

Abstract
Preintegration complexes (PICs) mediate retroviral integration, and recent results indicate an important role for the inner nuclear membrane protein emerin in orienting human immunodeficiency virus type 1 (HIV-1) PICs to chromatin for integration. Two other host cell proteins, the barrier-to-autointegration factor (BAF) and lamina-associated polypeptide 2α (LAP2α), seemed to play a similar preintegrative role for Moloney murine leukemia virus (MMLV) in addition to HIV-1. In contrast, we determined efficient HIV-1 and MMLV infection of HeLa-P4 cells following potent down-regulation of emerin, BAF, or LAP2α protein by using short interfering RNA. Mouse embryo fibroblasts ablated for emerin protein through gene knockout support the same level of HIV-1 infection as cells derived from wild-type littermate control animals. As the expression of human emerin in mouse knockout cells fails to affect the level of infectivity achieved in its absence, we conclude that HIV-1 efficiently infects cells in the absence of emerin protein and, by extension, that emerin is not a universally important regulator of HIV-1 infectivity.
Retroviruses are unique among animal viruses in that their replication requires the covalent integration of their genetic material into a cell chromosome. The early events of the retroviral life cycle up to and including integration take place within the context of large nucleoprotein structures that are derived from the virion core. After entering a cell, the core partially disassembles or uncoats, a process required for the successful initiation of reverse transcription. DNA synthesis ensues within the resultant reverse transcription complex (RTC). The RTC then matures into the preintegration complex (PIC), which houses viral and cellular components necessary for cDNA trafficking into the nucleus and to the chromosomes where the integrase protein catalyzes integration. See reference 10 for a thorough overview of the early events of the retroviral life cycle.
Large structural entities like retroviral RTCs and PICs preclude directive intracellular transport via passive diffusion (4). Thus, active mechanisms must be in play to ensure their net positive flow through the cytoplasm. Though mechanistic details remain largely unresolved, RTCs apparently engage microtubules to move toward the nucleus (1, 28). Lentiviruses like human immunodeficiency virus type 1 (HIV-1) can efficiently infect nondividing cells under conditions in which simpler viruses like the Gammaretrovirus Moloney murine leukemia virus (MMLV) are noninfectious (18). Hence, lentiviruses are proposed to harbor nuclear localization signals to drive the active transport of their PICs through intact nuclear pore complexes that persist in the absence of nuclear membrane breakdown (31). Conversely, MMLV infection requires cells to pass through the M phase of the cell cycle, provoking a model whereby these PICs access chromosomes after nuclear membrane breakdown, during nuclear/cytoplasmic mixing (19, 32). Despite a long history of intensive research, little is known about the exact mechanisms by which retroviral PICs gain access to the nuclear environment and chromosome targets for integration.
A recent study (13) suggests this might occur via differential engagement of specific host cell factors (reviewed in reference 20). The barrier-to-autointegration factor (BAF), a small, nonspecific DNA-binding protein, was first identified through its ability to suppress the autointegration activity of MMLV PICs in in vitro integration reactions (17). BAF was subsequently shown to be a component of MMLV (40) and HIV-1 (22) PICs and was proposed to play an important role in target DNA binding during MMLV integration in vitro (40). In cells, BAF interacts with a class of inner nuclear membrane (INM) proteins through an approximate 40-amino-acid residue LEM (for lamina-associated polypeptide 2 [LAP2], emerin, manin) domain, which helps to bridge the proteins and hence the INM to chromatin (reviewed in reference 35). LAP2α, a LAP2 isoform that lacks the transmembrane domain and thus is not integral to the INM, is a component of MMLV PICs and potentiates viral cDNA integration in vitro (41). RNA interference (RNAi)-mediated knockdown of LAP2α in mouse NIH 3T3 cells stalled MMLV replication, though it was not determined if the replication block occurred primarily before or after viral integration (41).
HeLa cells silenced for BAF or LAP2α expression via small interfering RNA (siRNA) significantly resisted transduction by pseudotyped MMLV vectors, indicating a preintegration or early postintegration block to infection under these conditions (13). Knocking down BAF, LAP2α, or emerin rendered HeLa cells as well as monocyte-derived macrophages (MDM) resistant to HIV-1 vector transduction (13). To address the underlying mechanism, infected MDM were fractionated, revealing that BAF or emerin silencing impaired integration via mislocalizing HIV-1 PICs away from their normal chromatin targets to a nonproductive nuclear matrix environment (13). These results therefore suggested that different retroviral genera depend upon BAF and perhaps LAP2α for proper localization to chromatin for integration, whereas lentiviruses in addition require an interaction with emerin to achieve this outcome. In contrast to these findings, we report here that HeLa cells potently down-regulated for emerin, BAF, or LAP2α expression support normal levels of HIV-1 and MMLV infectivity. To critically address the role of emerin in HIV-1 infection, cells derived from knockout mice were utilized in in vitro studies. Our results fail to support a significant role for emerin protein in regulating HIV-1 infectivity.
MATERIALS AND METHODS
Molecular clones.
Plasmids pTY-EFeGFP, pCEP4-Tat, and pHP-dI-N/A were previously described (5). Plasmids pCG-gagpol and pCG-VSV-G were kind gifts of Jonathon Walsh and Richard Mulligan (Harvard Medical School). Envelope expression vectors derived from HIV-1NL4-3 (21), HIV-189.6 (25), and a C-terminally mutated form of HIV-1HXB10 (27) were previously described.
