

Abstract
Flux distribution in central metabolic pathways of Desulfovibrio vulgaris Hildenborough was examined using 13C tracer experiments. Consistent with the current genome annotation and independent evidence from enzyme activity assays, the isotopomer results from both gas chromatography-mass spectrometry (GC-MS) and Fourier transform-ion cyclotron resonance mass spectrometry (FT-ICR MS) indicate the lack of an oxidatively functional tricarboxylic acid (TCA) cycle and an incomplete pentose phosphate pathway. Results from this study suggest that fluxes through both pathways are limited to biosynthesis. The data also indicate that >80% of the lactate was converted to acetate and that the reactions involved are the primary route of energy production [NAD(P)H and ATP production]. Independently of the TCA cycle, direct cleavage of acetyl coenzyme A to CO and 5,10-methyl tetrahydrofuran also leads to production of NADH and ATP. Although the genome annotation implicates a ferredoxin-dependent oxoglutarate synthase, isotopic evidence does not support flux through this reaction in either the oxidative or the reductive mode; therefore, the TCA cycle is incomplete. FT-ICR MS was used to locate the labeled carbon distribution in aspartate and glutamate and confirmed the presence of an atypical enzyme for citrate formation suggested in previous reports [the citrate synthesized by this enzyme is the isotopic antipode of the citrate synthesized by the (S)-citrate synthase]. These findings enable a better understanding of the relation between genome annotation and actual metabolic pathways in D. vulgaris and also demonstrate that FT-ICR MS is a powerful tool for isotopomer analysis, overcoming the problems with both GC-MS and nuclear magnetic resonance spectroscopy.
Sulfate-reducing bacteria (SRB) such as Desulfovibrio vulgaris Hildenborough are ubiquitous in nature and play an important role in global sulfur cycling and the mineralization of organic matter (21, 38, 39). The uncontrolled growth of D. vulgaris contributes to the biocorrosion of oil and gas pipelines and the souring of production wells (18, 37, 53). Conversely, the ability of D. vulgaris to reduce heavy metals and radionuclides to insoluble forms provides a unique microbe-oriented solution for bioremediation (25, 27, 28). In addition to its environmental importance, D. vulgaris has a unique energy metabolism that has the potential to be used for hydrogen or methane production in either pure or mixed cultures (7, 13, 38, 49).
The availability of an annotated genome sequence for D. vulgaris (25) makes it an ideal organism for investigating SRB physiology, and several functional genomics studies have described the transcriptome and proteome of this organism (10, 24, 35). Information from these analyses is critical for validating genome annotation and predictions for operons and regulons. Moreover, D. vulgaris metabolism has been studied for several decades, but the recently published annotated genomic sequence of D. vulgaris contained the following unresolved predictions related to key pathways (25). (i) Although the tricarboxylic acid (TCA) cycle lacks a typical 2-oxoglutarate dehydrogenase, a ferredoxin-dependent 2-oxoglutarate synthase (9) homolog (EC 1.2.7.3, 2-oxoglutarate↔succinyl coenzyme A) has been annotated for this step. (ii) While the annotation predicts pathways for respiration using sulfate and other terminal electron acceptors, it remains to be determined whether the TCA cycle functions oxidatively (via the ferredoxin-dependent 2-oxoglutarate synthase) or only reductively. (iii) Although citrate synthases have been reported for other deltaproteobacteria (6), neither D. vulgaris nor the closely related Desulfovibrio desulfuricans G20 contains a citrate synthase homolog in the annotated genome. However, these organisms are not auxotrophic for amino acids typically derived from citrate, and previous experiments have suggested the presence of an atypical enzyme that enables the production of citrate in Desulfovibrio spp. (19, 20, 34).
Metabolic flux analysis is an ideal method for linking genome annotation to cellular phenotypes (14), and isotopomer analysis is the in vivo method of choice for examination of cellular metabolic pathways (44). Analysis of isotopomer distributions in metabolites (often amino acids) requires advanced techniques such as nuclear magnetic resonance (NMR) spectroscopy or mass spectrometry (MS). Although NMR spectroscopy can be used to determine the location of the 13C label in individual isotopomers, not all isotopomers can be detected using this technique, since carbon atoms separated by more than one bond do not influence each other's resonance sufficiently (42). Further, labeled carbon sources that result in metabolites with the isotopic label solely on a carboxyl carbon are difficult to address using common 13C NMR techniques. Additionally, though NMR-based techniques are nondestructive, their sensitivity is low, necessitating a large amount of costly labeled culture. Among mass spectroscopic techniques, gas chromatography coupled to MS detection (GC-MS) is typically the technology of choice, since it requires much less sample, and isotopomer analysis software tools enable quick identification of the isotopomer pattern. By examining different mass fragments, one can determine certain labeled positions, such as the label on an α-carboxyl group. But GC-MS alone cannot locate all labeled positions in amino acids or organic acids.
In contrast to GC-MS and NMR spectroscopy, Fourier transform-ion cyclotron resonance MS (FT-ICR MS) provides, with direct injection (i.e., without chromatographic separation of the sample), an accurate mass determination of many of the metabolites in complex mixtures (8, 32). Furthermore, electrospray ionization (ESI) is amenable to polar compounds without the need for derivatization, eliminating the need to correct for isotope distributions in the derivatizing agent. An additional advantage of FT-ICR MS is its ability to detect metabolites in the low nanomolar range. As a trapping technique, FT-ICR MS performs multiple stages of mass spectrometric events (MSn) utilizing collision-induced dissociation (CID). Detection of mass/charge ratios as low as 30 can be achieved in the ICR cell (32), and mass spectrometric fragmentation patterns of amino acids are very well understood (4, 22, 23, 29, 51). This study outlines the utilization of ESI FT-ICR MS to localize the position of 13C atoms in metabolites of interest and represents an important complementary technique to GC-MS for pathway annotation and metabolic flux analysis of D. vulgaris.
MATERIALS AND METHODS
Bacterial growth and maintenance.
Desulfovibrio vulgaris Hildenborough (ATCC 29579) was obtained from the American Type Culture Collection (Manassas, VA). All experiments used defined lactate sulfate medium, LS4D (35), with the modification that titanium citrate was not used. The inoculum for all experiments was started from fresh frozen stock (unlabeled culture). Isotopic labeling experiments were run in triplicate using LS4D medium containing 99% l-[1-13C]lactate (Cambridge Isotope) with a 1:10 inoculation volume. A culture in the exponential-growth phase (optical density at 600 nm [OD600], 0.4) was used as the inoculum for subcultures. To remove the effects of unlabeled lactate and glycerol from the initial stock culture, two sequential 10% subcultures into a labeled medium were performed to obtain the final culture. All incubations were performed at 30°C in an anaerobic chamber (Coy Laboratory Products Inc., Grass Lake, MI) under an atmosphere of 5% CO2, 5% H2, and 90% N2.
