

Abstract
NK cells are key effectors of innate immunity and host survival during cytomegalovirus (CMV) infection. Innate murine CMV (MCMV) resistance in MA/My mice requires Ly49H/m157-independent H-2k-linked NK cell control. Here we show that replacement of MA/My H-2k with C57L H-2b susceptibility genes led to a remarkable loss of innate virus immunity, though NK gamma interferon was induced in H-2b and H-2k strains shortly after infection. Thus, H-2b genes expressed in C57L or MA/My.L-H2b are sufficient in alerting NK cells to intrusion but fail to support NK restraint of viral infection. In addition, novel H-2 recombinant strains were produced and utilized in a further refinement of a critical genetic interval controlling innate H-2k-linked MCMV resistance. Importantly, this analysis excluded the gene interval from Kk class I through class II. The responsible gene(s) therefore resides in an interval spanning Dk class Ia and more-distal major histocompatibility complex (MHC) nonclassical class Ib genes. Recently, the NK activation receptor Ly49P and MHC class I Dk proteins were genetically implicated in MCMV resistance, in part because Ly49P-expressing reporter T cells could specifically bind Dk molecules on MCMV-infected mouse embryonic fibroblasts (MEFs). However, as we found that H-2k innate resistance differs in the C57L or MA/My backgrounds and because MCMV very efficiently downregulates H-2k class I proteins in L929 cells and primary MEFs shortly after infection, a Ly49P/Dk model should not fully explain H-2k-linked MCMV resistance.
Cytomegaloviruses (CMV) are betaherpesviruses that display species-specific tropism. Because human CMV (HCMV) and murine CMV (MCMV) cause severe infections in immunodeficient or immunologically immature hosts and share many biological features in their natural host settings, MCMV is an important model system for studies of innate viral immunity (27). Additionally, because NK cells provide critical innate immune defenses in CMV infections (25), the MCMV infection model is also useful for understanding the role of NK cells and their capacity to recognize virus targets.
Chalmer et al. noted a survival role for H-2k after high-dose MCMV infection in C3H, CBA, and BALB.K inbred mice (7). Further study revealed similar levels of MCMV infection and replication in mouse embryonic fibroblasts (MEFs) of different H-2 haplotypes, including H-2b and H-2k (7). NK cells were also implicated, since enhanced NK cytotoxicity is a general feature of MCMV infection in more-resistant H-2k mouse strains (3). Scalzo et al. (32) later documented a primary role for NK cells in innate MCMV immunity in MA/My through immunodepleting NK cells with anti-NK1.1 monoclonal antibody (mAb) before infection. Elsewhere, we confirmed the importance of NK cells in innate MA/My MCMV immunity and extended our previous findings by demonstrating that H-2k introgression onto the C57L genetic background in C57L.M-H2k (L.M-H2k) congenic mice was sufficient to convert MCMV susceptibility to resistance through NK control (11). An epistatic genetic interaction between Cmv3 (Ly49P) and H-2k was recently noted, and because Ly49P-transduced reporter T cells specifically bind class I Dk proteins on MCMV-infected MEFs, the receptor-ligand interaction is implicated in MA/My resistance (10). However, other unknown genetic factors clearly contribute, since H-2k substantially reduces spleen virus replication (32) and mortality (3) even when expressed on the BALB/c (Ly49P−) background. Further study is thus required to elucidate the mechanisms that govern the effectual NK recognition of MCMV-infected targets leading to innate major histocompatibility complex (MHC)-linked control in H-2k mice.
Because NK cells also express inhibitory receptors for MHC class I molecules and their effector functions are determined through integration of stimulatory and inhibitory signals (22), an alternate hypothesis also involves NK recognition of MHC class I deficiency, as often occurs in viral infection. Cell surface expression of H-2b and H-2d class I proteins are specifically and dramatically altered by MCMV in vitro (28, 38, 42). MCMV gp48 hijacks nascent MHC class I proteins for lysosomal degradation (28), gp40 blocks endoplasmic reticulum-Golgi MHC class I export (42), and gp34 escorts class I molecules to the cell surface to interfere with cytotoxic T-lymphocyte recognition (17, 20). Although the mechanisms are distinct, HCMV likewise utilizes several gene products to specifically alter HLA class I expression (27). However, HCMV also stabilizes HLA-E proteins with class I signal peptide-related UL40 leader peptide (35), presumably to bind inhibitory CD94/NKG2A receptors on NK cells and evade attack (5, 37, 39). Additionally, HCMV utilizes UL141 and UL142 to directly interfere with NK recognition and attack (36, 41). Taken together, the data show that H-2k resistance may require altered MHC class I or class I-related protein recognition through inhibitory receptor sensing in addition to direct stimulation.
To assess the role of H-2k in MCMV resistance, we replaced it in MA/My mice with the H-2b susceptibility locus of C57L. We examined MCMV infectivity and replication in MEFs derived from the resistant or susceptible strains, and innate MCMV immunity phenotypes were also analyzed. To also examine the role of NK cells, intracellular gamma interferon (IFN-γ) was quantified for comparison in H-2k and H-2b strains over a 3.5-day (d) time course following infection. Further, since the regulation of H-2k class I proteins by MCMV has not been previously characterized, we also examined Kk and Dk class I proteins on L929 cells and primary (C57L × MA/My)F1 MEFs shortly after virus infection. We provide evidence indicating that a Ly49P/Dk model is inadequate to fully account for MA/My MCMV resistance.
MATERIALS AND METHODS
Mice.
