multienzyme complex, which is an assemblage that plays a pivotal role in cellular carbohydrate metabolism, catalyzing the oxidative decarboxylation of pyruvate and the subsequent acetylation of coenzyme A (CoA) to form acetyl-CoA (5, 19). During the process of PDH enzyme complex reactions, PDH E1 is responsible for the first step of the multistep process and catalyzes pyruvate decarboxylation, followed by transferring the hydroxyethyl group to thiamine diphosphate (ThDP), which together with Mg2+ acts as the reaction cofactor (7). Subsequent gene deletion and biochemical analyses showed that pirinSm regulated (inhibited) PDH E1 and PDH enzyme complex activities. In accordance, the cellular ATP concentration and NADH/NAD+ ratio increased in the pirinSm gene-deleted S. marcescens mutant grown to late logarithmic phase. These results show a new role of pirinSm involving in the regulation of pyruvate catabolism to acetyl-CoA. This may subsequently affect cellular central carbohydrate metabolism to go towards the tricarboxylic acid (TCA) cycle or fermentation pathway.
MATERIALS AND METHODS
Bacterial strains, plasmids, primers, and culture conditions.
S. marcescens CH-1 (28) is a clinical isolate routinely maintained at 37°C on Luria-Bertani (LB) plates. The chromosomal DNA sequence of S. marcescens Db11 was determined at the Sanger Institute (http://www.sanger.ac.uk/cgi-bin/BLAST/submitblast/s_marcescens). The bacterial strains, plasmids, and primers used in this study are described in Table 1.
TABLE 1.
Bacterial strains, plasmids, and primers used in this study
| Strain, plasmid, or primer | Relevant characteristics | Source or reference |
|---|---|---|
| Strains | ||
| Serratia marcescens | ||
| CH-1 | Clinical isolate | 28 |
| PC103 | CH-1 pirinSm::Sm, Smr | This study |
| Escherichia coli | ||
| CC118(λpir) | λ−-pir lysogen of CC118 [Δ(ara-leu) araD ΔlacX 74 galE galK phoA20 thi-1 rpsE rpoB argE(Am) recA1]; permissive host for suicide plasmids requiring the Pir protein | 23 |
| S17-1(λpir) | λ−-pir lysogen of S17-1 [thi pro hsdR hsdM+recA RP4 2-Tc::Mu-Km::Tn7 (Tpr Smr)]; permissive host able to transfer suicide plasmids requiring the Pir protein by conjugation to recipient cells | 23 |
| BL21(DE3) | E. coli B F−ompT hsdS (rB− mB−) dcm+ Tetrgal (DE3) endA Hte | Stratagene |
| Plasmids | ||
| pUT-Sm | Suicide plasmid requiring the Pir protein for replication and containing a mini-Tn5 cassette containing Smr genes | 23 |
| pKT25 | Encodes the T25 fragment of B. pertussis adenylate cyclase, corresponding to the first 224 amino acids of CyaA | 14 |
| pKT25-zip | Leucine zipper of GCN4 genetically fused in frame to the T25 fragment | 14 |
| pUT18 | Encodes the T18 fragment (amino acids 225 to 399 of CyaA) | 14 |
| pUT18C-zip | Leucine zipper of GCN4 genetically fused in frame to the T18 fragment; pKT25-zip and pUT18C-zip serve as positive controls for complementation | 14 |
| pBAD18-Kan | Cloning and expression vector, arabinose regulation; Kmr | 28 |
| pGEX | Expression vector, GST Tag | Pharmacia Biotech |
| pSC10 | pGEX::pirinSm; Ampr | This study |
| pSC11 | pGEX::nlpBSm; Ampr | This study |
| pSC12 | pGEX::aceESm; Ampr | This study |
| pSC15 | pBAD18-Kan::pirinSm; Kmr | This study |
| pSC16 | pUT-Sm derivative carrying pirinSm gene inserted with Smr omega cassette; Smr | This study |
| pSC17 | pKT25::pirinSm; Kmr | This study |
| pSC18 | pUT18::aceESm; Ampr | This study |
| pSC19 | pGEX::aceFSm; Ampr | This study |
| pSC20 | pGEX::lpdASm; Ampr | This study |
| pRSET | Expression vector, His Tag | Invitrogen |
| pBG20 | pRSET::His-aceESm; a 2.7-kb DNA fragment containing the PDH E1 gene (aceESm) from S. marcescens CH-1 was PCR amplified and cloned into pRSET vector; Ampr | This study |
| Primers | ||
| Db11-pir-F | 5′-ATGAGTCAGCCACGTCCT-3′ | |
| Db11-pir-R | 5′-GGCAGGCGTGGTGCGGTCA-3′ | |
| Pirk-1 | 5′-GTCGACGGCCGGAGCGACCAAGT-3′ | |
| Pirk-2 | 5′-AAGCTTAACACAGGACAGCTCCC-3′ | |
| Pirk-3 | 5′-AAGCTTGCCATACCAAGGCTGA-3′ | |
| Pirk-4 | 5′-GATATCGACATCTGACTAACGGAGC-3′ |
Enzymes and chemicals.
DNA restriction and modification enzymes were purchased from Roche (Mannheim, Germany). Taq polymerase and PCR-related products were obtained from either Perkin-Elmer (Boston, MA) or Takara Biomedicals (Shiga, Japan). Other laboratory-grade chemicals were purchased from Sigma Chemical Company (St. Louis, MO) or Merck (Schwabach, Germany).
Recombinant DNA techniques.
Unless otherwise indicated, standard protocols were used for DNA/DNA hybridization, plasmid and chromosomal DNA preparation, transformation, electroporation, PCR, restriction digestion, agarose gel electrophoresis, DNA recovery from agarose gels, DNA ligation, and conjugation. Southern blotting analysis of chromosomal DNA was performed using nylon membranes (Hybond N+; Amersham, Piscataway, NJ) and a DIG High Prime labeling kit (Roche) according to the recommendations of the manufacturer. PCR DNA amplicons were cloned using pCR 2.1 and the TA cloning kit (Invitrogen, Carlsbad, CA). DNA sequencing and analysis were performed using a Perkin-Elmer Autosequencer model 377 with a Taq DyeDeoxy terminator cycle sequencing kit (Applied Biosystems, Foster, CA). The DNA sequences of PCR products were confirmed by sequencing both strands from two or three independent reactions.
Cloning the pirinSm gene locus in S. marcescens CH-1.
The primers Db11-pir-F and Db11-pir-R (Table 1), designed from the S. marcescens Db11 pirin DNA sequence, were used to amplify a 924-bp partial pirin DNA fragment showing high nucleic acid sequence identity (99%) to the Db11 pirin gene from S. marcescens strain CH-1. This DNA fragment was subsequently labeled and used as a probe to clone the pirinSm gene locus from S. marcescens CH-1. Conventional restriction digestions, Southern blot hybridizations, cloning, and sequencing identified a 5-kb DNA fragment containing the pirinSm gene (Fig. 1).
FIG. 1.

Genetic map of the S. marcescens pirin gene locus and construction of the pirinSm gene deletion mutant S. marcescens PC103. (A) Genetic maps of S. marcescens Db11 and CH-1 pirin gene loci. The LysM domain (2) and AraC family (29) are proteins encoded, respectively, by the two genes identified upstream and downstream of the pirinSm gene. (B) Construction of the pirinSm gene knockout mutant strain PC103 by insertion of a Sm resistance gene Ω cassette into the S. marcescens CH-1 pirin gene through homologous recombination (28). pSC16 is a recombinant suicide vector for insertion-deletion mutagenesis of the pirinSm gene. (C) Western blot analysis of pirinSm production. Panel i, whole-cell crude extracts were separated by 12.5% SDS-PAGE and subjected to Coomassie brilliant blue staining. Panel ii, results of pirinSm detection using anti-pirinSm polyclonal antibodies. Lane 1, S. marcescens CH-1; lane 2, S. marcescens PC103; lane 3, S. marcescens PC103 transformed with pSC15(pBAD18-Kan::pirinSm); lane M, protein marker.