Analysis of extracellular metabolites and biomass composition.
Cell growth was monitored by measuring both OD600 and total protein concentration using the Bradford protein assay (catalog no. 500-0006; Bio-Rad Laboratories, Hercules, CA). The concentrations of pyruvate (PYR), succinate, lactate, and acetate in the medium were measured using enzymatic kits (r-Biopharm, Darmstadt, Germany). To measure biomass weight, 50 ml of cells was centrifuged at 4,800 × g and 4°C for 20 min, the cell pellet was dried in a lyophilizer (catalog no. 7420020; Labconco, Kansas City, MO) for 24 h, and the dry weight was determined by gravimetry. Fatty acids in the dried biomass were quantified, using previously described methods (47), by Microbial ID (Newark, Delaware). Amino acids in the dried biomass were quantified, using the Beckman 6300 amino acid analyzer (Beckman Coulter, CA), by the Molecular Structure Facility at the University of California, Davis. The sulfate concentration was determined by reaction with barium chloride and measurement of absorbance at a wavelength of 450 nm (13). All measurement methods for biomass constituents (carbohydrates, RNA, and DNA) were conducted using previous reported protocols (15, 31). Briefly, carbohydrate was quantified using the phenol reaction, RNA was assayed using the orcinol reaction method, and DNA was measured using a colorimetric procedure that involves the reaction of DNA with diphenylamine in a mixture of perchloric acid. Glucose, pure Escherichia coli RNA (catalog no. 7940; Ambion, Austin, TX), and deoxyribose were used as standards for the carbohydrate, RNA, and DNA measurements, respectively.
GC-MS procedure.
In order to measure amino acid labeling patterns in cellular protein, the biomass was harvested by centrifugation at 10,000 × g and 4°C for 20 min and then lysed via sonication in deionized water. Sonication was conducted using a microtip for 3 min with pulses of 3 s on and 1 s off. The protein from the resulting lysate was precipitated using trichloroacetic acid and then hydrolyzed in 6 M HCl at 100°C for 24 h. In the resulting amino acid mixture, cysteine and tryptophan were lost due to oxidation, and glutamine and asparagine were deaminated. GC-MS samples were prepared in 100 μl of tetrahydrofuran (THF) and 100 μl of N-(tert-butyldimethylsilyl)-N-methyl-trifluoroacetamide (Sigma-Aldrich, St. Louis, MO). All samples were derivatized in a water bath at 65 to 80°C for 1 h, producing tert-butyldimethlysilyl derivatives. One microliter of the derivatized sample was injected into an Agilent (Wilmington, DE) 6890 gas chromatograph equipped with a DB5-MS column (J&W Scientific, Folsom, CA) and analyzed using an Agilent 5973 mass spectrometer. The GC operation conditions were as follows: the GC column was held at 150°C for 2 min, heated at 3°C per min to 280°C, heated at 20°C per min to 300°C, and held for 5 min at that temperature.
To prepare GC-MS samples using bis(trimethylsilyl)trifluoroacetamide (BSTFA) as the derivatization reagent (measure lactate, pyruvate, and succinate labeling pattern), 0.5 ml of supernatant was frozen in liquid nitrogen and then lyophilized overnight. The dried samples were prederivatized with a solution (0.3 ml) of 2% hydroxylamine hydrochloride (Fluka, Milwaukee, WI) in pyridine (Sigma-Aldrich, St. Louis, MO) overnight at room temperature. Following this, each sample was derivatized at room temperature for 25 min using 0.5 ml BSTFA (Sigma-Aldrich) before measurement by GC-MS. This derivatization added trimethylsilyl groups to carboxyls and converted oxoacids (e.g., pyruvate) to oximes for greater MS suitability. Decane (Aldrich Chemical) was used as an internal standard. One microliter of the derivatized sample was injected into the GC-MS column. The column was held at 60°C for 1 min after injection and then heated 20°C/min to 130°C, 4°C/min to 150°C, and finally 40°C/min to 260°C, where it was held for 3 min. Helium carrier gas was used at a column flow rate of 1.2 ml/min with a 1:20 split ratio at injection.
FT-ICR spectrometry.
For ESI FT-ICR MS analysis, the supernatant or the dried hydrolyzed mixture sample was prepared in 1 ml of a methanol-H2O (1/1) mixture plus 1% formic acid. The resulting solution was amenable to ionization with an electrospray source. The analysis was performed on an Apex III FT-ICR MS (Bruker Daltonics, Billerica, MA) equipped with a 9.4 T actively shielded magnet. Ions were generated using the Apollo I ESI source in positive-ion mode at a flow rate of 120 μl/min, nebulizing gas pressure of 40 lb/in2, and a dry gas temperature of 175°C, with source voltages of −4.4 kV on the atmospheric side of the capillary, −4.0 kV on the end cap shield, and −2.0 kV on the cylinder shield. After this stage, ions were accumulated for 2 s in an external hexapole ion guide and transferred to the cell with a background pressure of ∼5 × 10−9 Pa for detection. The operating software was XMASS version 6.0 (Bruker Daltonics, USA). Each spectrum was composed of 32 scans. Multiple-stage mass spectrometry experiments were carried out by isolating the ions of interest and activating them by sustained off-resonance irradiation (SORI)-CID in the ICR cell (29).
Annotated pathway map and algorithm for flux calculation.
The key biochemical pathways included in the D. vulgaris model were glycolysis, the TCA cycle, and the pentose phosphate (PP) pathway (1) (Fig. 1). Each reaction and its corresponding gene are listed in Table S1 in the supplemental material. Extracellular concentrations of pyruvate, acetate, and succinate were measured directly using enzymatic methods. The remaining unknown fluxes were determined based on network stoichiometries and isotopomer data (44). The fluxes through the pool of amino acids, carbohydrate, and RNA/DNA are dependent on biomass production and the measured average biomass composition (see Table S2 in the supplemental material) (44).
FIG. 1.

Flux distribution in D. vulgaris. Fluxes were estimated from measurements taken at mid-log phase (30 to 40 h) and normalized by the average lactate uptake rate in log phase (13.8 ± 1.8 mmol h−1 g [dry weight]−1). Solid arrows indicate reactions that are active. Dashed arrows indicate that the reaction has zero flux based on enzyme and isotopomer data. An “x” indicates the absence of an annotated gene for the step. Data in brackets are the exchange coefficients of reactions that are significantly reversible based on isotopomer model analysis. Based on the flux distribution, partial oxidation of 1 mol of lactate produces ∼2.0 mol of NADH/NADPH and ∼0.9 mol of ATP; sulfate reduction (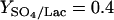 ) (mol/mol) consumes 1.6 mol of NADH and uses 0.8 mol of ATP per mol of lactate to activate sulfate groups.
) (mol/mol) consumes 1.6 mol of NADH and uses 0.8 mol of ATP per mol of lactate to activate sulfate groups.