MA/My and C57L breeder pairs were purchased from The Jackson Laboratory (Bar Harbor, ME) and housed in the MR-5 specific-pathogen-free vivarium at the University of Virginia, which is fully accredited by the American Association for Accreditation of Laboratory Animal Care. A simple sequence length polymorphism (SSLP) marker-assisted genome selection method was used to produce MA/My.L-H2b (M.L-H2b) mice as we have described previously (11, 30, 33). Briefly, (C57L × MA/My)F1 hybrid mice were first backcrossed to MA/My. Backcross offspring were selected for H-2b-linked D17Mit16 and D17Mit10 donor alleles on a genetic background enriched for MA/My alleles. Selected backcross mice were then backcrossed to MA/My, and further selective backcrossing followed by brother-sister crossing for H-2b homozygosity yielded the M.L-H2b (N6 [sixth backcross generation]) mice tested as shown in Fig. 1. Notably, homozygous M.L-H2b (N9) mice display comparable MCMV susceptibilities (not shown). (C57L × MA/My)F1 hybrids, L.M-H2k (N7), and MA/My.L-H2bR1 (M.L-H2bR1 [N9]) recombinant congenic mice were bred in the same facility. All animal studies were approved by and conducted in accordance with Animal Care and Use Committee oversight.
FIG. 1.

Deficient innate MCMV immunity in MA/My.L-H2b. (A) The chromosome 17 interval from D17Mit57 to D17Mit93 in M.L-H2b is depicted at top with informative SSLP markers used in genetic screening and selection also shown. Physical map locations (in megabases [Mb]) are indicated. The H-2 complex and K and D class I genes are also shown. Virus levels in MCMV-infected (d 3.5) M.L-H2b, L.M-H2k, and control mice were quantitated by QPCR (B) or virus plaque assay (C). The plaque assay detection level is indicated by the broken line in panel C. Four to eight animals were studied per group. (D) Spleen (filled symbols) and liver (open symbols) virus levels also were quantitated for similarly infected (MA/My × C57L)F1 and M.L-H-2b/k mice and their noncongenic H-2k littermates (LM control), and for C57L control mice, by QPCR. (E) Spleen virus levels in MCMV-infected (M.L-H2bR1 × MA/My) × C57L hybrids segregated by H-2 genotype (as designated) and control strains quantitated by QPCR are shown. Data are representative of two independent experiments.
Cell lines and tissue culture.
NIH 3T12 (ATCC CCL-164), NIH 3T3 (ATCC CRL1658), and L929 (ATCC CCL-1, H-2k) were grown in Dulbecco's modified Eagle medium supplemented with 10% (vol/vol) newborn calf serum, penicillin-streptomycin (100 U/ml:100 μg/ml), and glutamine (2.0 mM). C57L, MA/My, (C57L × MA/My)F1, and C57L.M-H2k MEFs were prepared on d 16 to 18 postcoitus as described previously (31). After expansion in Dulbecco's modified Eagle medium plus 10% fetal bovine serum in T175 flasks, passage 2 MEFs were stored under liquid nitrogen. Only passage 2 to 5 MEFs were utilized in experiments. For IFN induction of MHC class I cell surface expression, MEFs were incubated in recombinant mouse IFN-γ (Research Diagnostics, Inc.) or IFN-β (Fitzgerald Industries, Concord, MA) for 24 to 30 h before virus infection.
Antibodies and flow cytometry.
Anti-mouse CD3ɛ (145-2C11) peridinin chlorophyll protein (PerCP), CD49b (DX5) phycoerythrin (PE), H-2Db (28-14-8) PE, H-2-Kb-Db (20-8-4S) biotinylated, control immunoglobulin G2a (IgG2a) (eBM2a), and rat anti-mouse CD71 and IgG2a were purchased from eBioscience. Anti-mouse NK1.1 (PK136) PE, IFN-γ fluorescein isothiocyanate, H-2Kk (36-7-5) PE, H-2Dk (15-5-5) biotinylated and H-2Kb (AF6-88.5) PE, and streptavidin-PerCP were purchased from BD Pharmingen. Anti-mouse H-2Dk (15-5-5) PE was purchased from BioLegend. Anti-H-2-Kb-Db (20-8-4S; kindly provided by V. Engelhard, University of Virginia, Charlottesville, VA) antibodies were purified from spent hybridoma supernatant by use of a HiTrap Protein G Hp column (Amersham Pharmacia Biotech, Sweden) and subsequently biotinylated using EZ-Link sulfo-N-hydroxysuccinimide-LC-biotin, 2-(4′-hydroxyazobenzene) benzoic acid (HABA), and avidin (Pierce). All antibody staining was performed on ice. For analysis of spleen leukocytes, cells were first blocked with mAb 2.4G2 (5 μg/ml) on ice for 30 min before primary antibody staining. For intracellular IFN-γ staining, cells were incubated for 2 h at 37°C in media containing either monensin (2.5 μM) or brefeldin A (5 μg/ml). Afterwards, cells were first stained for cell surface markers and then fixed and permeabilized using a CytoFix/CytoPerm kit (BD Pharmingen). Labeled cells were analyzed by flow cytometry on a FACScan instrument (BD Biosciences), and data were subsequently analyzed using FlowJo (TreeStar, version 4.3.1).
Virus assays.
MCMV (Smith strain, ATCC VR 194) salivary gland stock virus was prepared after serial passage in BALB/c as described previously (29). Experimental mice (8 to 12 weeks of age) were intraperitoneally infected with MCMV (1 × 105 PFU). On d 3.5 postinfection (84 to 90 h), spleen and liver virus levels were quantified using quantitative real-time PCR (QPCR) as described previously (29, 40). All sample measurements were performed in triplicate. Spleen and liver homogenate infectious MCMV PFU were also measured on NIH 3T3 monolayers essentially as described previously (6, 40), but with a slight modification. Briefly, serially 10-fold-diluted tissue homogenates (100 μl) were plated on NIH 3T3 cell monolayers (70 to 90% confluent) in duplicate in 24-well tissue culture plates for 1 h at 37°C with frequent rocking. Afterwards, MCMV-infected cell monolayers were fully resuspended in D10 medium (1 ml). Virus plaques were counted microscopically on d 3. In confirmation, infectious virus particles can be efficiently and reliably distributed through limiting dilution based on viral titers determined using this strategy. In particular, when NIH 3T3 monolayers are infected with 0.67 PFU per well (10 wells of a 24-well plate), 20 to 70% of wells contain at least one plaque by d 3 after cellular infection. Most wells without viral plaques on d 3 after infection remain plaque free through d 7 after infection.