Construction of S. marcescens CH-1 pirinSm gene insertion-deletion mutant PC103.
For construction of the pirinSm gene mutant, a protocol was designed for specific insertion of a 2-kb streptomycin (Sm)-resistant Ω cassette, excised from pHP45Ω, into the pirinSm gene in S. marcescens CH-1 (23, 28). Briefly, the 5′ region of the pirinSm gene was amplified by PCR using primer pair Pirk1 and Pirk2, TA cloned into pCR 2.1 (Invitrogen), and excised as a SalI/HindIII fragment. A second PCR product encompassing the 3′ region of the pirinSm gene was generated using primer pair Pirk3 and Pirk4, TA cloned into pCR 2.1 (Invitrogen), and excised as a HindIII/EcoRI fragment. The two DNA fragments together with the Ω cassette were ligated with the SalI/EcoRI-digested pUT-mini-Tn5-Km1 suicide vector (8) to form plasmid pUT-pirinSm::Sm.
For gene inactivation by homologous recombination, pUT-pirinSm::Sm was transferred from E. coli S17-1(λ pir) to S. marcescens CH-1 by conjugation (15). Transconjugants were spread on LB plates containing streptomycin (100 μg/ml) and tetracycline (13 μg/ml). Mutant candidates were screened by colony PCR. Southern blot hybridization using the pirinSm gene as a probe was performed to confirm the mutant genotype in which a double-crossover event had occurred (data not shown). The resultant pirinSm mutant strain was designated S. marcescens PC103.
Western blot analysis.
The Western blot procedures were modified from those described by Sambrook et al. (24). In brief, bacterial cells harvested were washed once in phosphate-buffered saline (PBS) (pH 7.5) and resuspended in cell lysis buffer [20 mM piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES) (pH 7.2), 100 mM NaCl, 1 mM phenylmethylsulfonyl fluoride). Samples were left for 30 min on ice and centrifuged at 14,000 rpm for 30 min at 4°C. The spent supernatants were then concentrated and analyzed by 12.5% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The separated proteins were transferred to a polyvinylidene difluoride membrane (Amersham) and incubated in blocking buffer (5% milk, 0.1% Tween 20) for 1 h. Further incubations with anti-His monoclonal antibody or anti-pirinSm polyclonal antibodies were then performed in blocking buffer for 1 h at room temperature, followed by treatment with horseradish peroxidase-conjugated anti-rabbit second antibody for another 1 h before development and X-ray film exposure (28).
GST pull-down assay.
The ProFound pull-down glutathione S-transferase (GST) protein-protein interaction kit (Pierce, Rockford, IL) was used for GST pull-down assay. Oversynthesis of GST fusion proteins was achieved by culturing E. coli DH5α cells containing recombinant plasmids pSC10 (GST-tagged pirinSm fusion protein in pGEX) in 3 ml of LB broth medium to the mid-logarithmic phase at 37°C, followed by addition of IPTG (isopropyl-β-d-thiogalactopyranoside) at a final concentration of 0.5 mM for induction. After further culture for 3 h, cells were centrifuged and then suspended in 100 μl of lysis buffer (20 mM Tris-HCl (pH 8.0), 100 mM NaCl, 1 mM EDTA, 0.5% NP-40) containing leupeptin (1 μg/ml), pepstatin A (1 μg/ml), and phenylmethylsulfonyl fluoride (1 mM). Glutathione-Sepharose 4B beads (20 μl) (Amersham) were then added to the spent supernatant, and the mixture was incubated with mild shaking for 2 h at 4°C. Beads washed three times with PBS (pH 7.4)-1% Triton X-100 buffer were subsequently added to 500 μl of spent cell lysates of S. marcescens CH-1 or 0.5 mM IPTG-induced E. coli BL21(DE3)(pBG20). pBG20 is a recombinant plasmid containing His-tagged S. marcescens PDH E1 (AceESm) fusion proteins. The reaction mixture was incubated at 4°C for 1 h to allow interaction between GST-pirinSm and the His-tagged fusion proteins. Beads were subsequently washed with PBS (pH 7.4)-1% Triton X-100 buffer, and proteins were eluted with 10 mM glutathione before separation by 12.5% SDS-PAGE and detection by immunoblotting.
Bacterial two-hybrid screening.
The bacterial two-hybrid system used in this study is based on interaction-mediated reconstitution of adenylate cyclase activity in an E. coli host (14). Briefly, interaction between T25-pirinSm and PDH-E1-T18 fusion proteins leads to the cytoplasmic production and assembly of functional adenylate cyclase in E. coli DHM1. This was detected qualitatively by the ability of E. coli cells to ferment maltose on maltose-MacConkey agar plates (Difco, Franklin, NJ) to form red colonies (17). IPTG (0.5 mM) was included in the medium to induce full expression and synthesis of hybrid proteins when necessary.
Protein identification.
In-gel digestion and mass spectrometric analysis were performed at EverNew Biotechnology, Inc. (Taipei, Taiwan), using procedures described previously (30, 34). Briefly, the gel piece was washed in 1 ml of 25 mM NH4HCO3 for 10 min and then in 1 ml of 25 mM NH4HCO3-50% acetonitrile for 10 min. After drying in a SpeedVac (ThermoSavant, Waltham, MA), the gel was incubated with 50 ml of 2% (vol/vol) β-mercaptoethanol in the dark for 20 min. Following incubation, an equal volume of 10% (vol/vol) vinylpyridine in 25 mM NH4HCO3-50% acetonitrile was added. After 20 min of incubation, the gel was washed several times in 1 ml of 25 mM NH4HCO3 and then dehydrated in 25 mM NH4HCO3 with 50% acetonitrile. The SpeedVac-dried gel was then treated with 50 ng of modified trypsin (Promega, Madison, WI) in an adequate volume of 25 mM NH4HCO3 at 37°C overnight. The resultant peptides were extracted with 200 μl of 0.1% formic acid, dried in the SpeedVac, and then stored at −20°C until further use.
Electrospray ionization-tandem mass spectrometry was performed using a ThermoFinnigan LCQ Deca ion trap mass spectrometer (Waltham, MA) interfaced with an Agilent 1100 high-pressure liquid chromatography (HPLC) system (Agilent Technologies, Palo Alto, CA). A 150- by 0.3-mm Agilent ZORBAX 300SB C18 column (3-μm particle diameter, 300-Å pore size) with mobile phases A (0.1% formic acid in water) and B (0.085% formic acid in acetonitrile) was used. The peptides were eluted at a flow rate of 5 μl/min, with gradients that consisted of 5% to 16% B over 5 min, 16% to 20% B over 40 min, and 20% to 65% B over 40 min.
The spectra for the eluates were acquired as successive sets of three scan modes, i.e., MS, zoom, and MS scans, as described previously (30, 34). The MS scan determined the intensity of the ions in the m/z range of 395 to 1605, and a specific ion was selected for the zoom and MS/MS scans. The former examined the charge number of the selected ion, and the latter acquired the spectrum (collision-induced dissociation [CID] spectrum or MS/MS spectrum) for the fragment ions derived by CID. In the first analysis, the most abundant ion in an MS spectrum was selected for the CID experiment; in the second analysis, only the ions with m/z values corresponding to the potential phosphopeptides were selected for the CID experiment. The acquired CID spectra were interpreted using TurboSequest software (ThermoFinnigan), which matched tandem mass spectra against a nonredundant protein database.