The reversible reactions are characterized by their net flux, vi, and their exchange flux, viexch. The net flux is defined as the difference between forward and backward fluxes, (vi→ − vi←). The exchange flux, viexch, is the smaller of the forward and backward fluxes, min(vi→, vi←), and is used to calculate the exchange coefficient, exchi, according to reference 56:
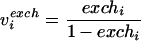 |
(1) |
Exchange coefficient values for key reactions were searched globally in the range [0 1]. All reactions could potentially be reversible and make the system highly underdetermined (44). To simplify model calculation, reactions in the PP pathway were considered to be unidirectional because they are used only for biosynthesis; the reversibility of these reactions had little impact on the isotopomer distribution. Additionally, the isotopomer data were not sensitive to the reversibility of some reactions (e.g., PYR→malate and PYR→oxaloacetate, and thus, they were also not considered to be reversible (2, 57).
By using the concept of atomic mapping matrices (see Table S3 in the supplemental material) (41, 42), the steady-state isotopomer distributions in the intracellular metabolite pools were obtained (MATLAB 6.0; The Mathworks); these isotopomer distributions were used to simulate MS data (m/z = M0, M1, M2, etc.). The optimal solution was found based on an objective function defined as
 |
(2) |
where vn is the unknown fluxes to be optimized in the program, Mi,m is the measured MS data, and Mi,c is the corresponding model-simulated MS data. The flux estimations can be calculated using the simulated annealing concept for all unknown fluxes to achieve a minimal ɛ (2). In order to verify whether the TCA cycle functions oxidatively or is incomplete, two model programs were constructed, one for a complete TCA cycle and one for an incomplete TCA cycle. By comparing the simulation results from two independent programs, only the model with the correct assumption for the TCA cycle resulted in final predictions consistent with all the measurement data and thus clarified the actual operation of the TCA cycle reactions in D. vulgaris. The MATLAB programs for calculation of flux and exchange coefficients can be obtained at http://vimss.lbl.gov/DvHFlux/.
RESULTS
D. vulgaris growth kinetics in LS4D medium.
D. vulgaris was grown in LS4D medium with l-[1-13C]lactate as the sole carbon and energy source; acetate was the main product (Table 1; see also Fig. S1 and S2 in the supplemental material). When grown in medium containing isotopically labeled lactate, the D. vulgaris doubling time was 8 to 9 h (see Fig. S1 in the supplemental material), with a mid-log phase density of ∼4 × 108 cells/ml (OD600, 0.35), and the corresponding biomass weight was 132 ± 22 mg/liter. The final cell density was ∼ 109 cells/ml (OD600, 0.7) after 45 h. The elemental composition of D. vulgaris was reported to be CH1.64N0.23O0.33S0.01P0.014 (50). The weight fractions of biomass components were determined to be 0.39 ± 0.04 protein, 0.14 ± 0.03 RNA/DNA, 0.19 ± 0.05 carbohydrate, 0.16 ± 0.02 ash, 0.05 ± 0.01 fatty acids, and 0.07 ± 0.03 other. Due to precipitation of metals by D. vulgaris during the experimental growth period, the total protein concentration in the culture reflected the total biomass more accurately than the OD600 (see Fig. S1 in the supplemental material). The growth kinetics could be described using a typical Monod model, and the fitted model parameters (using nonlinear least-square fitting) (see Fig. S1 and S2 in the supplemental material) in this experiment were compared with reported values (Table 1) as follows:
 |
(3) |
where μmax is the maximum specific growth rate, CL is the lactate concentration, and KS is the Monod saturation constant for lactate (5).
TABLE 1.
Kinetic parameters for growth of Desulfovibrio vulgaris Hildenborough in LS4D lactate medium
| Source | YX/C (g/mol) |
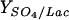 (mol/mol) (mol/mol) |
t1/2 (h) | μmax (h−1) | Ks (mM) | Products (mol/mol lactate) |
|---|---|---|---|---|---|---|
| This study | 6.1 ± 0.8 | 0.40 ± 0.05 | 8.2 ± 2.0 | 0.10 ± 0.01 | 27 ± 2 | Acetate (0.84 ± 0.03), alcohol (<0.002), pyruvate (∼0.02), succinate (∼0.01) |
| Previous worka,b | 5.3-6.8a,b | 0.4-0.57b | 2.7-4.3a | 0.22-0.27a,b | 29b | Acetate (0.97),a ethanol (∼0.02),a H2 (up to 0.5) |
Growth kinetics (Table 1) showed that the minimal doubling time was 8.2 h, the fitted specific growth rate was approximately 0.10 h−1, and the Monod constant (Ks) was 27 mM. An overall gross biomass yield (Yx/c) for lactate was 6.1 g/mol lactate. Complete oxidization of 1 mol of lactate requires consumption of 12 mol e−, i.e., 1.5 mol sulfate/mol of lactate, but the sulfate/lactate yield coefficient in this study was only 0.4. Trace amounts of pyruvate and succinate were detected, but neither formate nor ethanol accumulated in the medium. Compared to results from D. vulgaris growth experiments in lactate medium with amino acid supplements (50), the minimal medium used in this study resulted in lower acetate production (only 84% of the lactate was partially oxidized to acetate rather than the 97% found in amino acid-supplemented medium). It was assumed that the lactate not converted to acetate was assimilated into biomass or was completely oxidized.
Mass spectrometry techniques for profiling isotopomer distribution pattern.
In order to investigate anaerobic pathways and fluxes under steady-state growth conditions, cells were grown in batch cultures and harvested in the exponential growth phase from batch cultures (a quasi-steady state); additionally, this approach is less expensive than continuous culture methods (16, 40, 43). Two types of positively charged species were clearly observed by GC-MS in this study: unfragmented molecules (M-57) and fragmented species that had lost one carboxyl group (M-159) (see Fig. S3 in the supplemental material). For amino acids containing two carboxylic groups, namely, aspartic acid and glutamic acid, the loss of the α-carboxyl is preferred due to the α-cleavage initiated by the radical site on the nitrogen atom of the amino group (33). The two fragmented molecules (M-57 and M-159) were used to determine if the α-carboxyl group was labeled. The natural abundance of heavy isotopes common in organic molecules as well as the derivatization agents, including 13C (1.13%), 18O (0.20%), 29Si (4.70%), and 30Si (3.09%), complicates the resulting mass isotopomer spectrum. The effects of these isotopes on mass fragment distributions of key metabolites were corrected using published algorithms before the data were used to calculate the lactate-derived 13C label distribution (26, 54). The correction program (Steve Van Dien, Genomatica Company, San Diego, CA) can be found at http://vimss.lbl.gov/DvHFlux/. Table 2 lists isotopomer distributions for nine targeted amino acids as well as lactate and succinate. Complete information for the isotopomer distribution of each amino acid and the standard errors from GC-MS measurement is provided in Table S4 in the supplemental material.