For in vitro MCMV infectivity and replication studies, MEFs growing in 24-well plates were infected with MCMV (multiplicity of infection [MOI] = 0.08). Supernatants were collected daily through d 8 and subsequently titered on 3T12 cells. DNA was also isolated from the remaining cell monolayers by use of a Puregene DNA isolation kit as described previously (34). Recombinant green fluorescent protein (GFP)-expressing Δm157-MCMV (11), wild-type K181, and Smith strain MCMV viruses were utilized after passage and thorough titer determinations with NIH 3T3 cells. Cell lines or primary MEFs were infected with a range of MCMV PFU (MOI, 0.01 to 10).
RESULTS
H-2b linked genes fail to support innate MCMV immunity through NK cells.
We previously noted that multiple genes contribute vital Ly49H-independent immunity shortly after MCMV infection in New Zealand White or MA/My mice (11, 29). Because H-2k-linked genes were shown to contribute substantially to MA/My virus resistance (10, 11) and also converted susceptibility to full resistance in C57L.M-H2k congenic mice (11), we examined whether they are also essential to innate immunity in this model system. We replaced H-2k in MA/My with H-2b from MCMV-susceptible C57L by use of SSLP marker-assisted selection to obtain H-2b donor alleles on a MA/My background (Fig. 1A). H-2b introgression was confirmed using allele-specific anti-MHC class I mAbs to stain M.L-H2b splenocytes (not shown).
We assessed innate MCMV immunity in M.L-H2b by measuring virus levels in spleens and livers. These mice displayed remarkable susceptibility that was even more severe than that of C57L (Fig. 1B and C). By comparison, similarly infected L.M-H2k/b and control MA/My mice were fully protected by H-2k. Thus, H-2k-linked gene expression is essential for innate MCMV protection through NK cells in MA/My, whereas H-2b linked gene expression is not adequate for effective NK control. While previous work indicated that an additional genetic factor beyond the MHC or the NK gene complex can contribute to innate MCMV control (11), we also evaluated immunity in (MA/My × C57L)F1 hybrids and their heterozygous congenics, M.L-H2b/k mice (Fig. 1D). As expected, virus levels in littermate control (H-2k) spleens were comparable with those for MA/My. Intriguingly, however, virus levels in M.L-H2b/k spleens were actually intermediate between those of the progenitor strains. This finding indicates that H-2k is not fully protective on the MA/My genetic background. Instead, H-2k homozygosity is required to fully establish innate MCMV resistance on the MA/My background. Moreover, since differences in MCMV control in H-2 heterozygous mice strictly correlated with their genetic background (compare L.M-H2k/b and M.L-H2b/k in Fig. 1B and D, respectively), an additional factor(s) coming from C57L must increase the extent of innate virus resistance afforded by the H-2k haplotype.
To further delimit an H-2k-linked critical region, novel H-2 recombinant mice were produced from the M.L-H2b and L.M-H2k strains. One strain, M.L-H2bR1, was identified as containing an informative recombination breakpoint between the H-2 K and D genes. Importantly, this strain retains the expression of Kb and C57L-derived MHC class II proteins (not depicted) but also expresses MA/My-derived class I Dk molecules. We crossed (MA/My × M.L-H2bR1)N9 backcross mice with C57L and studied MCMV control in their hybrid offspring. As expected, a 1:1 Mendelian distribution of homozygous (Kb to I-Eb region) and fully heterozygous (H-2k,b) progeny on F1 genetic backgrounds was observed. The corresponding innate MCMV immunities in the two groups shown in Fig. 1E indicate that Kk expression is not needed to limit MCMV in the spleen. The H-2k-linked critical region should therefore reside distal to SSLP marker 17Uva12 and the class II genes while overlapping Dk class I and more-distal H-2 nonclassical class Ib genes.
Similar levels of MCMV infectivity and replication in susceptible C57L and resistant C57L.M-H2k strain-derived MEFs.
Though previous work has shown that MCMV infectivity and replication kinetics are similar in H-2b- and H-2k-derived MEFs (7), Harnett and Shellam (16) noted high MCMV susceptibility in BALB/c (H-2d) and B6 (H-2b) strain-derived MEFs compared with that for MEFs from H-2k strains C3H and CBA. Thus, we also analyzed viral growth kinetics in MEFs prepared from embryos of MCMV-resistant and -susceptible strains. Figure 2A shows that MCMV grew exponentially from d 1 to d 5 with similar kinetics in MEFs derived from either strain. In addition, comparable levels of infectious virions were released into the MEF supernatants after d 2 and through d 6. Furthermore, we did not observe significant differences in viral replication, plaque size, or viral cytopathic effect in comparison with MA/My MEFs (not shown). In agreement with Chalmer et al. (7), we conclude that H-2-linked genetic differences in innate MCMV immunity are not controlled at the level of MCMV infectivity or replication within host cells.
FIG. 2.

Multistep virus growth curves in H-2b and H2k MEFs. C57L and C57L.M-H2k MEFs were infected with MCMV at an MOI of 0.08 PFU per cell. The virus titers in the supernatants were detected daily by plaque assay on 3T12 cells (A), and quantitative real-time PCR was performed using DNA isolated from the remaining monolayers (B). MCMV shows similar levels of virus productivity in C57L (closed symbols) and C57L.M-H2k (open symbols) MEFs.
NK cell effector functions in M.L-H-2b and L.M-H-2k.