Plasmid pTY-CMVLuc for expressing the self-inactivating (SIN) HIV-1 vector HIV-SIN-Luc was derived from pTY-EFeGFP as follows. The cytomegalovirus (CMV) immediate-early promoter was amplified from pcDNA6/V5-HisB (Invitrogen Corp., Carlsbad, CA) using primer pair AE2592 (5′-GACTATCGATGTTGACATTGATTATTGACTAG; ClaI site underlined) and AE2593 (SexAI-tagged 5′-GACTACCAGGTGAGCTCTGCTTATATAGACCTCC) and PfuUltra DNA polymerase (Stratagene, La Jolla, CA), while at the same time the luciferase open reading frame (ORF) was amplified from pGL3-Basic (Promega Corp., Madison, WI) using SexAI-tagged AE2594 (5′-GACTACCAGGTACCACCATGGAAGACGCCAAAAAC) and EcoRI-tagged AE2595 (5′-GACTGAATTCTTACACGGCGATCTTTTC) primers. Following digestion, the two fragments were ligated in a three-fragment ligation with ClaI/EcoRI-cut pTY-EFeGFP.
The human emerin ORF was amplified from HsCD00003114 template DNA (Harvard Institute of Proteomics, Cambridge, MA) using primers AE2727 (5′-GCGGCAGGATCCGCCACCATGGACAACTACGCAGATCTTCG) and AE2728 (5′-GCGAGCAAGCTTCTAGAAGGGGTTGCCTTCTTCAGC). BamHI/HindIII-digested DNA was ligated with BglII/HindIII-cut pLPCX (Clontech, Mountain View, CA) to yield pLPCX-emerin. Regions of all plasmid constructs that were derived via PCR were sequenced to verify the absence of unwanted secondary changes.
Cells.
Low-passage-number mouse embryo fibroblasts (MEFs) derived from lamin A/C (39) and emerin (15) knockout mice, together with matched littermate control cells, were a generous gift from Colin Stewart (National Cancer Institute, Frederick, MD). MEFs, HeLa-P4 (6), and 293T cells were cultured in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum, 100 IU/ml penicillin, and 100 μg/ml streptomycin. Primary MEFs were transformed following transduction with a pLB(N)CX-based retroviral vector expressing the simian virus 40 (SV40) large T antigen (3) (a generous gift from Darrel Borger and James DeCaprio, Dana-Farber Cancer Institute) and selection in blasticidin. Peripheral blood mononuclear cells isolated using Ficoll-Paque Plus (GE Healthcare Bio-Sciences Corp., Piscataway, NJ) were propagated in RPMI medium (Invitrogen Corp.) containing penicillin (50 IU/ml), streptomycin (50 μg/ml), glutamine (2 mM), and 10% human AB serum (Gemini Bio-Products, Woodland, CA). Nonadherent cells were removed following 1 night of adsorption (2 × 106 cells per well) onto 24-well Primaria tissue culture plates (Becton Dickinson Labware, Franklin Lakes, NJ). Cells were further incubated for 6 days, changing medium every other day.
siRNA design and transfection.
Targeting siRNAs (Dharmacon Research, Boulder, CO) derived in this study contained the following plus-strand sequences: EMD5, 5′-GACCUGUCCUAUUAUCCUAdTdT; TMPO2, 5′-GCAAAUAUCCAGUUUCUUCdTdT; and BANF1-1, 5′-GAAGCUGGAGGAAAGGGGUdTdT. Control sequences harboring internal base changes were as follows: EMD5C, 5′-GACCUGUAUCCUUAUCCUAdTdT; TMPO2C, 5′-GCAAAUAACGACUUUCUUCdTdT; and BANF1-1C, 5′-GAAGCUGCACGUAAGGGGUdTdT (mutated bases underlined). Previously-described sequences were as follows: EMDJS, 5′-GAGGAGUGCAAGGAUAGGGdTdT; TMPOJS, 5′-GACAAGUUGAAGAGUGAGUdTdT; BANF1-JS, 5′-GGCCUAUGUUGUCCUUGGCdTdT (13); and CD4, 5′-GAUCAAGAGACUCCUCAGUdGdA (30).
HeLa-P4 cells (2 × 105) plated in six-well trays the day before transfection were transfected twice on successive days using 10 nM siRNA and oligofectamine as recommended by the manufacturer (Invitrogen Corp.). Cells trypsinized the following day were either plated in 12-well plates (5 × 104 cells/well) for infection, lysed for quantitative reverse transcription-PCR (qRT-PCR) or Western blot analysis, or processed for fluorescence-activated cell sorting (FACS). Oligofectamine was also used with MDM, which were transfected once with 0.5 to 2.0 μM siRNA (38). Two days posttransfection, MDM were either infected or processed for qRT-PCR or Western blotting.
Western blotting and flow cytometry.
Cells were extracted on ice for 15 min in buffer containing 50 mM Tris-HCl, pH 7.5, 300 mM NaCl, 0.5% Triton X-100, 1 mM EDTA, and complete protease inhibitor (Roche Diagnostics, Indianapolis, IN), and protein concentration in clarified lysates was quantified using the Dc protein assay kit (Bio-Rad Laboratories, Hercules, CA). Ten micrograms of total protein electrophoresed through 4 to 20% Tris-glycine gels (Invitrogen Corp.) was transferred to polyvinylidene difluoride membrane using a Trans-Blot SD semidry transfer cell apparatus (Bio-Rad Laboratories). Emerin and LAP2α antibodies were from Abcam, Inc. (Cambridge, MA) and ImmuQuest, Ltd. (Cleveland, United Kingdom), respectively; BAF antibodies were a generous gift from Katherine Wilson (Johns Hopkins University School of Medicine) (34). Blots were developed using secondary antibodies from Dako North America, Inc. (Carpinteria, CA), and the ECL Plus kit (GE Healthcare Bio-Sciences Corp.). Knockdown (fold) was assessed using Quantify One version 4.1.1 software (Bio-Rad Laboratories) by comparing signals to standard curves generated by end-point dilution of mock-transfected cell extracts.
HeLa-P4 cells (5 × 105) washed once in FACS buffer (phosphate-buffered saline supplemented with 0.1% [wt/vol] bovine serum albumin and 20 mM Na azide) were resuspended in 100 μl FACS buffer and stained with fluorescein isothiocyanate (FITC)-conjugated anti-CD4 or isotype-matched immunoglobulin G1 (IgG1) antibodies (BD PharMingen, San Diego, CA). Following two washes, cells were resuspended in 300 μl FACS buffer. Data (104 events) were collected on a flow cytometer (Beckman Coulter Inc, Fullerton, CA); FL1 color analysis was performed using WinMDI 2.8 software.