TABLE 2.
Measured and model-predicted fragment mass distributions for 13C-labeled metabolites from D. vulgaris hydrolysates or supernatanta
| Amino acid (precursor[s]b) | Fragment | Measured (model-predicted) mass distribution for fragment:
|
13C-enriched position | |||
|---|---|---|---|---|---|---|
| M0 | M1 | M2 | M3 | |||
| Glycine (PEP) | No loss | 0.17 (0.15) | 0.82 (0.82) | 0.01 (0.02) | Carboxyl group | |
| Serine (PEP) | No loss | 0.17 (0.15) | 0.81 (0.82) | 0.02 (0.02) | Carboxyl group | |
| Loss of COOH | 0.98 (0.97) | 0.02 (0.03) | 0 (0) | |||
| Alanine (pyruvate) | No loss | 0.17 (0.16) | 0.81 (0.82) | 0.02 (0.03) | Carboxyl group | |
| Loss of COOH | 0.97 (0.96) | 0.03 (0.04) | 0.0 (0) | |||
| Leucinec (pyruvate and acetyl-CoA) | No loss | 0.75 (0.89) | 0.23 (0.10) | 0.02 (0.01) | Carboxyl group | |
| Loss of COOH | 0.95 (0.94) | 0.03 (0.02) | 0.01 (0.01) | |||
| Glutamate (OXO) | No loss | 0.21 (0.20) | 0.73 (0.74) | 0.06 (0.06) | C-5 carboxyl group | |
| Loss of COOH | 0.22 (0.21) | 0.77 (0.76) | 0.01 (0.02) | |||
| Asparate (OAA) | No loss | 0.04 (0.03) | 0.29 (0.30) | 0.66 (0.65) | 0.01 (0.02) | C-1 or C-4 carboxyl group |
| Loss of COOH | 0.20 (0.21) | 0.79 (0.76) | 0.01 (0.02) | |||
| Methionine (OAA) | No loss | 0.04 (0.03) | 0.27 (0.30) | 0.65 (0.65) | 0.02 (0.02) | Carboxyl group |
| Loss of COOH | 0.20 (0.21) | 0.75 (0.76) | 0.03 (0.02) | 0.02 (0.0) | ||
| Histidine (C5P) | No loss | 0.16 (0.12) | 0.62 (0.64) | 0.11 (0.12) | 0.11 (0.12) | The carboxyl group is not enriched |
| Loss of COOH | 0.17 (0.12) | 0.62 (0.64) | 0.11 (0.12) | 0.12 (0.12) | ||
| Phenylalanine (PEP + E4P) | No loss | 0.03 (0.0) | 0.12 (0.06) | 0.35 (0.31) | 0.50 (0.57) | Carboxyl group |
| Loss of COOH | 0.04 (0.02) | 0.32 (0.25) | 0.60 (0.65) | 0.03 (0.05) | ||
| Lactate | No loss | 0.08 (0.10) | 0.91 (0.89) | 0.02 (0.01) | Carboxyl group | |
| Loss of COOH | 0.96 (0.96) | 0.03 (0.04) | 0.01 (0.0) | |||
| Succinate | No loss | 0.07 (0.03) | 0.26 (0.30) | 0.67 (0.65) | 0 (0.01) | C-1 and/or C-4 carboxyl group |
| Predicted CO2 | (0.22) | (0.78) | ||||
13C-labeled biomass was sampled in the mid-log phase in LS4D medium (n = 4). The complete data for isotopomer distributions and standard deviations for all amino acids are given in Table S4 in the supplemental material.
PEP, phosphoenolpyruvate; OXO, 2-oxoglutarate; OAA, oxaloacetate; E4P, erythrose-4-phosphate.
FT-ICR MS cannot distinguish the mass distributions between leucine and isoleucine, because both molecules have the same mass molecular weight. GC-MS cannot measure the ion fragment (M-47)+ (no loss) accurately because of the overlay of mass peaks by (f302)+ (fragment with only the α and β carbons of leucine).
Profiling a complex mixture of metabolites with MS typically requires the use of chromatographic separation prior to detection. With the high-precision mass determination provided by FT-ICR MS, all the amino acids, corresponding isotopomers, and other components of the sample could be identified without separation. With external calibration the FT-ICR MS gave, in some cases, mass errors of <1 ppm (e.g., [13C]alanine versus [13C]proline) and an average error of <2 ppm. Unlike raw data from GC-MS, which are heavily skewed by the natural abundance of isotopes in the derivatization agent, the isotopomer abundances of the key metabolites from FT-ICR MS can be used directly without correction. Furthermore, FT-ICR MS data matched results obtained by GC-MS for most of amino acids (see Table S4 in the supplemental material), which indicates that FT-ICR MS is another very valuable technique for isotopomer analysis.
The experimental isotopomer distributions (Table 2; see also Table S4 in the supplemental material) were used to check whether the labeling patterns detected were consistent with the pathways deduced from the annotated genome. The similarity of isotopomer patterns in some amino acids confirmed that these amino acids were derived from the same precursor. Examples are threonine and aspartate from oxaloacetate and tyrosine and phenylalanine from phosphoenolpyruvate and erythrose-4-phosphate. Since this is redundant isotopomer information (17), only one amino acid from each precursor was used in the model calculation (Table 2). For some key amino acids, including glycine, alanine, serine, and aspartate, the isotopomer distribution after the loss of the first carboxyl group showed that the 13C label was localized to the carboxyl group, indicating that the carbon backbone of these amino acids is from pyruvate, which is also labeled mainly at the carboxyl group.
Confirmation of an atypical citrate synthase pathway via FT-ICR MS.
The presence of an atypical citrate synthase has been shown in several anaerobic bacteria, including Desulfovibrio spp. and Clostridium kluyveri, and was named (R)-citrate synthase because it produces citrate with a stereochemistry opposite to that found in most organisms (19, 20). This atypical pathway (Fig. 2) was determined by Gottschalk et al. using in vitro or in vivo radioactive 14C tracer experiments (19, 20) and required enzymatic cleavage and release of the glutamate carboxyl groups as 14CO2 to pinpoint the location of the labeled carbon in glutamate.
FIG. 2.

Transition of carbons in the synthesis of citrate from lactate reflects the prochiral nature of citrate in the TCA cycle (a symmetrical molecule that reacts asymmetrically). Oxaloacetate is the precursor of aspartic acid. 2-Oxoglutarate is the precursor of glutamate. The FT-ICR MS spectrum of glutamic acid confirms that the C-5 (#) originated from the C-4 of oxaloacetate and that the C-1 (^) of glutamic acid originated from the C-1 of acetyl-CoA.