To delineate whether an inherent defect in H-2b NK virus sensing might explain an inability to restrain viral replication, we examined NK IFN-γ shortly after infection, as this cytokine is rapidly induced in NK cells of MCMV-resistant C57BL/6 mice during MCMV infection (26). Spleen cellularity generally corresponded with either the MA/My or the C57L genetic background through the first 39 h of infection (Fig. 3). By 90 h after infection, however, splenocyte numbers dropped precipitously in homozygous H-2b strains and also in M.L-H2b/k littermate controls (Fig. 3A). Thus, splenocyte numbers were not absolutely correlated with high or low virus levels, but in mice without the H-2k haplotype, substantial losses were always noted. Also consistent with previous findings (8, 11, 12), NK cell numbers drop similarly through the first 39 h after infection in resistant and susceptible strains (not depicted). Moreover, similar numbers of spleen NK cells were producing IFN-γ with maximal induction in strains studied by the same time point (Fig. 3B). Notably, NK cells did not produce IFN-γ after injection of heat- or UV-inactivated MCMV into the H-2k or H-2b strains under study (not depicted). After 90 h, spleen NK cell IFN-γ levels were diminished considerably in resistant and susceptible mice and did not correlate with MCMV control traits.
FIG. 3.

Quantification of NK IFN-γ in MCMV-infected MA/My.L-H-2b and C57L.M-H-2k congenic strains. (A) Spleen leukocytes of uninfected (open) and 39-h (gray) and 90-h (black) MCMV-infected mice (strains designated) were compared. (B) Gated CD3− NK1.1+ IFN-γ+ spleen cell levels from uninfected (open) and 39-h (gray) and 90-h (black) MCMV-infected mice are also shown. Shown are average values (two to seven mice/group) compiled from three independent experiments. Error bars indicate standard deviations.
Role of H-2k class I expression in MCMV infection.
H-2-linked control in innate MCMV immunity suggested that MHC class I molecules might have a role in NK cell control. As regulation of Kk and Dk class I proteins by MCMV has not been previously characterized, we also examined their expression on L929 (H-2k) cells and primary (C57L × MA/My)F1 MEFs shortly after virus infection. We utilized a reporter MCMV virus (MCMV-Δm157/EGFP [11]) to directly visualize infected cells by flow cytometry. Although more virus is required to infect L929 (H-2k) than NIH 3T3 cells, flow cytometric analysis revealed that 24% of L929 cells (MOI = 10) were GFP+ MCMV-infected cells by 24 h after infection (not shown). Under these conditions, Kk and Dk proteins on L929 cells were significantly decreased only on infected (GFP+) cells (Fig. 4A). Similar decreases in class I display were observed when L929 cells were infected with the wild-type K181 strain (not depicted). Fine differences in Kk or Dk proteins on MCMV-infected L929 prompted us to further investigate potential gene- or allele-specific viral regulation of MHC class I expression. We measured class I expression on primary (C57L × MA/My)F1 MEFs as an independent means to establish that the diminishment of class I proteins of either haplotype is a general feature of MCMV infection. Due to low constitutive class I levels on the MEFs, IFN-γ induction was utilized to boost expression before MCMV infection. In the same way, we found that MCMV downregulated Kk, Dk, and H-2b class I molecules by ∼90% on F1 MEFs (Fig. 4B). MCMV control of class I expression is specific, since CD71, a common surface protein, was not affected (Fig. 4C). We obtained similar results using Smith strain MCMV (data not shown). H-2b and H-2k class I proteins are therefore efficiently and specifically decreased by MCMV on cells expressing alleles of either haplotype.
FIG. 4.
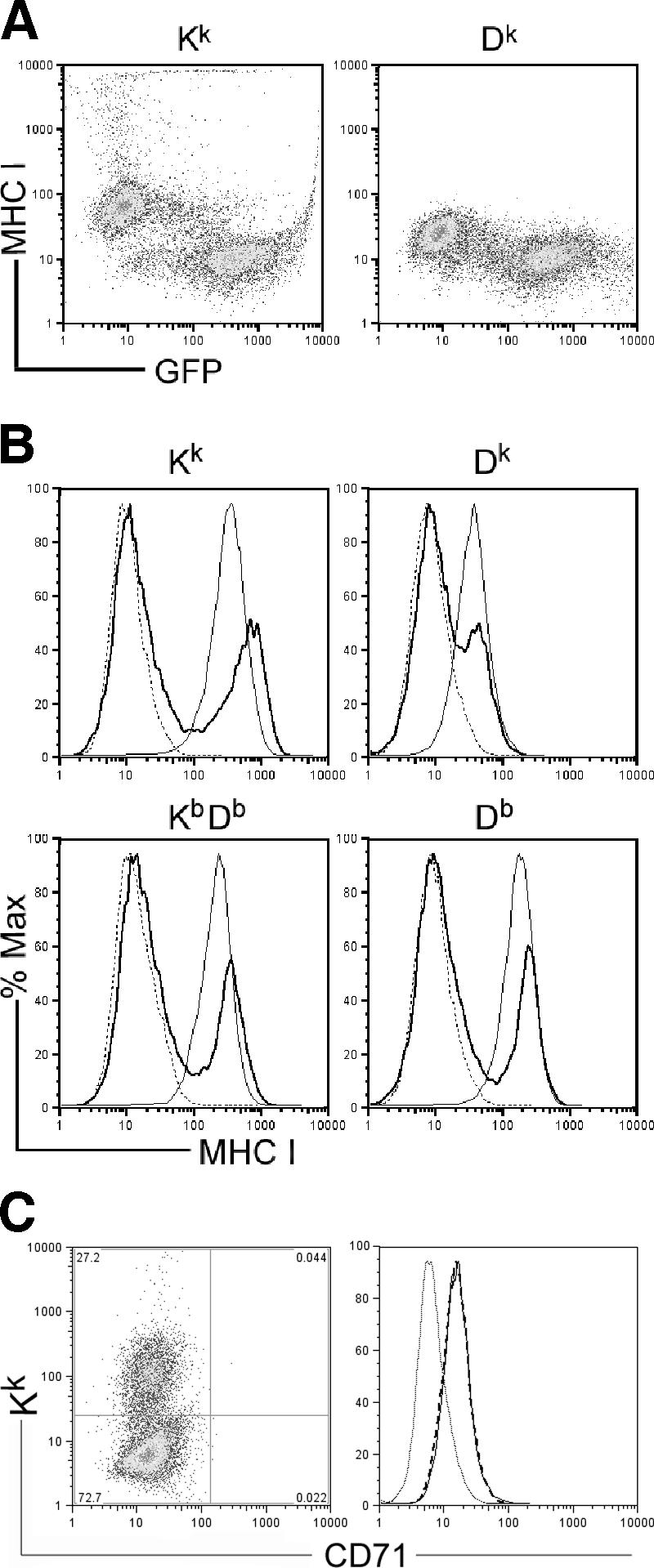