Synthesis and purification of S. marcescens PDH E1, E2, and E3 subunits.
PDH subunits E1, E2, and E3 were overproduced in E. coli DH5α cells containing pSC12, pSC19, and pSC20, respectively. Briefly, overnight cultures of E. coli cells transformed with each recombinant plasmid were diluted 1:10 in fresh LB medium and grown for 2 h at 37°C before addition of 0.1 mM IPTG. After another 3 h of growth, cells were centrifuged and resuspended in lysis buffer (20 mM Tris-HCl [pH 8.0], 100 mM NaCl, 1 mM EDTA, 1% TritonX-100). Cells were lysed by sonication followed by centrifugation at 10,000 × g for 20 min at 4°C. The supernatant was then subjected to glutathione-Sepharose purification (26).
PDH E1 and PDH enzyme complex activity assays.
Determination of PDH E1 was achieved by an assay that measures the rate of reduction of the artificial electron acceptor 2,6-dichlorophenolindophenol (DCPIP) by the E1 component, with pyruvate as a substrate (DCPIP assay) (12). The decrease in absorbance at 600 nm was monitored at 30°C in a suspension comprising 0.2 mM ThDP, 2 mM MgCl2, 50 μM DCPIP, 100 mM potassium phosphate (pH 7.0), and spent crude protein extracts (20 to 50 μg), purified E1 protein, or other protein reactants at of 15 μg each. After incubation at 30°C for 10 min, reactions were initiated by addition of pyruvate (final concentration of 400 μM), followed by monitoring the amount of DCPIP reduced over the reaction period of up to 2 h. DCPIP reduction was calculated by the absorbance change, using a molar absorption constant of ɛ600 = 11,000 M−1 cm−1 for DCPIP (35).
PDH enzyme complex catalytic activity was measured using the PDH assay (5). The PDH assay measured the rate of NADH formation by absorbance at 340 nm at 30°C (5, 10). The reaction was started by addition of final concentrations of 2 mM pyruvate and 0.13 mM CoA to an assay mixture containing 0.2 mM ThDP, 1 mM MgCl2, 2.6 mM cysteine HCl, 2.5 mM NAD+, 50 mM potassium phosphate (pH 7.0), and 20 to 50 μg of the reconstituted PDH enzyme complex (E1/E2/E3 molar ratio = 2:2:1) or spent crude cellular extracts. Specific activities were expressed as units (micromoles of NADH formed per minute) per milligram of whole-cell lysate in the assay.
Determination of acetyl-CoA concentration.
Procedures for determination of acetyl-CoA concentrations followed the protocols of Deutsch et al. (9). Briefly, acetyl-CoA was separated by HPLC using a C18 column (Mightysil RP-18 GP; Kanto Chemical Co., Tokyo, Japan) and eluted with an interrupted linear gradient of acetonitrile in 0.1 mol/liter potassium phosphate (pH 5.0). Initial conditions were 81% solvent A (0.1 mol/liter potassium phosphate, pH 5.0) and 19% solvent B (40% acetonitrile in solvent A); a constant flow rate of 0.25 ml/min was used during separation. The column was equilibrated at 3% of B for 10 min between injections. Detection was by absorbance at a wavelength of 254 nm. Retention times of the acetyl-CoA standards were used to identify the peaks on the HPLC chromatograms of each reaction sample. To see the effect of pirinSm on production of acetyl-CoA, the reaction samples, which contained 0.2 mM ThDP, 1 mM MgCl2, 2.6 mM cysteine HCl, 2 mM pyruvate, 0.13 mM CoA, 2.5 mM NAD+, 50 mM potassium phosphate (pH 7.0), and crude cellular extracts containing 50 μg of protein, were incubated for 1 h at 30°C. Each sample was then applied to HPLC for quantification of acetyl-CoA concentration by peak height with reference to standards of known acetyl-CoA concentrations.
Cellular ATP concentration assay.
The BacTiter-Glo microbial assay kit (Promega) together with a photon-counting Autolumat luminometer LB953 (Berthold, Dortmund, Germany) for detection of light emission were used for determination of cellular ATP concentration. Briefly, spent cell lysates (100 μl) prepared from fixed amounts of bacteria (volume [milliliters] × optical density at 600 nm [OD600] = 0.8) were mixed thoroughly with 100 μl of substrate solution. This was followed by incubation for 2 min at room temperature. The luminescence was then measured at 5-min intervals, and light emission was recorded for 10 seconds. Results were obtained in triplicate as ATP concentrations obtained after calculation against a standard curve at time intervals of 10 min.
Measurement of NAD+ and NADH concentrations.
Dinucleotide extraction was modified from previously described methods (27). Equal numbers of bacterial cells (determined by volume [milliliters] × OD600 = 5) were harvested at 5 h postinoculation in the late log phase by centrifugation for 15 min at 2,500 × g. After washing of the bacterial pellet in PBS, either 1 ml of 0.2 M HCl (NAD+ extraction) or 1 ml of 0.2 M KOH (NADH extraction) was added. Samples were boiled for 10 min. After centrifugation (5 min, 10,000 × g, 4°C), cell-free lysates were neutralized. Dinucleotide levels were measured with an enzymatic cycling assay described by Bernofsky and Swan (3). Briefly, 400 μl of cycling buffer [2.2 ml of 623 mM bicin, 2.2 ml of 2.6 mM 3-(4,5-dimethyl-thiazoyl-2)-2,5-diphenyltetrazolium bromide (MTT), 2.2 ml of 26 mM EDTA, 1.75 ml of 10.4 mM phenazine ethosulfate, and 0.4 ml ethanol] were added to 400 μl of neutralized extraction solution. After incubation for 5 min at room temperature in the dark, the reaction was initiated by adding 160 μl of 1.3-mg/ml yeast alcohol dehydrogenase (Sigma), and the rate of MTT reduction was monitored spectrophotometrically at 570 nm. The intracellular concentrations of dinucleotides were determined by known concentrations of NAD+ and NADH (0, 2.5, 5, 25, and 50 μM). All assays were performed in triplicate from three independent cultures.
Determination of acetate concentration.
The amount of acetate was determined as previously described (31). Briefly, equal numbers of bacterial cells (determined by volume [milliliters] × OD600 = 5) were centrifuged for 15 min at 2,500 × g. The bacterial pellets were suspended in PBS before incubation at 80°C for 15 min to stop the enzymatic reactions. The cell lysates were centrifuged for 5 min at 10,000 × g at 4°C, and acetate concentrations in the supernatants were determined using a kit purchased from R-Biopharm (Marshall, MI).
RESULTS
A pirin ortholog in S. marcescens.
During the process of selecting for S. marcescens precocious-swarming mutants by transposon mutagenesis assay (15), a pirin gene homolog in a mutant strain was identified to be interrupted by the transposon (Soo and Lai, unpublished data). The pirin gene in S. marcescens CH-1 was subjected to genetic and functional analyses. Through conventional restriction, cloning, and sequencing analyses, a 5-kb DNA fragment containing a putative pirinSm gene was identified in S. marcescens CH-1 (GenBank accession number DQ288954). Genetic maps flanking the pirinSm gene were similar for CH-1 and Db11 (Fig. 1A). Upstream of the pirinSm gene was an open reading frame predicted to encode a LysM protein domain (2), and downstream of the pirinSm gene was an open reading frame encoding an AraC family protein (29) (Fig. 1A).