Here we describe a swift and precise method to confirm the atypical pathway via an in vivo nonradioactive 13C tracer experiment. This method relied solely on FT-ICR MS, because GC-MS can identify only the labeled position on the C-1 carboxyl group, and because it is difficult to use 1-dimensional or 2-dimensional 13C NMR spectroscopy to ascertain if one or both of the second- and third-position carbons in the two amino acids are labeled (46). The structures of product ions generated by fragmentation of glutamic and aspartic acid during FT-ICR MS are well documented (22, 23). For this reason, any shift in the product ions allows an unambiguous assignment of the position of the 13C in the amino acid backbone. The fragmentation of glutamic acid (Fig. 3a) showed a combined loss of H2O and CO (with nominal mass equal to m/z 103), involving the C-1 carboxyl group, in line with the known instability of α-aminoacylium ions (4). Furthermore, the appearance of the peak corresponding to m/z 85 (Fig. 3b) involves, in some sequence, the overall loss of 2 H2O molecules and 1 CO molecule from the C-1 position. This result accurately matches the fragmentation that gives rise to the m/z 103 ion. Although these results rule out the presence of the 13C in the C-1 position, they still do not pinpoint the labeled atom on the amino acid backbone, necessitating another fragmentation event, i.e., an MS3 experiment. The product ion at m/z 85 has a high abundance and consequently is a good candidate for fragmentation to obtain the diagnostic product ions. In fact, the fragmentation of m/z 85 shows a 13CO loss (Fig. 3b), unequivocally identifying the position of the labeled carbon at the C-5 position. All mass errors were below 1 ppm, and in some cases even below 0.1 ppm, enabling the isolation and fragmentation of the ions of interest with accurate mass measurements. The FT-ICR MS method was also used for the localization of 13C incorporated into aspartic acid (see Table S5 in the supplemental material). In aspartic acid, the result indicates that the labeled C always localized to either one or both of the carboxylic groups. The transitions of the carbons in the TCA cycle metabolites of D. vulgaris are diagramed in Fig. 2.
FIG. 3.

FT-ICR MS to locate the 13C carbon position in glutamate. (a) SORI-CID (MS2) of [13C-Glu+H]+ generated by ESI. The fragmentation includes no loss (m/z 149), loss of H2O (m/z 131), loss of H2O and CO (α) (m/z 103), and loss of 2 H2O and CO (α) (m/z 85). Data in parentheses are measurement errors, expressed in parts per million. (b) SORI-CID (MS3) of m/z 85 generated in the ICR cell. The ion (m/z 56) indicates that the remaining carbon bone was not labeled after the loss of the second carboxyl group.
Determination of the flux distribution using the isotopomer model and reported enzyme activities.
Isotopomer analysis is a useful approach for determining the fluxes through branching pathways that converge later (such as glycolysis and the PP pathway) or the fluxes through metabolic cycles (such as the TCA cycle) (Fig. 1). The lactate flux (taken to be 100) branched into three pathways at pyruvate. The major flow was into the TCA cycle; the second flow, into gluconeogenesis and the PP pathway; and the third flow, toward biomass production (e.g., synthesis of alanine, valine, etc.). Approximately 84% of the lactate was partially oxidized to acetate via acetyl coenzyme A (acetyl-CoA), and this route was a main source of ATP production for D. vulgaris. About 5% of the lactate was oxidized completely via a carbon monoxide dehydrogenase pathway (48, 52).
The flux results indicated the absence of flux through 2-oxoglutarate↔succinate; that is, the TCA cycle is branched and ends with glutamate and succinate. The flux through the glyoxylate shunt also converged to zero when the model was optimized, consistent with the absence of isocitrate lyase in this pathway (30). In a branched TCA cycle, four reactions (pyruvate→malate, pyruvate→oxaloacetate, phosphoenolpyruvate↔oxaloacetate, and malate↔oxaloacetate) are not distinguishable by isotopomer analysis, because two pairs of molecules ([i] pyruvate and phosphoenolpyruvate and [ii] malate and oxaloacetate) have the same carbon backbones. In order to have a unique solution, the pathway was further simplified using the assumption that there were no fluxes through oxaloacetate↔malate and phosphoenolpyruvate↔oxaloacetate, because of a lack of key enzymes for the two reactions: phosphoenolpyruvate carboxylase is not found in the annotated genome, while malate dehydrogenase activity has been found to be absent in D. vulgaris (30).
The flux distribution results and the reversibility of major fluxes are presented in Fig. 1. The predicted labeling patterns of all metabolites, based on calculated fluxes and exchange coefficients, reasonably match the measured data (Fig. 1; Table 2). Deviations between the modeled and measured isotopomer data could arise from several sources: (i) differences in biological replicates and in the measurements of the biosynthetic fluxes based on the biomass composition; (ii) the noise affecting the accuracy of MS data for low-abundance ions; (iii) the effect of overlay of certain mass peaks [for example, in GC-MS, the (f302)+ peak may reduce the measurement accuracy for the leucine (M-57)+ peak, while the isoleucine and leucine peaks are indistinguishable by FT-ICR MS]; and (iv) possible reactions affecting some amino acid pathways [for example, the precursor of leucine (2-oxoisocaproate) may exchange its carboxyl group with 12CO2 or 13CO2 generated by the decarboxylation of labeled metabolites (36)].
DISCUSSION
Though initial genome annotation provided a preliminary overview for D. vulgaris metabolism, misannotation is possible due to a lack of homology between genes/enzymes in the target organism and those in related or unrelated organisms. For instance, the published, annotated genome sequence indicates that several amino acid synthesis pathways (e.g., those for lysine, methionine, histidine, and alanine) are incomplete (1). However, D. vulgaris is able to grow in minimal medium and therefore must contain complete amino acid synthesis pathways. The reverse is also true; i.e., having an annotated gene or genes for a metabolic pathway does not guarantee the presence of the pathway. For example, although the annotated genome sequence for D. vulgaris has genes encoding all of the enzymes in glycolysis, our previous experiments showed that D. vulgaris is not able to grow on glucose or fructose (unpublished data). Therefore, accurate genome annotation requires further curation or biochemical confirmation via enzymatic or tracer experiments.