RNA extraction and qRT-PCR.
RNA extracted from cells using the RNeasy mini kit (QIAGEN, Valencia, CA) was quantified by spectrophotometry. The primers, designed to span intron/exon boundaries where applicable, were AE2616 (5′-TCCATCACCAGGTGCATGATGAC) and AE2617 (5′-GGTAGTGCGTGATGCTCTGGTAGG) for human emerin, AE2163 (5′-GAATCAAGATCTTCTACTCC) and AE2164 (5′-TAGTGGACTTCACTTTCTTG) for LAP2α, and AE1012 (5′-CGAGACTTCGTGGCAGAGCC) and AE1013 (5′-AGCACCAGAAACTGGCCAAG) for BAF. Cycling and quantitative normalization to cyclophilin A expression levels were performed as described previously (43).
Viruses and infections.
The pNLX.Luc(R−)-based single-round HIV-1 reporter virus was produced by cotransfecting 293T cells with an envelope expression vector as described previously (24), whereas four plasmids (pTY-CMVLuc, pCEP4-Tat, pHP-dI-N/A, and pCG-VSV-G) were used to generate HIV-SIN-Luc. MMLV reporter viruses based on pFB-Luc (Stratagene) were produced by cotransfection with pCG-gagpol and an envelope expression vector. Viral yields were determined by reverse transcriptase activity in cell supernatants as described previously (9, 29). Cells in 12-well plates were infected in 350 μl for 8 h, virus was removed, and cells were processed 36 h (MEFs and HeLa-P4) or 56 h (MDM) thereafter for determination of luciferase activity. Background luciferase levels, which ranged from 0.2% to 5.0% of positive signals depending on the multiplicity of infection, were determined from parallel infections with virions lacking envelope glycoproteins. Luciferase values are reported as relative light units (RLU) per μg of total cell protein concentration in cell lysates. MEFs transduced with pLPCX-based vectors were selected in puromycin.
RESULTS AND DISCUSSION
Potent gene silencing in HeLa-P4 cells using siRNA.
A recent study showed that HeLa cells knocked down for BAF or LAP2α expression using siRNA resisted MMLV infection and that HeLa cells or MDM down-regulated for BAF, LAP2α, or emerin expression potently resisted HIV-1 as well (13). Due to our long-standing interest in BAF and HIV-1 PIC function (7, 12, 22) and the more recent finding that LAP2α is a component of MMLV PICs and can work with BAF to potentiate MMLV integration in vitro (41), we too had generated and characterized relatively potent siRNAs directed against BAF and LAP2α (Table 1). Our initial studies focused on HeLa-P4 cells, a CD4-positive HeLa cell derivative that can be efficiently infected by HIV-1 (6) and efficiently transfected with siRNA (43).
TABLE 1.
siRNA sequences used in this study
| Human gene and siRNA | Sequence (position)a | Source or reference |
|---|---|---|
| BANF1 | ||
| BANF1-1 | GAAGCUGGAGGAAAGGGGUdTdT (96) | This study |
| BANF1-1C | GAAGCUGCACGUAAGGGGUdTdT | This study |
| BANF1-JS | GGCCUAUGUUGUCCUUGGCdTdT (123) | 13 |
| TMPO | ||
| TMPO2 | GCAAAUAUCCAGUUUCUUCdTdT (1553) | This study |
| TMPO2C | GCAAAUAACGACUUUCUUCdTdT | This study |
| TMPO2JS | GACAAGUUGAAGAGUGAGUdTdT (40) | 13 |
| EMD | ||
| EMD5 | GACCUGUCCUAUUAUCCUAdTdT (532) | This study |
| EMD5C | GACCUGUAUCCUUAUCCUAdTdT | This study |
| EMDJS | GAGGAGUGCAAGGAUAGGGdTdT (433) | 13 |
| CD4 | GAUCAAGAGACUCCUCAGUdGdA (1302) | 30 |
Sequence of plus-sense siRNA strands. Nucleotide mismatches in control sequences are in boldface. Numbers in parentheses indicate the nucleotide position of the 5′ base within the open reading frame.
HeLa-P4 cells transfected with our BAF-targeting siRNA BANF1-1 displayed on average a 30-fold reduction in steady-state mRNA levels as determined by qRT-PCR (data not shown), equating to an approximate 15-fold decrease in BAF protein levels in repeat experiments (Fig. 1A, lane 1). The recently published siRNA referred to here as BANF1-JS also potently knocked down BAF protein under these conditions (∼11-fold as compared to mock-transfected cells; Fig. 1A, lanes 2 and 4). Some siRNAs can elicit pleiotropic effects: for example, by inducing the cellular interferon response (37). To control for potential off-site effects, mismatched control siRNAs that differ from our targeting sequences by 3 to 4 bp were designed. Altering three positions within the BANF1-1 sequence counteracted the knockdown (Fig. 1A, lane 3), thus defining BANF1-1C control siRNA (Table 1). Our TMPO2 siRNA reduced the steady-state level of LAP2α protein about 11-fold in repeat experiments, wherein the published sequence elicited about a 10-fold effect (Fig. 1B, lanes 1 and 2) (data not shown). Three base pair changes were used to counteract the LAP2α knockdown, defining mismatched control sequence TMPO2C (Fig. 1B, lane 3; Table 1).
FIG. 1.