Extensive computational searches using programs such as NCBI-BLAST and FASTA were performed to identify the S. marcescens pirin-like proteins from the data banks. S. marcescens pirin homologs are highly conserved, from many bacterial species to human pirin (26% identity). Pirin orthologs identified in bacteria include those from Pseudomonas aeruginosa PA14 (54% identity), an Acinetobacter sp. (49% identity), Corynebacterium glutamicum (43% identity), Brucella melitensis (37% identity), E. coli (YhhW protein, 37% identity), Ralstonia solanacearum (31% identity), Agrobacterium tumefaciens (27% identity), and Clostridium perfringens (24% identity). The amino acid sequence alignments of S. marcescens CH-1 pirinSm with these pirin orthologs are shown in Fig. S1 in the supplemental material. Based on the results of protein sequence pattern analysis from the MOTIFS (http://motif.genome.jp/) and SMART (http://smart.embl-heidelberg.de/) programs, the S. marcescens pirin protein displays a typical bicupin fold comprising a single N-terminal metal coordination site (pirin domain) and a C-terminal pirin-C conserved domain.
Interaction of S. marcescens pirin with the PDH E1 subunit.
Current data cannot directly indicate the function of pirinSm in S. marcescens. To characterize pirinSm function, the potential proteins interacting with pirinSm in S. marcescens CH-1 were targeted. A recombinant plasmid, pSC10, in which the pirinSm gene was N-terminally fused with GST to form a GST-pirinSm fusion protein was constructed. pSC10 was subsequently transformed into E. coli DH5α for oversynthesis of the GST-pirinSm protein. After purification, GST protein pull-down assay was performed. Separation of captured proteins by SDS-PAGE highlighted three major bands with predicted molecular masses of 99 kDa (band a), 76 kDa (band b), and 50 kDa (band c) after comparison with the negative controls (GST-nlpBSm [28] fusion and GST protein only) (Fig. 2).
FIG. 2.
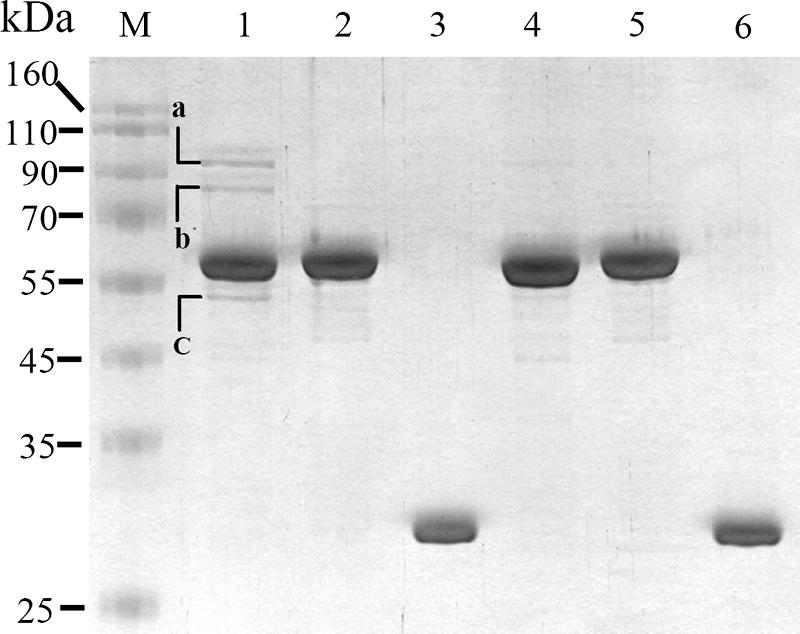
Use of protein pull-down assay for identification of S. marcescens proteins interacting with pirinSm. Glutathione-Sepharose 4B beads were used to capture protein complexes comprising GST-pirinSm fusion protein and the potential pirinSm interactors in CH-1. After elution, SDS-PAGE analysis was performed for protein separation. The cell lysate of E. coli DH5α containing either pSC10 (GST-pirinSm fusion protein was oversynthesized) (lane 1), pSC11 (GST-NlpBSm [28] fusion protein was oversynthesized) (lane 2), or pGEX (GST Tag only) (lane 3) was mixed with CH-1 lysate at a volume ratio of 1:1. Lane 4, GST-pirinSm fusion protein; lane 5, GST-NlpBSm fusion protein; lane 6, GST Tag protein; lane M, protein markers. Identification of the separated proteins was achieved by amino acid sequence analysis using electrospray ionization-MS/MS and comparison of the partial amino acid sequences with nonredundant protein databases in the SEQUEST Browser (http://fields.scripps.edu/sequest/). The three proteins with highest identity scores were E. coli PDH subunit E1 (band a, 99 kDa), E. coli PDH subunit E2 (band b, 76 kDa), and E. coli succinate dehydrogenase subunit E2 (band c, 50 kDa).
Amino acid sequence analyses using ESI-MS/MS were subsequently performed for protein identification. Comparison of the partial amino acid sequences of each protein with nonredundant protein databases in the SEQUEST Browser (http://fields.scripps.edu/sequest/) highlighted three proteins with the highest identity to the respective bands. They were E. coli PDH subunit E1 (band a), E. coli PDH subunit E2 (band b), and E. coli 2-oxoglutarate dehydrogenase (ODH) complex subunit E2 (band c) (see Fig. S2 in the supplemental material). PDH E1 was subsequently selected for further study.
The GST pull-down assay was further performed to see whether oversynthesized PDH E1 protein subunit interacted with oversynthesized pirinSm. A recombinant plasmid, pBG20, encoding a His-tagged PDH E1Sm (AceESm) fusion protein was constructed. The pBG20 and pSC10 plasmids were transformed into E. coli BL21(DE3) and E. coli DH5α, respectively, followed by induction with 0.5 mM IPTG for protein oversynthesis. The spent supernatants of whole-cell lysates prepared from both cell types at a volume ratio of 1:1 were mixed thoroughly and incubated at 4°C for 2 h before addition of glutathione-Sepharose 4B beads for capturing protein complexes. SDS-PAGE followed by Western blot analysis using anti His-Tag antibody confirmed that the S. marcescens PDH E1 subunit was pulled down by GST-pirinSm (Fig. 3A).
FIG. 3.