MCMV downregulation of H-2k and H-2b class I proteins on L929 cells and primary (C57L × MA/My)F1 MEFs. (A) Shown is a representative dot plot of H-2k class I protein levels on L929 cells by 24 h after infection with a MCMV-Δm157 reporter virus (MOI = 10). (B) Histograms of MHC class I protein levels on (C57L × MA/My)F1 MEFs (broken line), IFN-γ-induced (20 U, 24 h) (C57L × MA/My)F1 MEFs (thin line), and IFN-γ-induced (C57L × MA/My)F1 MEFs (bold line) infected with GFP-expressing MCMV (MOI = 4) assessed by use of flow cytometry at 12 h postinfection are shown. (C) CD71 cell surface staining on Kk high (thin line) and Kk low (broken line) IFN-γ-induced (C57L × MA/My)F1 MEFs infected with wild-type K181 (MOI = 4). Control Ig staining (thin dotted line) is also shown.
Because type I interferons are indispensable in innate immunity and MCMV resistance, we also studied class I downregulation by MCMV on IFN-β-stimulated F1 MEFs. Though IFN-β directly affected the percentage of cells infected in a dose-dependent manner, presumably through direct inhibition of viral gene expression (24), MCMV again efficiently decreased H-2k and H-2b class I proteins on F1 MEFs (Fig. 5A). Additionally, though uninfected F1 MEFs have similar levels of Kk and Dk display, the amount of Dk class I proteins was consistently less than half that of Kk proteins on infected cells. Similar results were obtained over a large range of IFN-β doses (Fig. 5B). Furthermore, Dk mean fluorescence intensity decreased sharply, whereas Kk mean fluorescence intensity levels remained fairly constant throughout the study, indicating that MCMV infection under certain conditions can selectively regulate MHC class I proteins in a gene-specific manner.
FIG. 5.

Efficient class I Dk protein downregulation in IFN-β-induced primary (C57L × MA/My)F1 MEFs. (A) Histograms of class I proteins on (C57L × MA/My)F1 MEFs (broken line), IFN-β-induced (50 to 1,000 units; 30 h) (C57L × MA/My)F1 MEFs (thin line), and IFN-β-induced and MCMV-infected (MOI = 4) (C57L × MA/My)F1 MEFs (bold line) are shown. (B) Comparison of Kk and Dk protein levels on gated GFP+ MCMV-infected cells at 12 h postinfection. cntrl, control.
DISCUSSION
An H-2k association with host survival following MCMV infection was long ago recognized, though a mechanistic understanding is still elusive. We show that dominant innate virus resistance was converted to profound susceptibility in MA/My.L-H2b congenic mice, since infectious MCMV freely replicates to very high levels in M.L-H2b spleens within days after infection. Thus, MA/My H-2k-linked loci are indeed necessary components of innate viral immunity. This difference cannot be explained by H-2-controlled differences in infectivity or replication, as we observe similar MCMV growth kinetics in MEFs derived from embryos from resistant or susceptible strains. Another intriguing and unexpected finding in the current report is seen in Fig. 1, since H-2k-linked resistance is fully competent in H-2 heterozygous or homozygous mice on the C57L background, whereas a single H-2k haplotype in H-2 heterozygous mice on the MA/My background did not result in full resistance. An additional polymorphic genetic factor(s) in C57L therefore further limits MCMV spread and replication when H-2k is available, perhaps through augmentation of NK sensing, but this factor by itself is not sufficient in innate virus resistance because C57L NK cells fail to thwart a comparable MCMV infection. We further show that proximal H-2 Kk class I and class II genes do not explain the observed genetic variation, since our novel H-2 recombinant strain is fully competent to limit virus replication in the spleen at early times after infection. Thus, a genetic locus distal to the MHC class II gene region and overlapping with Dk class Ia and more-distal nonclassical class Ib genes should contain the responsible gene(s) required in H-2k-linked innate MCMV resistance through NK cells. Because multiple H-2 loci were previously implicated in the control of MCMV-induced mortality (15), we were also prompted to study mortality in the novel strains. Recent preliminary studies with sublethal or lethal doses of MCMV have so far indicated similar mortality rates for our H-2k recombinant strain and littermate control tested as shown in Fig. 1E (X. Xie and M. G. Brown, unpublished data).