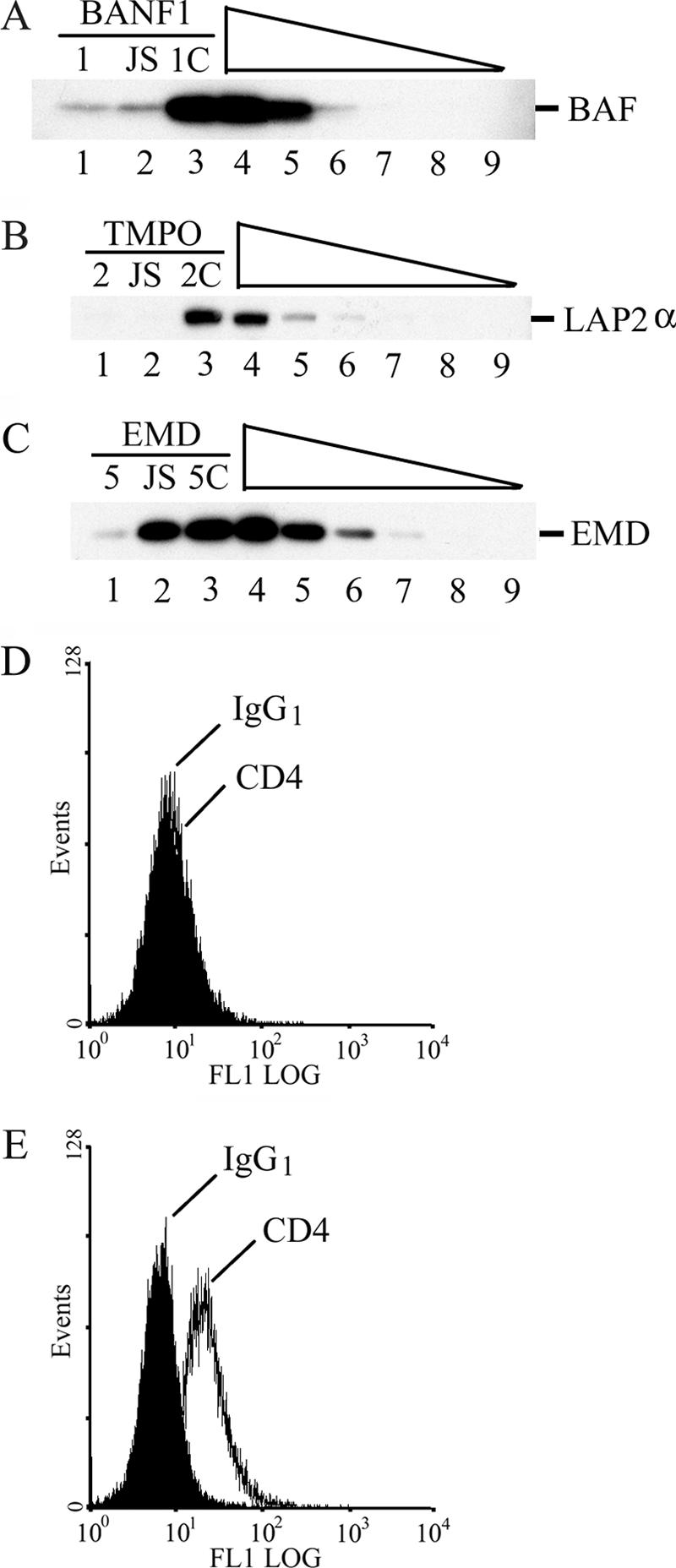
Characterization of knockdown and control siRNA sequences. (A) Proteins (10 μg) from HeLa-P4 cells transfected with the indicated siRNAs (lanes 1 to 3) were analyzed by Western blotting using anti-BAF antibodies. A twofold dilution series of mock-transfected cell extract was loaded in lanes 4 (undiluted) to 9 (32-fold dilution). BANF1-1 and BANFJS each elicited an approximate fivefold knockdown in this experiment, whereas approximate 15- and 11-fold knockdowns, respectively, were achieved in repeat experiments. (B and C) Same as panel A, except that blots were probed with anti-LAP2α and emerin antibodies, respectively. Of note, the EMD5 and EMDJS knockdowns in panel C (∼sevenfold and 30%) were the weakest and strongest effects observed for these siRNAs, respectively. In repeat experiments, EMD5 yielded 15-, 26-, 27-, and 33-fold knockdowns under conditions in which EMDJS failed to impact emerin steady-state levels. (D) HeLa-P4 cells transfected with CD4 siRNA and stained with FITC-conjugated isotype control IgG1 (filled histogram) or anti-CD4 antibodies (line) were analyzed by FACS. (E) Same as panel D, except that cells were mock transfected. Results are representative of a minimum of two independent experiments.
Knockdown efficiencies of in-house-generated versus published siRNAs were also evaluated for human emerin. Screening four different siRNAs obtained via Dharmacon SMARTpool technology identified the highly potent EMD5 sequence, which, on average, elicited a 22-fold reduction in emerin protein (Fig. 1C, lane 1) (data not shown). Changing 4 bp within the EMD5 sequence counteracted the knockdown (Fig. 1C, lane 3), defining the control sequence EMD5C (Table 1). Unlike our results with BAF and LAP2α, the published sequence, referred to here as EMDJS, exhibited a relatively weak ∼10% capacity to reduce steady-state emerin protein levels in repeat experiments (Fig. 1C, lane 2). As this was unexpected, an independent synthetic batch of EMDJS was analyzed. This siRNA behaved similarly to the initial batch in that it failed to appreciably reduce steady-state emerin protein levels when compared alongside cells transfected with EMD5 or EMD5C (data not shown).
HeLa-P4 cells knocked down for BAF, LAP2α, or emerin support normal levels of HIV-1 and MMLV infectivities.
As BAF and emerin are reportedly necessary to complete the integration step of the HIV-1 life cycle (13), siRNA-transfected cells were challenged with envelope-deleted viral vectors carrying the luciferase reporter gene (HIV-Luc and MLV-Luc) to quantify levels of HIV-1 and MMLV transduction. This experimental design restricts the infections to single rounds and affords quantitative readouts of proviral gene expression. As a control, the CD4 receptor, a cellular protein known to be important for HIV-1 glycoprotein-mediated infection (8, 14, 26), was targeted for knockdown using a previously-characterized siRNA (30). This siRNA (Table 1) potently down-regulated cell-surface CD4 levels (Fig. 1D) as compared to those on mock-transfected cells (Fig. 1E) or cells transfected with EMD5C siRNA (data not shown). To analyze CD4-dependent entry, HIV-Luc particles were engineered to carry the HIV-1NL4-3 envelope glycoprotein, whereas MLV-Luc particles were pseudotyped with an appropriately truncated gp41 variant (27).