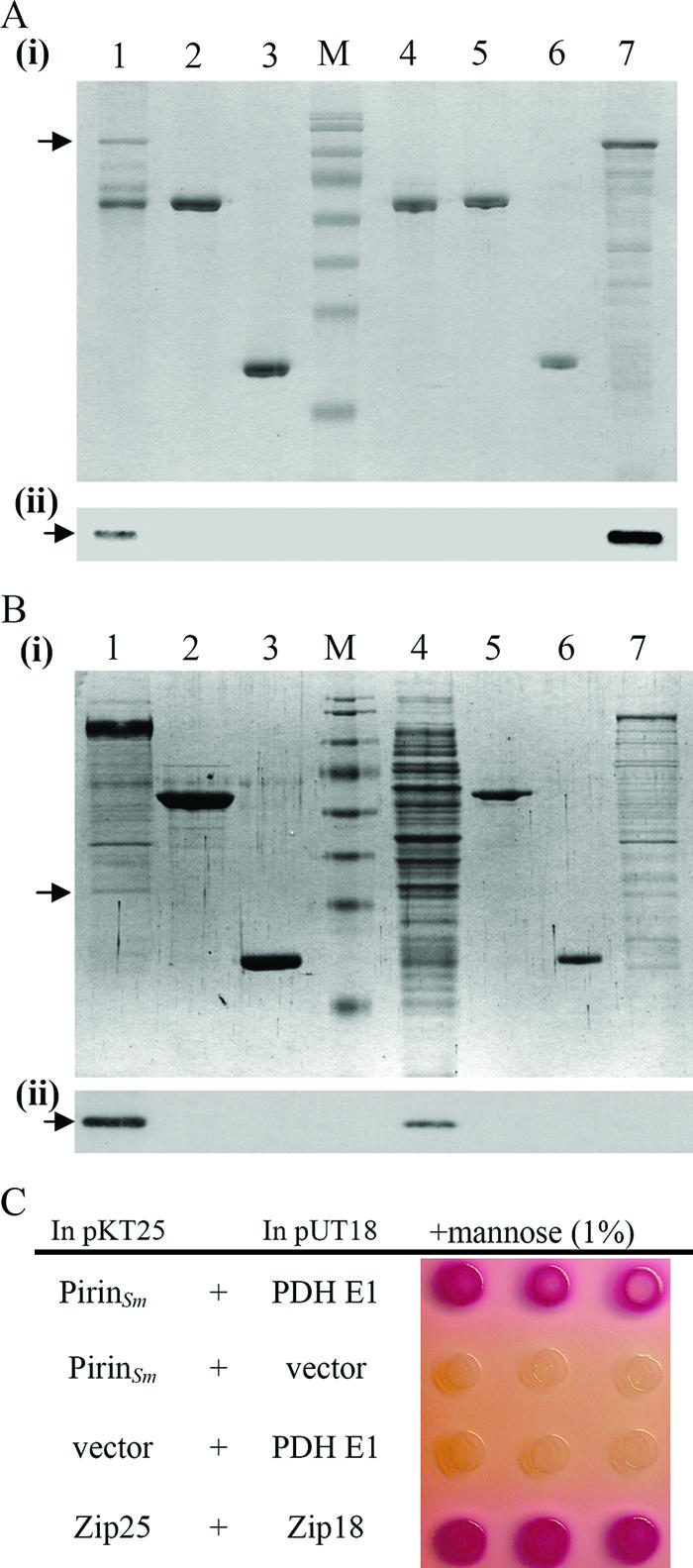
Confirmation of interaction between S. marcescens pirin and PDH E1. (A) In vitro GST pull-down assay followed by SDS-PAGE (panel i) and Western blot analysis (panel ii) using anti-His-Tag antibody was performed to confirm the interaction between GST-pirinSm and His-PDH E1. GST-pirinSm and His-PDH E1 were oversynthesized in E. coli before the assay. Lane 1, GST-pirinSm with His-PDH E1; lane 2, GST-NlpBSm with His-PDH E1; lane 3, GST Tag with His-PDH E1; lane 4, GST-pirinSm only; lane 5, GST-NlpBSm only; lane 6, GST Tag only; lane 7, E. coli(pBG20) spent crude extract containing His-PDH E1; arrow, His-PDH E1; lane M, protein markers (same as those in Fig. 2). (B) Oversynthesized GST-PDH E1 was used as the bait to confirm its interaction with pirinSm. After interaction, SDS-PAGE (panel i) and Western blot analysis using anti-pirinSm polyclonal antibody (panel ii) were performed. Lane 1, GST-PDH E1 with CH-1 lysate; lane 2, GST-NlpBSm with CH-1 lysate; lane 3, GST Tag with CH-1 lysate; lane 4, spent CH-1 cell lysate; lane 5, GST-NlpBSm only; lane 6, GST Tag protein only; lane 7, spent crude extract containing GST-PDH E1 from E. coli DH5α; arrow, pirinSm. (C) Bacterial two-hybrid assay. Colony color changed from colorless to pink-red after transformation of both pSC17 (pKT25 plasmid containing the pirinSm gene) and pSC18 (pUT18 plasmid containing the PDH E1 aceESm gene) into E. coli DHM1, indicating specific activation of maltose catabolic genes and interaction between GST-pirinSm and His-PDH E1 (14). Zip25 and Zip18 were used as positive controls.
Oversynthesized PDH E1 was subsequently used as the bait to confirm its interaction with pirinSm in S. marcescens CH-1. The recombinant plasmid pSC12 encoding the GST-PDH E1Sm fusion protein was transformed into S. marcescens CH-1, followed by 0.5 mM IPTG induction and pull-down assay to see whether pirinSm was captured. SDS-PAGE analysis followed by Western blot analysis using monospecific polyclonal anti-pirinSm antibody confirmed pirinSm as the interactor with PDH E1 (Fig. 3B).
To confirm the interaction between pirinSm and PDH E1, a bacterial two-hybrid assay was further performed. A change of colony color from colorless to pink-red after transforming both pSC17 (plasmid pKT25 [14] containing the pirinSm gene) and pSC18 (plasmid pUT18 [14] containing the PDH E1 aceESm gene) into E. coli DHM1 indicated specific activation of maltose catabolic genes (Fig. 3C), thus confirming a positive interaction between these two proteins. In brief, the PDH E1 subunit interacted with S. marcescens pirin.
Inhibition of PDH E1 activity by pirinSm.
To see whether the PDH E1 activity was affected by oversynthesis of pirinSm, the DCPIP assay (12) was performed. Spent cell lysates of E. coli BL21(DE3) cells containing either pBG20 (PDH E1 subunit), pSC10 (pirinSm), or the pGEX vector were prepared after cells were grown to late logarithmic phase at an OD600 of 0.6 under induction with 0.5 mM IPTG. Measurements of PDH E1 activities were achieved by mixing both cell lysates (25 μg PDH E1 lysate with 50 μg GST-pirinSm or GST Tag lysate) with externally added pyruvate at a final concentration of 400 μM as the substrate. As shown in Fig. 4A, the PDH E1 activities of mixed cell lysates from E. coli BL21(DE3)(pSC10) and E. coli BL21(DE3)(pBG20) were 30% lower than those from E. coli BL21(DE3)(pGEX) and E. coli BL21(DE3)(pBG20).
FIG. 4.

PirinSm inhibits PDH E1 and PDH enzyme complex activities. (A) Effect of pirinSm on PDH E1Sm specific activity. E. coli BL21(DE3) was used as the host for protein synthesis. (B) Effect of pirinSm gene overexpression (0.02% arabinose induction) from pSC15 on PDH E1Sm specific activity in S. marcescens CH-1. (C) Effect of pirinSm gene deletion on PDH E1Sm specific activity in S. marcescens CH-1. (D) Effect of pirinSm gene overexpression (0.02% arabinose induction) from pSC15 on PDH E1Sm specific activity in S. marcescens PC103. (E) Purification of S. marcescens PDH E1, E2, and E3 subunits in E. coli DH5α cells. Lane 1, GST-E1 subunit (pSC12); lane 2, GST-E2 subunit (pSC19); lane 3, GST-E3 subunit (pSC20); lane 4, GST-pirin (pSC10); lane 5, GST-NlpBSm (pSC11); 6, GST protein (pGEX vector). (F) Effect of purified pirinSm, NlpBSm, and GST (15 μg each) on specific activity of purified PDH E1 subunit (15 μg). (G) PDH enzyme complex activity in S. marcescens CH-1 and PC103. (H) Effect of pirinSm gene overexpression (0.02% arabinose induction) from pSC15 on specific PDH enzyme complex activity in S. marcescens PC103. (I) Effect of pirinSm on reconstituted PDH enzyme complex at an E1/E2/E3 molar ratio of 2:2:1. The DCPIP assay (12) was used to measure PDH E1 activity; the rate of NADH formation was measured spectrophotometrically at a wavelength of 340 nm (5) to measure PDH enzyme complex activity. Black and white bars indicate reactions with or without pyruvate added as the substrate. PBS (pH 7.4) was used as the negative control for all assays. CH-1, S. marcescens CH-1; PC103, a pirinSm gene insertion-deletion mutant strain derived from S. marcescens CH-1; pBAD18, pBAD18-Kan control vector; pSC15, pBAD18-Kan::pirinSm gene; NlpBSm, a membrane lipoprotein identified in S. marcescens CH-1 (28). a, change in OD600 per minute per milligram of protein. b, change in OD340 per minute per milligram of protein. Values are means and standard deviations from three independent experiments. *, P < 0.05.