To further examine the role of NK cells in innate H-2k control, IFN-γ was quantified for comparison in H-2k and H-2b strains over a 3.5-d time course following infection. Deficient NK control in C57L and M.L-H2b is not due to an intrinsic defect in virus sensing, since active viral replication alerted their NK cells to enhance IFN-γ production to levels similar to those for NK cells in MA/My or L.M-H2k shortly after infection. Similar diminished responses in all strains were also noted by later time points and did not directly correlate with innate virus immunity. Whether this response is due to virus sensing through innate receptors (i.e., TLR3 and TLR9) leading to enhanced cytokine stimulation of NK function (4, 34) or due to direct NK recognition of virus-infected targets is an important question. Recent reports indicate that NK IFN-γ release is severely impaired in mice without TLR9 or MyD88 and that dendritic cell-derived interleukin 18 (IL-18) is also important (1, 9). Here we show similar numbers of NK cells producing IFN-γ and comparable induction levels after MCMV infection; thus, these data suggest that an MCMV → TLR9 → MyD88 → IL-18 axis in dendritic cells and an IL-18 → IFN-γ axis in NK cells during the nonspecific phase of NK activation are fully competent in the strains tested in Fig. 3. Nevertheless, H-2b-linked gene expression fails to support NK control of viral replication on the C57L or MA/My genetic backgrounds.
While both MCMV and HCMV convergently acquired key gene products to manipulate MHC class I display (27), genetic variance in NK control may not be altogether surprising, since these cells readily respond to MHC-compatible targets with deficient class I expression. Because Wagner et al. noted before that class I Kb molecules are somewhat refractory to MCMV control (38), we reasoned that class I gene- or allele-specific regulation by MCMV might explain H-2k protection, at least in part. However, minor variations in class I display might best be observed only under certain circumstances, since Kk (decreased by ∼95%) and Dk (decreased by ∼50%) proteins were differently downregulated on MCMV-infected L929 cells. Dk display, on the other hand, was affected more than Kk by MCMV in IFN-β-induced F1 MEFs, since a substantially greater number of infected (GFP+) cells continued to display Kk proteins even after Dk had seemingly been lost from the cell surface. IFN-β may conceivably have an impact on Kk induction greater than that of other class I proteins. Alternately, MCMV proteins known to bind and downregulate class I display may more avidly bind Dk when class I synthesis is strongly induced by type I interferons. Taken together, our findings suggest that only very minimal levels of Dk ligands that could potentially stimulate Ly49P receptors on NK cells in the course of MCMV infection should be available on infected host cells and that even fewer might be expected to hold a particular relevant virus peptide, as suggested recently (10).
Alternately, MCMV might stabilize some cell surface class I displays, including that of Dk proteins, while at the same time interfering with cytotoxic T-lymphocyte recognition. MCMV gp34 is notable in this regard, since it binds endoplasmic reticulum-resident class I proteins destined for expression at the cell surface. Interestingly, Dk but not Kk surface expression is selectively affected by gp34 in infected L929 cells (Xie and Brown, unpublished data). An intriguing possibility is that Ly49P+ NK cells might specifically recognize gp34-associated Dk proteins. However, innate H-2k resistance is likely more complex, since significant protection has been observed even on the BALB/c background, where Ly49P receptors are not expressed (32). Finally, it is possible that NK recognition and control become fully competent only in mice that express H-2k class I ligands during development. While NK cells in C57L and M.L-H2b mice do recognize viral intrusion and IFN-γ expression is stimulated shortly afterward, these cells could be hyporesponsive (14) or unlicensed (19) in terms of their capacity to attack infected cells. NK receptors in H-2k mice, on the other hand, might recognize viral signatures more efficiently than those in other H-2 haplotypes, in addition to undergoing potential stimulation through Ly49P/Dk interactions during NK encounters with MCMV-infected cells. While inhibitory KIR/MHC interactions also insure functional competency in human NK cells (2) and can regulate antiviral immune responses in infected individuals (13, 18), this model should enhance our understanding of the role of NK cells in the early detection and control of viral pathogens.
Acknowledgments
We thank V. Engelhard for reagents provided.
This work was supported by Public Health Service grant AI50072 from the National Institute of Allergy and Infectious Diseases.
Footnotes
Published ahead of print on 18 October 2006.
REFERENCES
- 1.Andoniou, C. E., S. L. H. van Dommelen, V. Voigt, D. M. Andrews, G. Brizard, C. Asselin-Paturel, T. Delale, K. J. Stacey, G. Trinchieri, and M. A. Degli-Esposti. 2005. Interaction between conventional dendritic cells and natural killer cells is integral to the activation of effective antiviral immunity. Nat. Immunol. 6:1011-1019. [DOI] [PubMed] [Google Scholar]
- 2.Anfossi, N., P. Andre, S. Guia, C. S. Falk, S. Roetynck, C. A. Stewart, V. Breso, C. Frassati, D. Reviron, D. Middleton, et al. 2006. Human NK cell education by inhibitory receptors for MHC class I. Immunity 25:331-342. [DOI] [PubMed] [Google Scholar]