CD4 knockdown potently inhibited HIV-Luc and MLV-Luc transduction of HeLa-P4 cells (Fig. 2A and B). In stark contrast, BAF, LAP2α, or emerin knockdown elicited much weaker antiviral effects, and, notably, these effects did not consistently reflect the levels of the targeted protein. For example, EMD5-transfected cells infected at a relatively high multiplicity of infection supported about half as much HIV-Luc transduction compared to mock-transfected cells (Fig. 2A, open bars). However, EMDJS siRNA, which failed to appreciably knockdown emerin protein (Fig. 1C), elicited a similar ∼twofold reduction in HIV-Luc titer and also similarly impacted MLV-Luc infectivity. Furthermore, the MLV-Luc titer was reduced ∼twofold as compared to mock transfection by TMPOJS, yet cells transfected with TMPO2, which reduced LAP2α protein levels to the same extent as TMPOJS (Fig. 1B), affected MLV-Luc less than twofold and had no affect on HIV-Luc infectivity (Fig. 2A and B). Analyzing the effects of BAF knockdown on HIV-Luc and MLV-Luc titers yielded the same overall picture. We conclude that HIV-Luc and MLV-Luc titers can be significantly reduced in HeLa-P4 cells upon potent knockdown of cell-surface CD4. In contrast, HeLa-P4 cells potently knocked down for BAF, LAP2α, or emerin support HIV-Luc and MLV-Luc titers that differ at most by 2- to 2.5-fold from mock or control siRNA-transfected cells, and moreover, these rather modest differences do not consistently reflect the levels of targeted proteins (Fig. 1 and 2) (data not shown).
FIG. 2.

HIV-1 and MMLV infectivities in siRNA-treated HeLa-P4 cells. (A) Normalized HIV-Luc infectivities following transfection with the indicated siRNAs. Values in empty bars were derived from cells infected with 2 × 106 reverse transcriptase cpm, whereas the filled-in bars were from infections initiated with 4 × 105 reverse transcriptase cpm. (B) Normalized luciferase values in cell extracts following infection with an equal volume (350 μl) of MLV-Luc. Mock, cells infected following treatment with oligofectamine reagent in the absence of siRNA; HeLaP4, infection of unmanipulated cells. Prior to infection, cell cultures were processed to generate the data presented in Fig. 1. Results are representative of a minimum of two independent experiments.
As the recent study that highlighted preintegrative roles for BAF and emerin in HIV-1 function in large part focused on MDM (13), significant effort was put into knocking down gene expression levels in primary cell cultures. One important consideration, for example, was that EMDJS might knock down emerin expression in a cell-type-dependent manner. In general, we found it much more difficult to perturb cell protein levels using siRNA in MDM as compared to HeLa-P4 cells. For example, our most potent sequence, EMD5, reduced emerin protein levels 22 ± 10-fold in HeLa-P4 cells (n = 5), yet elicited only an ∼1.6-fold effect using cells derived from four different blood donors. Cells derived from a fifth donor, however, responded particularly well to EMD5, revealing an approximately 11-fold reduction in emerin protein levels (Fig. 3A, lane 1). HIV-Luc pseudotyped with the dualtropic HIV-189.6 envelope glycoprotein infected these cells marginally (∼30%) less well than EMD5C- or mock-transfected cells, indicating a potential role for emerin protein in HIV-1 infection under these conditions (Fig. 3B). Of note, this culture, as well as other MDM cultures, was unresponsive to EMDJS siRNA (data not shown).
FIG. 3.

A modest HIV-1 defect in MDM following emerin protein knockdown. (A) Lanes 1 and 2, levels of emerin protein in MDM cell extracts following transfection with the indicated siRNA (2 μM). Lanes 3 to 7, twofold dilution series of mock-transfected extract from undiluted (lane 3) to 16-fold diluted (lane 7). (B) Normalized luciferase activity levels in extracts of cells transfected with the indicated siRNA (or mock transfected) and infected with 350 μl of HIV-Luc. Black bars, infections initiated with a 1:5 dilution of virus inoculum.
Emerin and A-type lamin knockout cells support normal levels of HIV-1 and MMLV infectivities.
Recent results highlight a potential limitation of RNAi technology when targeting host factors that may function at the preintegration/integration step in the HIV-1 life cycle. Reducing lens epithelium-derived growth factor (LEDGF)/p75 levels ∼15-fold in HeLa-P4 cells yielded a much less drastic (2- to 3-fold) infectivity defect, suggesting that residual protein levels might suffice for near wild-type levels of cofactor function under these conditions (43). Indeed, recent results using optimized short-hairpin RNA (shRNA)-expressing lentiviral vectors that specifically targeted residual LEDGF/p75 levels unveiled a significant role for the host cell factor in HIV-1 integration (23). In this sense, genetic knockout cells represent the optimal tissue culture model, as they completely lack the protein of interest.
Pseudotyped HIV-1 vectors can efficiently transduce mouse fibroblasts (2, 36), and the murine emerin gene has been knocked out (15). Moreover, MEF cells derived from knockout embryos proliferate in vitro (15). Thus, to critically address the role of emerin protein in HIV-1 preintegration biology, infectivities were quantified using primary cells derived from emerin knockout embryos (Emd−/y) versus those from matched littermate control (Emd+/+) animals. A self-inactivating HIV-1-based vector that expresses luciferase from an internal CMV promoter (HIV-SIN-Luc) was used in these experiments to bypass the relatively poor activity of the HIV-1 promoter in mouse cells (11). MEFs derived from A-type lamin knockout embryos (Lmna−/−) were analyzed in parallel, as the lack of lamin A/C proteins in large part mislocalizes emerin from the nuclear periphery to the cell cytoplasm (39).