Inhibition of PDH E1 activity in S. marcescens CH-1 by pirinSm gene overexpression.
As S. marcescens CH-1(pSC15) (pirinSm gene expression under the control of the pBAD promoter) cells were grown to an OD600 of 0.6 in LB broth culture, the amount of pirinSm protein was induced up to fivefold in the presence of 0.02% arabinose (data not shown), and the PDH E1 activity was reduced up to 40% compared with that from S. marcescens CH-1(pBAD18) (Fig. 4B).
We reasoned that the PDH E1 activity might be increased in the S. marcescens CH-1 pirinSm mutant strain. To evaluate this possibility, the pirinSm gene was knocked out by insertion-deletion homologous recombination through an Sm resistance Ω cassette (23) in S. marcescens CH-1 to form the mutant strain S. marcescens PC103 (Fig. 1B). Southern blot hybridization (data not shown) and Western blot analysis (Fig. 1C) confirmed deletion of the pirinSm gene in S. marcescens CH-1. No significant difference in growth dynamics in LB broth cultures was observed between CH-1 and PC103 (data not shown). Spent cellular crude extracts of CH-1 and PC103 cells grown to an OD600 of 0.6 were prepared and used in the DCPIP assay. PDH E1 activity in PC103 cells was 250% higher than that in CH-1 cells (Fig. 4C). Complementation of S. marcescens PC103 by transforming pSC15 into S. marcescens PC103 cells inhibited the PDH E1 activity (Fig. 4D), while transforming the control vector pBAD18 into S. marcescens PC103 cells did not. Thus, S. marcescens PDH E1 activity was inhibited by pirinSm.
To characterize the specific inhibition of PDH E1 activity by pirinSm, GST-tagged PDH E1 and GST-tagged pirinSm were purified from E. coli DH5α(pSC12) and E. coli DH5α(pSC10), respectively (Fig. 4E). The DCPIP assay showed that GST-PDH E1 activity was 30% lower in the presence of GST-pirinSm than that of GST alone or GST-NlpBSm fusion protein (28) (Fig. 4F).
PirinSm inhibition of PDH enzyme complex activity in S. marcescens.
Inhibition of PDH E1 activity by pirinSm strongly suggested that PDH enzyme complex activity would be inhibited. To confirm this supposition, a total of 50 μg of spent cellular crude extract each was prepared from S. marcescens CH-1 and PC103 and was used in the PDH activity assay. The cellular PDH activity in S. marcescens PC103 cells was 40% higher than that in S. marcescens CH-1 (Fig. 4G). On the other hand, oversynthesis of pirinSm in the S. marcescens PC103 mutant strain led to inhibition of PDH activity by 30% compared with S. marcescens PC103 containing the control vector (Fig. 4H). To rule out possible background interferences, the in vitro-reconstituted PDH enzyme complex comprising E1, E2, and E3 at a molecular ratio of 2:2:1 was further used for measurement of PDH activity. As shown in Fig. 4I, the reconstituted PDH activity was inhibited about 35% by GST-pirinSm compared with the GST-NlpBSm and GST-alone controls.
Increased PDH activity in the pirinSm gene-deleted mutant strain S. marcescens PC103 should result in increased acetyl-CoA production. To evaluate this supposition, spent cellular crude extracts of S. marcescens CH-1 and PC103 were prepared, followed by C18 reverse-phase HPLC separation for determination of acetyl-CoA concentrations. The acetyl-CoA concentration increased about 50% in S. marcescens PC103 in comparison to that in S. marcescens CH-1 (Table 2). Complementation of the pirinSm gene in S. marcescens PC103 by transformation of pSC15 into S. marcescens PC103 cells reduced the acetyl-CoA concentration up to 40% compared with that of S. marcescens PC103 (Table 2). Thus, pirinSm inhibited PDH enzyme complex activity in S. marcescens CH-1.
TABLE 2.
PDH enzyme complex activities and acetyl-CoA concentrations in S. marcescens CH-1 and PC103 cells
| Bacterial strain | PDH enzyme complex activity
|
Acetyl-CoA concn
|
||
|---|---|---|---|---|
| μmol NADH formed/mg/min (mean ± SD)a | % | nmol/mg protein (mean ± SD)a | % | |
| CH-1 | 0.701 ± 0.035 | 100 | 0.750 ± 0.055 | 100 |
| PC103 | 0.983 ± 0.053 | 140 | 1.125 ± 0.087 | 150 |
| PC103(pBAD18) | 1.189 ± 0.134 | 170 | 1.165 ± 0.076 | 155 |
| PC103(pSC15) | 0.794 ± 0.071 | 113 | 0.706 ± 0.042 | 94 |
Data are from three independent experiments.
Cellular ATP concentration and NADH/NAD+ ratio are increased in S. marcescens PC103.
Following the process of converting pyruvate into acetyl-CoA by the PDH enzyme complex, acetyl-CoA is further oxidized through TCA cycle intermediates, accompanied by synthesis of NADH and reduced flavin adenine dinucleotide (FADH2) (4). The majority of the cellular ATP is produced from these reduced NADH and FADH2 compounds after further respiration reactions (25). Alternatively, acetyl-CoA is converted to acetate through dissimilation via the PTA-ACKA (phosphotransacetylaseacetate kinase) pathway and produces less ATP (33). Thus, alteration of the acetyl-CoA concentration should result in a change in cellular ATP concentration. Indeed, the cellular ATP concentration in S. marcescens PC103 grown to an OD600 of 0.6 in the late log phase was increased 220% compared with that in S. marcescens CH-1 (Fig. 5A); complementation of S. marcescens PC103 with pSC15 reduced the ATP concentration to the normal level (Fig. 5B). The increase in ATP concentration in S. marcescens PC103 might have been due to activation of either the TCA cycle or the PTA-ACKA pathway or both. To clarify this, the NADH/NAD+ ratio, which shows TCA cycle activity and intracellular redox status (31), and cellular acetate concentration were determined. While there was no significant difference in acetate concentration (Fig. 5C), the NADH/NAD+ ratio in S. marcescens PC103 was significantly higher than that in S. marcescens CH-1 (0.759 versus 0.439; P < 0.05) (Fig. 5D), suggesting that increased TCA cycle activity is mainly responsible for increased ATP concentration in S. marcescens PC103.
FIG. 5.

Cellular ATP concentration and NADH/NAD+ ratio are increased in the pirinSm gene deletion mutant strain S. marcescens PC103. Bacterial cells were grown to an OD600 of 0.6, followed by determination of cellular ATP concentration (A and B), cellular acetate concentration (C), and intracellular redox status through measuring the concentrations and determining the ratio of NADH to NAD+ (D). CH-1, S. marcescens wild-type strain; PC103, pirin gene deletion mutant S. marcescens strain; PC103(pBAD18) and PC103(pSC15), S. marcescens PC103 containing the pBAD18-Kan control vector or pBAD18-Kan::pirinSm, respectively. All results are means and standard deviations from three independent experiments. *, P < 0.05.