Pyruvate↔acetyl-CoA is a key reaction in D. vulgaris metabolism. The genome annotation indicates that D. vulgaris has no pyruvate dehydrogenase, but D. vulgaris can convert pyruvate to acetyl-CoA and CO2 via an oxidoreductase. D. vulgaris also contains a pyruvate formate lyase enzyme to convert pyruvate to acetyl-CoA and formate without generating NADH; then formate can be further oxidized by formate dehydrogenase to CO2, generating NADH. Although l-[13C]lactate was the sole carbon and energy source in the growth medium, much less pyruvate (81%) was labeled (precursor to alanine). This may be explained by the highly reversible reaction (pyruvate↔acetyl-CoA + CO2 or formate) (exch = 0.99 [see equation 1]) that exchanges unlabeled carbon from dissolved CO2 in the medium. Such high reversibility is not surprising, because this reaction can be catalyzed by several enzymes (25), including pyruvate synthase, pyruvate formate-lyase, pyruvate:ferredoxin oxidoreductase, and oxo-organic acid oxidoreductase, present in D. vulgaris. These enzymes may also be able to convert acetate to pyruvate if a significant amount of hydrogen and CO2 is present in the atmosphere of the anaerobic hood (5% H2 and 5% CO2) (3). Furthermore, this bacterium contains dihydrolipoyl dehydrogenase (EC 1.8.1.4), which acts on a sulfur group of donors and uses NAD+ or NADP+ as an electron acceptor. In this way, D. vulgaris can generate both NADH and NADPH via pyruvate→acetyl-CoA to satisfy energy and biosynthesis requirements. Since the TCA cycle in D. vulgaris is mainly for biosynthesis purposes, pyruvate→acetyl-CoA is thus a main step for NADH and NADPH production. The involvement of many different enzymes for this reaction may improve the flexibility and robustness of D. vulgaris metabolism under conditions of environmental uncertainty.
Sulfate reduction is an energetically poor process, because reduction of 1 mol of sulfate requires 2 mol of ATP to activate sulfate molecules and 4 mol of NADH to reduce them to H2S. Thus, sulfate reduction consumes more than 80% of the NADH/NADPH and 90% of the ATP generated from the lactate→pyruvate→acetate metabolism route (Fig. 1). However, an alternative pathway for oxidation of acetyl-CoA to 5,10-methyl-THF (5,10-Me-THF) and carbon monoxide has been reported for sulfate reducers (Fig. 4) (48, 52). In this pathway, both 5,10-Me-THF and carbon monoxide can be further oxidized to CO2 to generate ATP and NADH. Although most Desulfovibrio spp. are thought not to completely oxidize acetyl-CoA (55), key enzymes for such activities are annotated in D. vulgaris (Fig. 4). These include carbon monoxide dehydrogenase (DVU2098, DVU2099), formate dehydrogenase (e.g., DVU0587, DVU0588), and methylenetetrahydrofolate dehydrogenase/methenyltetrahydrofolate cyclohydrolase (DVU0323). Further, previous reports (52) have shown that a significant amount of carbon monoxide is produced during the growth of D. vulgaris in lactate/sulfate medium, consistent with cleavage of acetyl-CoA into 5,10-Me-THF and CO (Fig. 4). Results from our study support the existence of an acetyl-CoA oxidation pathway (relative flux, ∼5%). The fact that the measured relative carbon flow to acetate (84% [Fig. 1]) is lower than the expected value (>90%) can be explained by acetyl-CoA oxidation. Additionally, the reactions acetyl-CoA↔CO↔CO2 are highly reversible, and thus the C-1 of acetyl-CoA exchanges carbon with the labeled carbon in CO2 (removed from pyruvate) via carbon monoxide dehydrogenase (52, 55). The formation of C-1-labeled acetyl-CoA may explain why more than 20% of leucine's carboxyl group was 13C labeled, even though this carbon is derived mainly from the C-2 of pyruvate and therefore should not be labeled (Fig. 4). Furthermore, since sulfate reduction demands are high for NADH and ATP, acetyl-CoA oxidation may provide an alternate energy-producing pathway in D. vulgaris. Oxidization of the product [CO] (bound carbon monoxide) generates hydrogen, which may be used to reduce sulfate and generate ATP via a hypothesized hydrogen metabolic cycle (4 mol of H2 generate a net 2.7 mol of ATP per mol of sulfate reduced) (52); while the product 5,10-Me-THF can be oxidized to formate and then CO2, those steps also yield ATP and NADH (52, 55).
FIG. 4.

Acetyl-CoA oxidization and leucine biosynthesis. The first and second carbons of leucine originate from the first and second carbons of acetyl-CoA. Acetyl-CoA can be reversibly oxidized via an alternative pathway. C1 is 5,10-Me-THF. Annotated enzymes involved in acetyl-CoA oxidization are as follows: for reaction 1, methylenetetrahydrofolate dehydrogenase and methenyltetrahydrofolate cyclohydrolase; for reaction 2, formate dehydrogenase; for reaction 3, carbon monoxide dehydrogenase. *C represents the first carbon in lactate and is the labeled 13C in our experiment. #C represents the second C of lactate and is the one incorporated into the leucine COOH group via acetyl-CoA. [13C]acetyl-CoA is likely present via reversible reactions 3 and 4 from the 13CO2 and leads to some percentage of the labeled COOH in leucine.
The TCA cycle is missing two reactions: malate↔oxaloacetate, which was not annotated in the D. vulgaris genome, and 2-oxoglutarate↔succinate. The second reaction has been suggested to be catalyzed by a ferredoxin-dependent enzyme (annotated as EC 1.2.7.3) (1) and was found to be active in certain Desulfobacter spp. Flux results supported the genome annotation of two reactions (pyruvate→oxaloacetate [E.C.6.4.1.1] and pyruvate→malate [E.C.1.1.1.4]) that fix CO2. The 13CO2 in the medium was derived mainly from the loss of the carboxyl group from pyruvate to form acetyl-CoA, whereas unlabeled CO2 was derived mainly from the gas mix used in the anaerobic chamber. A majority of the aspartate had both carboxyl groups labeled with 13C (M2 = 0.66). The 13C-labeling pattern of succinate was similar to that of aspartate (i.e., oxaloacetate) but very different from that of glutamate, which was primarily singly labeled. The flux based on these isotopomer data indicates that succinate was derived from pyruvate and CO2 through the reductive branch of the TCA cycle, while glutamate was derived from citrate through the oxidative branch of the TCA cycle; i.e., there is no ferredoxin-dependent 2-oxoglutarate synthase (EC 1.2.7.3) activity (2-oxoglutarate + ferredoxinox + CoA↔succinyl-Co A + CO2 + ferredoxinred) (9).
Citrate is a symmetrical molecule, but aconitase is known to be stereospecific for the prochiral structure of citrate, providing the stereochemical bias of the reaction (36). The results from the FT-ICR MS analysis of glutamate support a unique carbon transition route (Fig. 2) and confirm previous observations that the citrate synthase in D. vulgaris has an atypical stereochemical propensity (19, 20). Although the presence of this enzyme has been known for decades, no sequence information is available. Interestingly, an ATP-citrate lyase gene has been annotated in D. vulgaris, but the question of whether the ATP-citrate lyase is involved in the unusual citrate synthesis requires further study (34).