HIV-SIN-Luc and MLV-Luc pseudotyped with the vesicular stomatitis virus G (VSV-G) envelope glycoprotein transduced lamin A knockout cells to a similar extent as matched littermate control cells over a 100-fold range in multiplicity of infection (Fig. 4A and B). These results are consistent with the observation that HIV-1 can efficiently infect MDM despite potent siRNA-mediated down-regulation of A-type lamin expression (13). Notably, HIV-SIN-Luc and MLV-Luc transduced the emerin knockout cells as efficiently as Emd+/+ littermate control cells (Fig. 4A and B). A lack of emerin protein in Emd−/y cell extracts confirmed the knockout cell phenotype (Fig. 4C, lane 2). These results demonstrate that normal levels of HIV-1 infection can occur under conditions of emerin protein mislocalization (39) or in its complete absence.
FIG. 4.

Efficient infection of emerin and A-type lamin knockout cells by MMLV- and HIV-1-based vectors. (A) The indicated cells infected with VSV-G-pseudotyped MLV-Luc (white bars, 2 × 105 reverse transcriptase cpm; black and gray bars, 10- and 100-fold dilutions of virus inoculum, respectively) were lysed, and cell extracts were processed to determine normalized levels of luciferase activity. (B) Same as panel A, except that cells were infected with HIV-SIN-Luc (white bars, 2 × 105 reverse transcriptase cpm). (C) Western blot analysis of emerin protein expression in the indicated primary MEF cells.
Mouse and human emerin are nonequivalent, displaying 75% identity and 91% amino acid sequence homology, considering conservative substitutions (Fig. 5A). Recent results implicate the most conserved part of the emerin protein, the N-terminal LEM domain that binds directly to BAF, as important for HIV-1 function (13). The mouse and human domains are 81% identical and 98% homologous (Fig. 5A), and, moreover, mouse and human BAF are 97% identical and 100% homologous (12). Thus, it seemed unlikely that the marginal difference in sequence identity between the mouse and human LEM domains could account for a requirement for emerin protein in human cells while at the same time dispense a requirement in mouse cells. To formally address this issue, human emerin was expressed in mouse knockout cells. To facilitate this analysis, primary MEFs transformed by the SV40 large T antigen were transduced with pLPCX-emerin or vector alone. The resulting Emd−/y pLPCX-EMD cells expressed human emerin protein at a level that approximated that of the endogenous murine protein (Fig. 5B, compare lane 2 to lane 3). Notably, Emd−/y pLPCX cells, which lack emerin protein (Fig. 5B, lane 1), supported slightly higher levels of HIV-SIN-Luc transduction than the engineered Emd−/y pLPCX-EMD cells (Fig. 5C). Thus, efficient HIV-1 infection of MEF cells occurs independent of cellular emerin protein.
FIG. 5.

Expression of human emerin in mouse knockout cells does not detectably alter their susceptibly to HIV-1 infection. (A) Primary sequence alignment of human (h_EMD) and mouse (m_Emd) emerin proteins. *, amino acid identity; :, homology through conservative substitution as defined by reference 33. Boxed areas indicate the LEM domain, lamin A binding region, and transmembrane domain (TMD) (16). The alignment was created using ClustalW (42). (B) Western blot analysis of emerin protein expression in transformed MEF cell lines. Lane 1, protein extracted from control Emd−/y cells transduced with empty vector; lane 2, Emd−/y pLPCX-EMD cells; lane 3, Emd+/+ pLPCX cells; lane 4, HeLa-P4 cells. Human emerin migrates slightly faster than the mouse protein under these electrophoresis conditions. Migration positions of molecular mass standards are indicated to the right. (C) Normalized levels of HIV-SIN-Luc infectivities in the indicated cell types. White bars, cells infected with 2 × 106 reverse transcriptase cpm; black and gray bars, cells infected with a 10- and 100-fold dilution of virus inoculum, respectively.
Conclusions.
Recent results implicate there are important roles for LAP2α (41) and BAF (13) during MMLV infection and for LAP2α, BAF, and emerin in determining HIV-1 infectivity (13). The brunt of the HIV-1 work was conducted in MDM to avoid potential complications of disrupting INM integrity during mitosis, though dividing HeLa cells knocked down for LAP2α, BAF, or emerin seemingly resisted HIV-1 transduction to the same extent as MDM (13). The work presented here would seem to refute these findings, as HeLa-P4 cells potently down-regulated for BAF, LAP2α, or emerin expression were readily infected with HIV-Luc and MLV-Luc single-round vectors (Fig. 1 and 2). To critically address the role of emerin protein in dividing target cells, primary knockout Emd−/y MEFs were challenged alongside matched Emd+/+ control cells with HIV-SIN-Luc. The results of these experiments fail to support an important role for emerin in regulating HIV-1 function (Fig. 4). As the expression of human emerin in mouse knockout cells did not detectably alter their susceptibility to HIV-1 infection (Fig. 5), we conclude that emerin plays little if any role in enabling HIV-1 to efficiently integrate into cell chromosomes when cells are actively dividing.
In contrast to HeLa-P4 cells, we failed to reproducibly knock down proteins to significant levels in human MDM. In one experiment, cells rather potently (∼11-fold) down-regulated for emerin protein revealed a relatively minor (∼30%) infection defect (Fig. 3). Though this result might seem to refute a rate-limiting role for emerin protein in determining HIV-1 infectivity in human MDM, it remains possible that greater knockdowns would unveil more significant defects, as has recently been demonstrated for the LEDGF/p75 cell factor in cycling T cells (23). Thus, it remains possible that emerin might play an important role in human MDM. We do note that results of an independent parallel study indicate that HIV-1-based vectors transduce primary macrophages derived from emerin knockout mice to the same extent as cells isolated from wild-type littermate control animals (V. KewalRamani, personal communication). Thus, it would seem that emerin protein is dispensable for normal levels of HIV-1 infection in at least some nondividing primary cell types. The generation of BAF and LAP2α knockout cells should help to clarify if either of these proteins plays an important role in retroviral preintegration trafficking and/or integration.