DISCUSSION
Very few studies on characterization of pirin orthologs in prokaryotes have been reported. Thus, whether pirinSm functions as an enzyme, a regulator involved in any signaling pathway, or a protein enhancing the interaction between DNA and protein complexes remains to be further characterized among bacterial species. The S. marcescens pirin shows sequential and structural similarity to E. coli YhhW (20); however, whether pirinSm is secreted outside the S. marcescens cells and degrades quercetin remains to be determined. In this report, through protein-protein interaction, genetic, and biochemical analyses, our results showed that pirinSm binds with PDH E1 and inhibits PDH E1 and PDH enzyme complex activities. Although PDH E1, PDH E2, and ODH E2 were identified as pirinSm interactor proteins, we still could not rule out the possibility that the PDH E3 subunit was also among the interacting components, as PDH E3 might be dissociated from E1 and E2 during the purification process. At the same time, the intriguing phenomenon that the estimated molecular mass of the interactor protein PDH E2 was 76 kDa after SDS-PAGE analysis was observed (Fig. 2, lane 1, band b). This is different from the predicted molecular mass of PDH E2 in E. coli (see Fig. S2 in the supplemental material) and S. marcescens CH-1, which is 66 kDa. One of the explanations is that posttranslational modification of PDH E2 might occur in S. marcescens CH-1. Furthermore, besides PDH E1 and E2, ODH E2 was also identified. ODH E2 is involved in the process of converting α-ketoglutarate to succinate-CoA and releasing CO2 and NADH, which is one of the enzymatic reactions belonging to the TCA cycle (4, 25). The composition of the ODH enzyme complex shows similarity to that of the PDH enzyme complex, as both complexes comprise E1, E2, and E3 subunits and functionally are components of the biochemical pathway leading to generation of NADH and FADH2 for subsequent ATP generation. Thus, interaction of pirinSm with components of these proteins strongly indicates that pirinSm is involved in the process of determination of cellular ATP concentration and energy production. Due to the significant increase in NADH/NAD+ ratio but not cellular acetate concentration in the pirinSm mutant (Fig. 5C and D), the effect of increased ATP concentration should be attributed mostly to increased TCA cycle activity. Comparatively, the PTA-ACKA pathway, which also produces ATP, seemed not to play a dominant role in the process.
On the basis of both sequential and structural similarities, pirin orthologs are classified as a subfamily of the cupin superfamily (11, 32). The cupin superfamily is one of the most functionally diverse protein classes and includes both enzymatic and nonenzymatic members, ranging from isomerases and epimerases that are involved in the modification of cell wall carbohydrates to nonenzymatic storage proteins in plant seeds and transcriptional cofactors in humans (11). Prediction of the secondary and tertiary structures of pirinSm through use of the JOY program (18) showed that the structure of pirinSm is similar to that of human pirin (Soo and Lai, unpublished data). These two proteins are predicted to contain a conserved N-terminal metal-binding domain and a C-terminal β-barrel domain, and they show three conserved histidine residues (His77, His79, and His121 for pirinSm; His56, His58, and His101 for human pirin) and a conserved glutamate residue (Glu123 for pirinSm; Glu103 for human pirin) in the N-terminal motifs (see Fig. S1 in the supplemental material), suggesting a functional conservation between the two proteins. However, differences are still evident, including the amino acids surrounding the predicted metal ion-binding site (data not shown). Thus, the conformation of pirin proteins might vary under different environments. This might explain the phenomenon that pirin family orthologs interact with different proteins and affect diverse biological processes.
How pirinSm interacts with and regulates PDH E1 and subsequently PDH enzyme complex activity remains undetermined. What we understand currently is that a compact pirinSm is essential for interaction with PDH E1, as no protein-protein interaction reaction was observed when we dissected the pirinSm protein into N-terminal and C-terminal domains (data not shown). Further deletion and functional analyses combined with surface plasmon resonance protein-protein interaction analysis should help us understand more about the pirinSm-PDH E1 interaction mechanism. As PDH is responsible for conversion of pyruvate into acetyl-CoA, increased PDH enzyme complex activity should mean that the direction of cellular pyruvate metabolism shunts towards the TCA cycle and subsequently the electron transport chain so that more cellular ATP is produced by oxidative phosphorylation through this aerobic respiratory pathway (25). This usually occurs when cells are growing aerobically or at the logarithmic phase in LB broth culture. In contrast, a reduction in PDH enzyme complex activity suggests that pyruvate metabolism is diverted towards the fermentation pathways, whereby less ATP is produced (25). This frequently occurs when cells are grown under anaerobic conditions or are growing into the stationary phase. While deletion of the pirinSm gene in S. marcescens CH-1 led to increased total cellular ATP production even though cells were grown to the early stationary phase at high cell density, the pirinSm concentration increased as cells grew into the transitional phase, and it peaked at the stationary phase (Soo and Lai, unpublished data). Together these results suggest that pirinSm might regulate S. marcescens central carbohydrate metabolism through diverting the reactions towards the fermentation pathways (thus producing less ATP and accumulating more fermentation products) as cells are grown to a high density. It is predicted that the ferrous ion Fe(II) is located within the N-terminal metal-binding motif of human pirin (22). If it is the same for pirinSm, then pirinSm might be involved in sensing the redox status (NADH/NAD+ ratio) within the cell through oxidation or reduction of ferrous ion. Such sensing might then affect pirinSm conformation and the subsequent interaction with and modulation of PDH enzyme complex activity. To confirm this, determination of the three-dimensional protein-protein interaction structure between pirinSm and PDH E1 by X-ray crystallography will have to be performed.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Science Council (NSC-94-2320-B-002-078 and NSC-95-2320-B-002-061) and the Aim for Top University Program Excellence Research Projects 2006, National Taiwan University.