- 3.Bancroft, G. J., G. R. Shellam, and J. E. Chalmer. 1981. Genetic influences on the augmentation of natural killer (NK) cells during murine cytomegalovirus infection: correlation with patterns of resistance. J. Immunol. 126:988-994. [PubMed] [Google Scholar]
- 4.Beutler, B., K. Crozat, J. A. Koziol, and P. Georgel. 2005. Genetic dissection of innate immunity to infection: the mouse cytomegalovirus model. Curr. Opin. Immunol. 17:36-43. [DOI] [PubMed] [Google Scholar]
- 5.Braud, V. M., D. S. Allan, C. A. O'Callaghan, K. Soderstrom, A. D'Andrea, G. S. Ogg, S. Lazetic, N. T. Young, J. I. Bell, J. H. Phillips, L. L. Lanier, and A. J. McMichael. 1998. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature 391:795-799. [DOI] [PubMed] [Google Scholar]
- 6.Brown, M. G., A. O. Dokun, J. W. Heusel, H. R. C. Smith, D. L. Beckman, E. A. Blattenberger, C. E. Dubbelde, L. R. Stone, A. A. Scalzo, and W. M. Yokoyama. 2001. Vital involvement of a natural killer cell activation receptor in resistance to viral infection. Science 292:934-937. [DOI] [PubMed] [Google Scholar]
- 7.Chalmer, J. E., J. S. Mackenzie, and N. F. Stanley. 1977. Resistance to murine cytomegalovirus linked to the major histocompatibility complex of the mouse. J. Gen. Virol. 37:107-114. [DOI] [PubMed] [Google Scholar]
- 8.Daniels, K. A., G. Devora, W. C. Lai, C. L. O'Donnell, M. Bennett, and R. M. Welsh. 2001. Murine cytomegalovirus is regulated by a discrete subset of natural killer cells reactive with monoclonal antibody to Ly49H. J. Exp. Med. 194:29-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Delale, T., A. Paquin, C. Asselin-Paturel, M. Dalod, G. Brizard, E. E. M. Bates, P. Kastner, S. Chan, S. Akira, A. Vicari, C. A. Biron, G. Trinchieri, and F. Briere. 2005. MyD88-dependent and -independent murine cytomegalovirus sensing for IFN-α release and initiation of immune responses in vivo. J. Immunol. 175:6723-6732. [DOI] [PubMed] [Google Scholar]
- 10.Desrosiers, M.-P., A. Kielczewska, J.-C. Loredo-Osti, S. G. Adam, A. P. Makrigiannis, S. Lemieux, T. Pham, M. Lodoen, K. Morgan, L. L. Lanier, and S. M. Vidal. 2005. Epistasis between mouse Klra and major histocompatibility complex class I loci is associated with a new mechanism of natural killer cell-mediated innate resistance to cytomegalovirus infection. Nat. Genet. 37:593-599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dighe, A., M. Rodriguez, P. Sabastian, X. Xie, M. A. McVoy, and M. G. Brown. 2005. Requisite H2k role in NK cell-mediated resistance in acute murine CMV infected MA/My mice. J. Immunol. 175:6820-6828. [DOI] [PubMed] [Google Scholar]
- 12.Dokun, A. O., S. Kim, H. R. Smith, H. S. Kang, D. T. Chu, and W. M. Yokoyama. 2001. Specific and nonspecific NK cell activation during virus infection. Nat. Immunol. 2:951-956. [DOI] [PubMed] [Google Scholar]
- 13.Fauci, A. S., D. Mavilio, and S. Kottilil. 2005. NK cell in HIV infection: paradigm for protection or targets for ambush. Nat. Rev. Immunol. 5:835-843. [DOI] [PubMed] [Google Scholar]
- 14.Fernandez, N. C., E. Treiner, R. E. Vance, A. M. Jamieson, S. Lemieux, and D. H. Raulet. 2005. A subset of natural killer cells achieves self-tolerance without expressing inhibitory receptors specific for self-MHC molecules. Blood 105:4416-4423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grundy, J. E., J. S. Mackenzie, and N. F. Stanley. 1981. Influence of H-2 and non-H-2 genes on resistance to murine cytomegalovirus infection. Infect. Immun. 32:277-286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harnett, G. B., and G. Shellam. 1982. Variation in cytomegalovirus replication in fibroblasts from different mouse strains in vitro: correlation with in vivo resistance. J. Gen. Virol. 62:39-47. [DOI] [PubMed] [Google Scholar]
- 17.Kavanagh, D. G., M. C. Gold, M. Wagner, U. H. Koszinowski, and A. B. Hill. 2001. The multiple immune-evasion genes of murine cytomegalovirus are not redundant: m4 and m152 inhibit antigen presentation in a complementary and cooperative fashion. J. Exp. Med. 194:967-977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khakoo, S. I., C. L. Thio, M. P. Martin, C. R. Brooks, X. Gao, J. Astemborski, J. Cheng, J. J. Goedert, D. Vlahov, M. Hilgartner, S. Cox, A. Little, G. J. Alexander, M. E. Cramp, S. J. O'Brien, W. M. C. Rosenberg, D. L. Thomas, and M. Carrington. 2004. HLA and NK cell inhibitory receptor genes in resolving hepatitis C virus infection. Science 305:872-874. [DOI] [PubMed] [Google Scholar]
- 19.Kim, S., J. Poursine-Laurent, S. M. Truscott, L. Lybarger, Y. Song, L. Yang, A. R. French, J. B. Sunwoo, S. Lemieux, T. H. Hansen, and W. M. Yokoyama. 2005. Licensing of natural killer cells by host major histocompatibility complex class I molecules. Nature 436:709-713. [DOI] [PubMed] [Google Scholar]
- 20.Kleijnen, M. F., J. B. Huppa, P. Lucin, S. Mukherjee, H. Farrell, A. E. Campbell, U. H. Koszinowski, A. B. Hill, and H. L. Ploegh. 1997. A mouse cytomegalovirus glycoprotein, gp34, forms a complex with folded class I MHC molecules in the ER which is not retained but is transported to the cell surface. EMBO J. 16:685-694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reference deleted.
- 22.Lanier, L. L. 2005. NK cell recognition. Annu. Rev. Immunol. 23:225-274. [DOI] [PubMed] [Google Scholar]
- 23.Reference deleted.