Acknowledgments
Plasmids pTY-EFeGFP, pCEP4-Tat, and pHP-dI-N/A were obtained through the AIDS Research and Reference Reagent Program, NIH, from Lung-Ji Chang (4). We are grateful to J. Walsh and R. Mulligan for PCG-gagpol and PCG-VSV-G plasmids; D. Borger and J. DeCaprio for the pLB(N)CX derivative expressing SV40 large T antigen; K. Wilson for anti-BAF antibodies; A. Schlesinger and J. Lieberman for advice on how to transfect human MDM; T. Sullivan and C. Stewart for their generous gifts of primary cells derived from Emd−/y, Emd+/+, Lmna−/−, and Lmna+/+ embryos; N. K. Raghavendra for help with figure preparation; and V. KewalRamani for sharing results prior to publication.
This work was supported by NIH grant AI52014.
Footnotes
Published ahead of print on 11 October 2006.
REFERENCES
- 1.Arhel, N., A. Genovesio, K. A. Kim, S. Miko, E. Perret, J. C. Olivo-Marin, S. Shorte, and P. Charneau. 2006. Quantitative four-dimensional tracking of cytoplasmic and nuclear HIV-1 complexes. Nat. Methods 3:817-824. [DOI] [PubMed] [Google Scholar]
- 2.Baumann, J. G., D. Unutmaz, M. D. Miller, S. K. J. Breun, S. M. Grill, J. Mirro, D. R. Littman, A. Rein, and V. N. KewalRamani. 2004. Murine T cells potently restrict human immunodeficiency virus infection. J. Virol. 78:12537-12547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Borger, D. R., and J. A. DeCaprio. 2006. Targeting of p300/CREB binding protein coactivators by simian virus 40 is mediated through p53. J. Virol. 80:4292-4303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Campbell, E. M., and T. J. Hope. 2005. Gene therapy progress and prospects: viral trafficking during infection. Gene Ther. 12:1353-1359. [DOI] [PubMed] [Google Scholar]
- 5.Chang, L.-J., V. Urlacher, T. Iwakuma, Y. Cui, and J. Zucali. 1999. Efficacy and safety analyses of a recombinant human immunodeficiency virus type 1 derived vector system. Gene Ther. 6:715-728. [DOI] [PubMed] [Google Scholar]
- 6.Charneau, P., M. Alizon, and F. Clavel. 1992. A second origin of DNA plus-strand synthesis is required for optimal human immunodeficiency virus replication. J. Virol. 66:2814-2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen, H., and A. Engelman. 1998. The barrier-to-autointegration protein is a host factor for HIV type 1 integration. Proc. Natl. Acad. Sci. USA 95:15270-15274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dalgleish, A. G., P. C. L. Beverly, P. R. Clapham, D. H. Crawford, M. F. Greaves, and R. A. Weiss. 1984. The CD4 (T4) antigen is an essential component of the receptor for the AIDS retrovirus. Nature 312:763-767. [DOI] [PubMed] [Google Scholar]
- 9.Deminie, C. A., and M. Emerman. 1993. Incorporation of human immunodeficiency virus type 1 Gag proteins into murine leukemia virus virions. J. Virol. 67:6499-6506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Engelman, A., and P. Cherepanov. 2006. Recent advances in retroviral replication: Cellular machines and novel anti-viral defense mechanisms, p. 91-129. In K. L. Hefferon (ed.), Recent advances in RNA virus replication. Transworld Research Network, Kerala, India.
- 11.Garber, M. E., P. Wei, V. N. KewalRamani, T. P. Mayall, C. H. Herrmann, A. P. Rice, D. R. Littman, and K. A. Jones. 1998. The interaction between HIV-1 Tat and human cyclin T1 requires zinc and a critical cysteine residue that is not conserved in the murine CycT1 protein. Genes Dev. 12:3512-3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harris, D., and A. Engelman. 2000. Both the structure and DNA binding function of the barrier-to-autointegration factor contribute to reconstitution of HIV type 1 integration in vitro. J. Biol. Chem. 275:39671-39677. [DOI] [PubMed] [Google Scholar]
- 13.Jacque, J. M., and M. Stevenson. 2006. The inner-nuclear-envelope protein emerin regulates HIV-1 infectivity. Nature 441:641-645. [DOI] [PubMed] [Google Scholar]
- 14.Klatzmann, D., E. Champagne, S. Chamaret, J. Gruest, D. Guetard, T. Hercend, J. C. Gluckman, and L. Montagnier. 1984. T-lymphocyte T4 molecule behaves as the receptor for human retrovirus LAV. Nature 312:767-768. [DOI] [PubMed] [Google Scholar]
- 15.Lammerding, J., J. Hsiao, P. C. Schulze, S. Kozlov, C. L. Stewart, and R. T. Lee. 2005. Abnormal nuclear shape and impaired mechanotransduction in emerin-deficient cells. J. Cell Biol. 170:781-791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee, K. K., T. Haraguchi, R. S. Lee, T. Koujin, Y. Hiraoka, and K. L. Wilson. 2001. Distinct functional domains in emerin bind lamin A and DNA-bridging protein BAF. J. Cell Sci. 114:4567-4573. [DOI] [PubMed] [Google Scholar]
- 17.Lee, M. S., and R. Craigie. 1998. A previously unidentified host protein protects retroviral DNA from autointegration. Proc. Natl. Acad. Sci. USA 95:1528-1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lewis, P., M. Hensel, and M. Emerman. 1992. Human immunodeficiency virus infection of cells arrested in the cell cycle. EMBO J. 11:3053-3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lewis, P. F., and M. Emerman. 1994. Passage through mitosis is required for oncoretroviruses but not for the human immunodeficiency virus. J. Virol. 68:510-516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li, M., and R. Craigie. 2006. HIV goes nuclear. Nature 441:581-582. [DOI] [PubMed] [Google Scholar]