Footnotes
Published ahead of print on 15 September 2006.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Adams, M. A., and Z. Jia. 2005. Structural and biochemical evidence for an enzymatic quinone redox cycle in Escherichia coli: identification of a novel quinol monooxygenase. J. Biol. Chem. 280:8358-8363. [DOI] [PubMed] [Google Scholar]
- 2.Bateman, A., and M. Bycroft. 2000. The structure of a LysM domain from E. coli membrane-bound lytic murein transglycosylase D (MltD). J. Mol. Biol. 299:1113-1119. [DOI] [PubMed] [Google Scholar]
- 3.Bernofsky, C., and M. Swan. 1973. An improved cycling assay for nicotinamide adenine dinucleotide. Anal. Biochem. 53:452-458. [DOI] [PubMed] [Google Scholar]
- 4.Buchanan, B. B., and D. I. Arnon. 1990. A reverse KREBS cycle in photosynthesis: consensus at last. Photosynth. Res. 24:47-53. [PubMed] [Google Scholar]
- 5.Danson, M. J., A. R. Fersht, and R. N. Perham. 1978. Rapid intramolecular coupling of active sites in the pyruvate dehydrogenase complex of Escherichia coli: mechanism for rate enhancement in a multimeric structure. Proc. Natl. Acad. Sci. USA 75:5386-5390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dechend, R., F. Hirano, K. Lehmann, V. Heissmeyer, S. Ansieau, F. G. Wulczyn, C. Scheidereit, and A. Leutz. 1999. The Bcl-3 oncoprotein acts as a bridging factor between NF-kappaB/Rel and nuclear co-regulators. Oncogene 18:3316-3323. [DOI] [PubMed] [Google Scholar]
- 7.de Kok, A., A. F. Hengeveld, A. Martin, and A. H. Westphal. 1998. The pyruvate dehydrogenase multi-enzyme complex from Gram-negative bacteria. Biochim. Biophys. Acta 1385:353-366. [DOI] [PubMed] [Google Scholar]
- 8.de Lorenzo, V., M. Herrero, U. Jakubzik, and K. N. Timmis. 1990. Mini-Tn5 transposon derivatives for insertion mutagenesis, promoter probing, and chromosomal insertion of cloned DNA in gram-negative eubacteria. J. Bacteriol. 172:6568-6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deutsch, J., S. I. Rapoport, and T. A. Rosenberger. 2002. Coenzyme A and short-chain acyl-CoA species in control and ischemic rat brain. Neurochem. Res. 27:1577-1582. [DOI] [PubMed] [Google Scholar]
- 10.Domingo, G. J., H. J. Chauhan, I. A. Lessard, C. Fuller, and R. N. Perham. 1999. Self-assembly and catalytic activity of the pyruvate dehydrogenase multienzyme complex from Bacillus stearothermophilus. Eur. J. Biochem. 266:1136-1146. [DOI] [PubMed] [Google Scholar]
- 11.Dunwell, J. M., A. Culham, C. E. Carter, C. R. Sosa-Aguirre, and P. W. Goodenough. 2001. Evolution of functional diversity in the cupin superfamily. Trends Biochem. Sci. 26:740-746. [DOI] [PubMed] [Google Scholar]
- 12.Fries, M., H. I. Jung, and R. N. Perham. 2003. Reaction mechanism of the heterotetrameric (alpha2beta2) E1 component of 2-oxo acid dehydrogenase multienzyme complexes. Biochemistry 42:6996-7002. [DOI] [PubMed] [Google Scholar]
- 13.Hihara, Y., M. Muramatsu, K. Nakamura, and K. Sonoike. 2004. A cyanobacterial gene encoding an ortholog of Pirin is induced under stress conditions. FEBS Lett. 574:101-105. [DOI] [PubMed] [Google Scholar]
- 14.Jobling, M. G., and R. K. Holmes. 2000. Identification of motifs in cholera toxin A1 polypeptide that are required for its interaction with human ADP-ribosylation factor 6 in a bacterial two-hybrid system. Proc. Natl. Acad. Sci. USA 97:14662-14667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lai, H. C., P. C. Soo, J. R. Wei, W. C. Yi, S. J. Liaw, Y. T. Horng, S. M. Lin, S. W. Ho, S. Swift, and P. Williams. 2005. The RssAB two-component signal transduction system in Serratia marcescens regulates swarming motility and cell envelope architecture in response to exogenous saturated fatty acids. J. Bacteriol. 187:3407-3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lapik, Y. R., and L. S. Kaufman. 2003. The Arabidopsis cupin domain protein AtPirin1 interacts with the G protein alpha-subunit GPA1 and regulates seed germination and early seedling development. Plant Cell 15:1578-1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miller, J. H. 1992. A short course in bacterial genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 18.Mizuguchi, K., C. M. Deane, T. L. Blundell, M. S. Johnson, and J. P. Overington. 1998. JOY: protein sequence-structure representation and analysis. Bioinformatics 14:617-623. [DOI] [PubMed] [Google Scholar]
- 19.Neidhardt, F. C. 1996. Respiration, p. 151-169. In F. C. Neidhardt, R. Curtiss III, J. L. Ingraham, E. C. C. Lin, K. B. Low, B. Magasanik, W. S. Reznikoff, M. Riley, M. Schaechter, and H. E. Umbarger (ed.), Escherichia coli and Salmonella: cellular and molecular biology. American Society for Microbiology, Washington, DC.
- 20.Oka, T., and F. J. Simpson. 1971. Quercetinase, a dioxygenase containing copper. Biochem. Biophys. Res. Commun. 43:1-5. [DOI] [PubMed] [Google Scholar]
- 21.Orzaez, D., A. J. de Jong, and E. J. Woltering. 2001. A tomato homologue of the human protein PIRIN is induced during programmed cell death. Plant Mol. Biol. 46:459-468. [DOI] [PubMed] [Google Scholar]
- 22.Pang, H., M. Bartlam, Q. Zeng, H. Miyatake, T. Hisano, K. Miki, L. L. Wong, G. F. Gao, and Z. Rao. 2004. Crystal structure of human pirin: an iron-binding nuclear protein and transcription cofactor. J. Biol. Chem. 279:1491-1498. [DOI] [PubMed] [Google Scholar]
- 23.Prentki, P., and H. M. Krisch. 1984. In vitro insertional mutagenesis with a selectable DNA fragment. Gene 29:303-313. [DOI] [PubMed] [Google Scholar]
- 24.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 25.Schagger, H. 2002. Respiratory chain supercomplexes of mitochondria and bacteria. Biochim. Biophys. Acta 1555:154-159. [DOI] [PubMed] [Google Scholar]
- 26.Smith, D. B., and K. S. Johnson. 1988. Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione S-transferase. Gene 67:31-40. [DOI] [PubMed] [Google Scholar]
- 27.Snoep, J. L., M. J. Teixeira de Mattos, P. W. Postma, and O. M. Neijssel. 1990. Involvement of pyruvate dehydrogenase in product formation in pyruvate-limited anaerobic chemostat cultures of Enterococcus faecalis NCTC 775. Arch. Microbiol. 154:50-55. [DOI] [PubMed] [Google Scholar]
- 28.Soo, P. C., J. R. Wei, Y. T. Horng, S. C. Hsieh, S. W. Ho, and H. C. Lai. 2005. Characterization of the dapA-nlpB genetic locus involved in regulation of swarming motility, cell envelope architecture, hemolysin production, and cell attachment ability in Serratia marcescens. Infect. Immun. 73:6075-6084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tobes, R., and J. L. Ramos. 2002. AraC-XylS database: a family of positive transcriptional regulators in bacteria. Nucleic Acids Res. 30:318-321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tsay, Y. G., Y. H. Wang, C. M. Chiu, B. J. Shen, and S. C. Lee. 2000. A strategy for identification and quantitation of phosphopeptides by liquid chromatography/tandem mass spectrometry. Anal. Biochem. 287:55-64. [DOI] [PubMed] [Google Scholar]
- 31.Vuong, C., J. B. Kidder, E. R. Jacobson, M. Otto, R. A. Proctor, and G. A. Somerville. 2005. Staphylococcus epidermidis polysaccharide intercellular adhesin production significantly increases during tricarboxylic acid cycle stress. J. Bacteriol. 187:2967-2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wendler, W. M., E. Kremmer, R. Forster, and E. L. Winnacker. 1997. Identification of pirin, a novel highly conserved nuclear protein. J. Biol. Chem. 272:8482-8489. [DOI] [PubMed] [Google Scholar]
- 33.Wolfe, A. J. 2005. The acetate switch. Microbiol. Mol. Biol. Rev. 69:12-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang, C. C., C. H. Huang, C. Y. Li, Y. G. Tsay, S. C. Lee, and C. W. Chen. 2002. The terminal proteins of linear Streptomyces chromosomes and plasmids: a novel class of replication priming proteins. Mol. Microbiol. 43:297-305. [PubMed] [Google Scholar]
- 35.Zahn, J. A., D. J. Bergmann, J. M. Boyd, R. C. Kunz, and A. A. DiSpirito. 2001. Membrane-associated quinoprotein formaldehyde dehydrogenase from Methylococcus capsulatus Bath. J. Bacteriol. 183:6832-6840. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.