- 24.Martinotti, M. G., G. Gribaudo, M. Gariglio, A. Angeretti, G. Cavallo, and S. Landolfo. 1992. Effects of interferon alpha on murine cytomegalovirus replication. Microbiologica 15:183-186. [PubMed] [Google Scholar]
- 25.Orange, J. S. 2002. Human natural killer cell deficiencies and susceptibility to infection. Microbes Infect. 4:1545-1558. [DOI] [PubMed] [Google Scholar]
- 26.Orange, J. S., B. Wang, C. Terhorst, and C. A. Biron. 1995. Requirement for natural killer cell-produced interferon gamma in defense against murine cytomegalovirus infection and enhancement of this defense pathway by interleukin 12 administration. J. Exp. Med. 182:1045-1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reddehase, M. J. 2002. Antigens and immunoevasins: opponents in cytomegalovirus immune surveillance. Nat. Rev. Immunol. 2:831-844. [DOI] [PubMed] [Google Scholar]
- 28.Reusch, U., W. Muranyi, P. Lucin, H. G. Burgert, H. Hengel, and U. H. Koszinowski. 1999. A cytomegalovirus glycoprotein re-routes MHC class I complexes to lysosomes for degradation. EMBO J. 18:1081-1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rodriguez, M., P. Sabastian, P. Clark, and M. G. Brown. 2004. Cmv1-independent antiviral role of NK cells revealed in murine cytomegalovirus infected New Zealand White mice. J. Immunol. 173:6312-6318. [DOI] [PubMed] [Google Scholar]
- 30.Scalzo, A. A., M. G. Brown, D. T. Chu, J. W. Heusel, W. M. Yokoyama, and C. A. Forbes. 1999. Development of intra-natural killer complex (NKC) recombinant and congenic mouse strains for mapping and functional analysis of NK cell regulatory loci. Immunogenetics 49:238-241. [DOI] [PubMed] [Google Scholar]
- 31.Scalzo, A. A., H. E. Farrell, and G. Karupiah. 2000. Techniques for studying murine natural killer cells in defense against viral infection, p. 163-177. In K. S. Campbell, and M. Colonna (ed.), Natural killer cell protocols: cellular and molecular methods, vol. 121. Humana Press Inc., Totowa, NJ. [DOI] [PubMed] [Google Scholar]
- 32.Scalzo, A. A., P. A. Lyons, N. A. Fitzgerald, C. A. Forbes, W. M. Yokoyama, and G. R. Shellam. 1995. Genetic mapping of Cmv1 in the region of mouse chromosome 6 encoding the NK gene complex-associated loci Ly49 and musNKR-P1. Genomics 27:435-441. [DOI] [PubMed] [Google Scholar]
- 33.Scalzo, A. A., R. Wheat, C. Dubbelde, L. R. Stone, P. Clark, C. A. Forbes, Y. Du, N. Dong, J. Stoll, W. M. Yokoyama, and M. G. Brown. 2003. Molecular genetic characterization of the distal NKC recombination hotspot and putative murine CMV resistance control locus. Immunogenetics 55:370-378. [DOI] [PubMed] [Google Scholar]
- 34.Tabeta, K., P. Georgel, E. Janssen, X. Du, K. Hoebe, K. Crozat, S. Mudd, L. Shamel, S. Sovath, J. Goode, L. Alexopoulou, R. A. Flavell, and B. Beutler. 2004. Toll-like receptors 9 and 3 as essential components of innate immune defense against mouse cytomegalovirus infection. Proc. Natl. Acad. Sci. USA 101:3516-3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tomasec, P., V. M. Braud, C. Rickards, M. B. Powell, B. P. McSharry, S. Gadola, V. Cerundolo, L. K. Borysiewicz, A. J. McMichael, and G. W. Wilkinson. 2000. Surface expression of HLA-E, an inhibitor of natural killer cells, enhanced by human cytomegalovirus gpUL40. Science 287:1031-1033. [DOI] [PubMed] [Google Scholar]
- 36.Tomasec, P., E. C. Y. Wang, A. J. Davison, B. Vojtesek, M. Armstrong, C. Griffin, B. P. McSharry, R. J. Morris, S. Llewellyn-Lacey, C. Rickards, A. Nomoto, C. Sinzger, and G. W. Wilkinson. 2005. Downregulation of natural killer cell-activating ligand CD155 by human cytomegalovirus UL141. Nat. Immunol. 6:181-188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ulbrecht, M., S. Martinozzi, M. Grzeschik, H. Hengel, J. W. Ellwart, M. Pla, and E. H. Weiss. 2000. Cutting edge: the human cytomegalovirus UL40 gene product contains a ligand for HLA-E and prevents NK cell-mediated lysis. J. Immunol. 164:5019-5022. [DOI] [PubMed] [Google Scholar]
- 38.Wagner, M., A. Gutermann, J. Podlech, M. J. Reddehase, and U. H. Koszinowski. 2002. Major histocompatibility complex class I allele-specific cooperative and competitive interactions between immune evasion proteins of cytomegalovirus. J. Exp. Med. 196:805-816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang, E. C. Y., B. McSharry, C. Retiere, P. Tomasec, S. Williams, L. K. Borysiewicz, V. M. Braud, and G. W. G. Wilkinson. 2002. UL40-mediated NK evasion during productive infection with human cytomegalovirus. Proc. Natl. Acad. Sci. USA 99:7570-7575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wheat, R. L., P. Y. Clark, and M. G. Brown. 2003. Quantitative measurement of infectious murine cytomegalovirus genomes in real-time PCR. J. Virol. Methods 112:107-113. [DOI] [PubMed] [Google Scholar]
- 41.Wills, M. R., O. Ashiru, M. B. Reeves, G. Okecha, J. Trowsdale, P. Tomasec, G. W. G. Wilkinson, J. Sinclair, and J. G. P. Sissons. 2005. Human cytomegalovirus encodes an MHC class I-like molecule (UL142) that functions to inhibit NK cell lysis. J. Immunol. 175:7457-7465. [DOI] [PubMed] [Google Scholar]
- 42.Ziegler, H., R. Thale, P. Lucin, W. Muranyi, T. Flohr, H. Hengel, H. Farrell, W. Rawlinson, and U. H. Koszinowski. 1997. A mouse cytomegalovirus glycoprotein retains MHC class I complexes in the ERGIC/cis-Golgi compartments. Immunity 6:57-66. [DOI] [PubMed] [Google Scholar]