- 21.Limón, A., N. Nakajima, R. Lu, H. Z. Ghory, and A. Engelman. 2002. Wild-type levels of nuclear localization and human immunodeficiency virus type 1 replication in the absence of the central DNA flap. J. Virol. 76:12078-12086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin, C.-W., and A. Engelman. 2003. The barrier-to-autointegration factor is a component of functional human immunodeficiency virus type 1 preintegration complexes. J. Virol. 77:5030-5036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Llano, M., D. T. Saenz, A. Meehan, P. Wongthida, M. Peretz, W. H. Walker, W. Teo, and E. M. Poeschla. 2006. An essential role for LEDGF/p75 in HIV integration. Science 314:461-464. (First published 7 September 2006; doi: 10.1126/science.1132319.) [DOI] [PubMed] [Google Scholar]
- 24.Lu, R., H. Z. Ghory, and A. Engelman. 2005. Genetic analyses of conserved residues in the carboxyl-terminal domain of human immunodeficiency virus type 1 integrase. J. Virol. 79:10356-10368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu, R., A. Limón, E. Devroe, P. A. Silver, P. Cherepanov, and A. Engelman. 2004. Class II integrase mutants with changes in putative nuclear localization signals are primarily blocked at a postnuclear entry step of human immunodeficiency virus type 1 replication. J. Virol. 78:12735-12746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maddon, P. J., A. G. Dalgleish, J. S. McDougal, P. R. Clapham, R. A. Weiss, and R. Axel. 1986. The T4 gene encodes the AIDS virus receptor and is expressed in the immune system and the brain. Cell 47:333-348. [DOI] [PubMed] [Google Scholar]
- 27.Mammano, F., F. Salvatori, S. Indraccolo, A. De Rossi, L. Chieco-Bianchi, and H. G. Göttlinger. 1997. Truncation of the human immunodeficiency virus type 1 envelope glycoprotein allows efficient pseudotyping of Moloney murine leukemia virus particles and gene transfer into CD4+ cells. J. Virol. 71:3341-3345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McDonald, D., M. A. Vodicka, G. Lucero, T. M. Svitkina, G. G. Borisy, M. Emerman, and T. J. Hope. 2002. Visualization of the intracellular behavior of HIV in living cells. J. Cell Biol. 159:441-452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakajima, N., R. Lu, and A. Engelman. 2001. Human immunodeficiency virus type 1 replication in the absence of integrase-mediated DNA recombination: definition of permissive and nonpermissive T-cell lines. J. Virol. 75:7944-7955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Novina, C. D., M. F. Murray, D. M. Dykxhoorn, P. J. Beresford, J. Riess, S. K. Lee, R. G. Collman, J. Lieberman, P. Shankar, and P. A. Sharp. 2002. siRNA-directed inhibition of HIV-1 infection. Nat. Med. 8:681-686. [DOI] [PubMed] [Google Scholar]
- 31.Piller, S. C., L. Caly, and D. A. Jans. 2003. Nuclear import of the pre-integration complex (PIC): the Achilles heel of HIV? Curr. Drug Targets 4:409-429. [DOI] [PubMed] [Google Scholar]
- 32.Roe, T., T. C. Reynolds, G. Yu, and P. O. Brown. 1993. Integration of murine leukemia virus DNA depends on mitosis. EMBO J. 12:2099-2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schwartz, R. M., and M. O. Dayhoff. 1978. Matrices for detecting distance relationships, p. 353-358. In M. O. Dayhoff (ed.), Atlas of protein sequence and structure, vol. 5, suppl. 3. National Biochemical Research Foundation, Washington, DC. [Google Scholar]
- 34.Segura-Totten, M., A. K. Kowalski, R. Craigie, and K. L. Wilson. 2002. Barrier-to-autointegration factor: major roles in chromatin decondensation and nuclear assembly. J. Cell Biol. 158:475-485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Segura-Totten, M., and K. L. Wilson. 2004. BAF: roles in chromatin, nuclear structure and retrovirus integration. Trends Cell Biol. 14:261-266. [DOI] [PubMed] [Google Scholar]
- 36.Siva, A. C., and F. Bushman. 2002. Poly(ADP-ribose) polymerase 1 is not strictly required for infection of murine cells by retroviruses. J. Virol. 76:11904-11910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sledz, C. A., M. Holko, M. J. de Veer, R. H. Silverman, and B. R. Williams. 2003. Activation of the interferon system by short-interfering RNAs. Nat. Cell Biol. 5:834-839. [DOI] [PubMed] [Google Scholar]
- 38.Song, E., S.-K. Lee, D. M. Dykxhoorn, C. Novina, D. Zhang, K. Crawford, J. Cerny, P. A. Sharp, J. Lieberman, N. Manjunath, and P. Shankar. 2003. Sustained small interfering RNA-mediated human immunodeficiency virus type 1 inhibition in primary macrophages. J. Virol. 77:7174-7181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sullivan, T., D. Escalante-Alcalde, H. Bhatt, M. Anver, N. Bhat, K. Nagashima, C. L. Stewart, and B. Burke. 1999. Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J. Cell Biol. 147:913-920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Suzuki, Y., and R. Craigie. 2002. Regulatory mechanisms by which barrier-to-autointegration factor blocks autointegration and stimulates intermolecular integration of Moloney murine leukemia virus preintegration complexes. J. Virol. 76:12376-12380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Suzuki, Y., H. Yang, and R. Craigie. 2004. LAP2alpha and BAF collaborate to organize the Moloney murine leukemia virus preintegration complex. EMBO J. 23:4670-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vandegraaff, N., E. Devroe, F. Turlure, P. A. Silver, and A. Engelman. 2006. Biochemical and genetic analyses of integrase-interacting proteins lens epithelium-derived growth factor (LEDGF)/p75 and hepatoma-derived growth factor related protein 2 (HRP2) in preintegration complex function and HIV-1 replication. Virology 346:415-426. [DOI] [PubMed] [Google Scholar]