In SRB, 5,10-Me-THF production controls the mercury methylation pathway, which produces a highly hazardous environmental pollutant (methylated mercury) prone to biomagnification (11, 12). Using 14C labeling, Choi et al. (12) proposed that the 5,10-Me-THF in SRB may originate from two possible sources: either from the C-3 of serine (serine→glycine + 5,10-Me-THF) or from formate via pyruvate formate-lyase (PYR→acetyl-CoA + formate→5,10-Me-THF) (5,10-Me-THF from the C-1 of pyruvate). Meanwhile, sulfate-reducing bacteria can also convert the C-2 of acetyl-CoA to 5,10-Me-THF by using the pathway shown in Fig. 4 (48). The labeling pattern of methionine (5 carbons) is similar to that of aspartate (4 carbons), both of which are derived from oxaloacetate. Our results indicate that the methyl of 5,10-Me-THF, which condensed with oxaloacetate to form methionine, was not labeled, demonstrating that 5,10-Me-THF production in D. vulgaris is not derived from the formate produced from pyruvate by pyruvate formate-lyase.
The measured biomass contains carbohydrates that are synthesized via glycolysis. However, the flux distribution, based on biomass growth and isotopomer data from histidine and phenylalanine/tyrosine, indicated that no appreciable flow was detectable through the glucose-6-phosphate→→C5P (ribose-5-phosphate, ribulose-5-phosphate, or xylulose-5-phosphate) pathway. This is consistent with the D. vulgaris gene annotation, where no candidates were found for two key enzymes (EC 1.1.1.49 [glucose-6-phosphate dehydrogenase] and EC 1.1.1.44 [phosphogluconate dehydrogenase]) in the PP pathway. This is further supported by the lack of any enzyme activity for glucose-6-phosphate dehydrogenase in the D. vulgaris lysate (i.e., no NADPH is produced via the pentose phosphate pathway [unpublished data]). Apparently, the incomplete PP pathway is used mainly for biosynthesis of amino acids and nucleic acids.
The D. vulgaris metabolism is a relatively energetically poor process and appears to be simpler than that of other bacteria, e.g., E. coli. From an evolutionary point of view, D. vulgaris is thought to present a simple but ancient metabolism developed in the primitive seas (19, 20, 45); understanding of its metabolism is potentially important for our understanding of the evolution of metabolic pathways in early life.
Conclusion.
We used nonradioactive tracer experiments and isotopomer analysis to examine the key annotated metabolic pathways in D. vulgaris as well as to quantify carbon flux through these pathways. Both GC-MS and FT-ICR MS techniques were used to obtain complete isotopomer information for the metabolites for subsequent isotopomer analysis. Data from this study confirmed several aspects of D. vulgaris metabolism, such as an incomplete pentose phosphate pathway, a branched and incomplete TCA cycle, the presence of an (R)-citrate synthase, and an acetyl-CoA oxidation route. This study demonstrates FT-ICR MS to be a potential tool for flux analysis with several advantages over conventional methods. Compared to GC-MS, samples do not need to be derivatized, and therefore the data can be used without correction for the natural isotopomer effect from the derivatized group. Compared to NMR spectroscopy, FT-ICR MS is a much more sensitive method with a very low detection limit (order of nanomoles). FT-ICR MS enables the localization of labeled carbon at locations not accessible via GC-MS or 13C NMR. ESI FT-ICR MS did not require the separation of metabolites by GC or high-performance liquid chromatography and thus is a high-throughput method for measuring the isotopomers compared to other methods that require separation prior to detection.
The isotopomer distribution data were essential to confirm the result that key central pathways in D. vulgaris are incomplete. However, due to the low flux (<5%) in the PP pathway and TCA cycle, the precise flux values and their reversibility through these pathways could not be ascertained using the isotopomer data. In this study, estimation of the fluxes toward central pathways, biomass synthesis, and other metabolites (acetate, pyruvate, and succinate) relied primarily on direct measurement of the production rates of these metabolites.
Supplementary Material
Acknowledgments
We thank Judy Wall (University of Missouri, Columbia) for helpful discussions on D. vulgaris pathways. Steve Van Dien (Genomatica Company) provided the isotopomer correction spreadsheet. Adam Meadows, Kenneth Kauffman, Jeannie Chu (University of California, Berkeley), Rick Huang (Lawrence Berkeley National Laboratory), and Gary Kruppa (Bruker Daltonics, CA) helped with experiments and isotopomer modeling.
This study is part of the work of the Virtual Institute for Microbial Stress and Survival (http://vimss.lbl.gov), supported by the U.S. Department of Energy, Office of Science, Office of Biological and Environmental Research, Genomics:GTL Program, through contract DE-AC02-05CH11231 between the Lawrence Berkeley National Laboratory and the U.S. Department of Energy.
Footnotes
Published ahead of print on 17 November 2006.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Alm, E. J., K. H. Huang, M. N. Price, R. P. Koche, K. Keller, I. L. Dubchak, and A. P. Arkin. 2005. The MicrobesOnline Web site for comparative genomics. Genome Res. 15:1015-1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arauzo-Bravo, M. J., and K. Shimizu. 2003. An improved method for statistical analysis of metabolic flux analysis using isotopomer mapping matrices with analytical expressions. J. Biotechnol. 105:117-133. [DOI] [PubMed] [Google Scholar]
- 3.Badziong, W., B. Ditter, and R. K. Thauer. 1979. Acetate and carbon dioxide assimilation by Desulfovibrio vulgaris (Marburg), growing on hydrogen and sulfate as sole energy source. Arch. Microbiol. 123:301-305. [Google Scholar]
- 4.Beranova, S., J. Cai, and C. Wesdemiotis. 1995. Unimolecular chemistry of protonated glycine and its neutralized form in the gas phase. J. Am. Chem. Soc. 117:9492-9501. [Google Scholar]
- 5.Blanch, H. W., and D. S. Clark. 1997. Biochemical engineering. Marcel Dekker Inc., New York, NY.
- 6.Bond, D. R., T. Mester, C. L. Nesbo, A. V. Izquierdo-Lopez, F. L. Collart, and D. R. Lovley. 2005. Characterization of citrate synthase from Geobacter sulfurreducens and evidence for a family of citrate synthases similar to those of eukaryotes throughout the Geobacteraceae. Appl. Environ. Microbiol. 71:3858-3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boopathy, R. 2002. Methanogenesis from furfural by defined mixed cultures. Curr. Microbiol. 44:406-410. [DOI] [PubMed] [Google Scholar]
- 8.Brown, S. C., G. Kruppa, and J. L. Dasseux. 2005. Metabolomics application of FT-ICR mass spectrometry. Mass Spectrom. Rev. 24:223-231. [DOI] [PubMed] [Google Scholar]
- 9.Buchanan, B. B. 1969. Role of ferredoxin in the synthesis of α-ketobutyrate from propionyl coenzyme A and carbon dioxide by enzymes from photosynthetic and nonphotosynthetic bacteria. J. Biol. Chem. 244:4218-4223. [PubMed] [Google Scholar]
- 10.Chhabra, S. R., Q. He, K. H. Huang, S. P. Gaucher, E. J. Alm, Z. He, M. Z. Hadi, T. C. Hazen, J. D. Wall, J. Zhou, A. P. Arkin, and A. K. Singh. 2006. Global analysis of heat shock response in Desulfovibrio vulgaris Hildenborough. J. Bacteriol. 188:1817-1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi, S. C., and R. Bartha. 1994. Environmental factors affecting mercury methylation in estuarine sediments. Bull. Environ. Contam. Toxicol. 53:805-812. [DOI] [PubMed] [Google Scholar]
- 12.Choi, S. C., T. Chase, and R. Bartha. 1994. Metabolic pathways leading to mercury methylation in Desulfovibrio desulfuricans LS. Appl. Environ. Microbiol. 60:4072-4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cooney, M. J., E. Roschi, I. W. Marison, C. Comninellis, and U. V. Stockar. 1996. Physiologic studies with the sulfate-reducing bacterium Desulfovibrio desulfuricans: evaluation for use in a biofuel cell. Enzyme Microb. Technol. 18:358-365. [DOI] [PubMed] [Google Scholar]
- 14.Cornish-Bowden, A., and M. L. Cardenas. 2000. From genome to cellular phenotype—a role for metabolic flux analysis? Nat. Biotechnol. 18:267-268. [DOI] [PubMed] [Google Scholar]
- 15.Daniels, L., R. S. Hanson, et al. 1994. Chemical analysis, p. 512-554. In P. Gerhardt, R. Murray, W. Wood, and N. Krieg (ed.), Methods for general and molecular bacteriology. American Society for Microbiology, Washington, DC.
- 16.Fischer, E., and U. Sauer. 2005. Large-scale in vivo flux analysis shows rigidity and suboptimal performance of Bacillus subtilis metabolism. Nat. Genet. 37:636-640. [DOI] [PubMed] [Google Scholar]
- 17.Fischer, E., and U. Sauer. 2003. Metabolic flux profiling of Escherichia coli mutants in central carbon metabolism using GC-MS. Eur. J. Biochem. 270:880-891. [DOI] [PubMed] [Google Scholar]
- 18.Gevertz, D., A. J. Telang, G. Voordouw, and G. E. Jenneman. 2000. Isolation and characterization of strains CVO and FWKO B, two novel nitrate-reducing, sulfide-oxidizing bacteria isolated from oil field brine. Appl. Environ. Microbiol. 66:2491-2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gottschalk, G. 1968. The stereospecificity of the citrate synthase in sulfate-reducing and photosynthetic bacteria. Eur. J. Biochem. 5:346-351. [DOI] [PubMed] [Google Scholar]
- 20.Gottschalk, G., and H. A. Barker. 1967. Presence and stereospecificity of citrate synthase in anaerobic bacteria. Biochemistry 6:1027-1034. [DOI] [PubMed] [Google Scholar]
- 21.Hadas, O., and R. Pinkas. 1995. Sulfate reduction processes in sediments at different sites in Lake Kinneret, Israel. Microb. Ecol. 30:55-66. [DOI] [PubMed] [Google Scholar]
- 22.Harrison, A. G., and Y. P. Tu. 1998. Ion chemistry of protonated aspartic acid derivatives. J. Mass Spectrom. 33:532-542. [DOI] [PubMed] [Google Scholar]
- 23.Harrison, G. A. 2001. Ion chemistry of protonated glutamic acid derivatives. Int. J. Mass Spectrom. 210/211:361-370. [Google Scholar]
- 24.He, Q., K. H. Huang, Z. He, E. J. Alm, M. W. Fields, T. C. Hazen, A. P. Arkin, J. D. Wall, and J. Zhou. 2006. Energetic consequences of nitrite stress in Desulfovibrio vulgaris Hildenborough, inferred from global transcriptional analysis. Appl. Environ. Microbiol. 72:4370-4381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heidelberg, J. F., R. Seshadri, S. A. Haveman, C. L. Hemme, I. T. Paulsen, J. F. Kolonay, J. A. Eisen, N. Ward, B. Methe, L. M. Brinkac, S. C. Daugherty, R. T. Deboy, R. J. Dodson, A. S. Durkin, R. Madupu, W. C. Nelson, S. A. Sullivan, D. Fouts, D. H. Haft, J. Selengut, J. D. Peterson, T. M. Davidsen, N. Zafar, L. Zhou, D. Radune, G. Dimitrov, M. Hance, K. Tran, H. Khouri, J. Gill, T. R. Utterback, T. V. Feldblyum, J. D. Wall, G. Voordouw, and C. M. Fraser. 2004. The genome sequence of the anaerobic, sulfate-reducing bacterium Desulfovibrio vulgaris Hildenborough. Nat. Biotechnol. 22:554-559. [DOI] [PubMed] [Google Scholar]
- 26.Hellerstein, M. K., and R. A. Neese. 1999. Mass isotopomer distribution analysis at eight years: theoretical, analytic, and experimental considerations. Am. J. Physiol.-Endocrinol. Metab. 276:E1146-E1170. [DOI] [PubMed] [Google Scholar]
- 27.Humphries, A. C., and L. E. Macaskie. 2002. Reduction of Cr(VI) by Desulfovibrio vulgaris and Microbacterium sp. Biotechnol. Lett. 24:1261-1267. [Google Scholar]
- 28.Humphries, A. C., K. P. Nott, L. D. Hall, and L. E. Macaskie. 2004. Continuous removal of Cr(VI) from aqueous solution catalysed by palladised biomass of Desulfovibrio vulgaris. Biotechnol. Lett. 26:1529-1532. [DOI] [PubMed] [Google Scholar]
- 29.Laskin, J., and J. H. Futrell. 2003. Collisional activation of peptide ions in FT-ICR mass spectrometry. Mass Spectrom. Rev. 22:158-181. [DOI] [PubMed] [Google Scholar]
- 30.Lewis, A. J., and J. D. Miller. 1977. The tricarboxylic and acid pathway in Desulfovibrio. Can. J. Microbiol. 23:916-921. [DOI] [PubMed] [Google Scholar]
- 31.Marshal, J. 2004. Production of secondary metabolites from acetyl Co-A precursors in bacterial and fungal hosts. Ph.D. thesis. University of California, Berkeley.
- 32.Marshall, A. G., C. L. Hendrickson, and G. S. Jackson. 1998. Fourier transform ion cyclotron resonance mass spectrometry: a primer. Mass Spectrom. Rev. 17:1-35. [DOI] [PubMed] [Google Scholar]
- 33.McLafferty, F. W., and F. Turecek. 1993. Interpretation of mass spectra, 4th ed. University Science Books, Sausalito, CA.
- 34.Moller, D., R. Schauder, G. Fuchs, and R. K. Thauer. 1987. Acetate oxidation to CO2 via a citric acid cycle involving an ATP-citrate lyase: a mechanism for the synthesis of ATP via substrate level phosphorylation in Desulfobacter postgatei growing on acetate and sulfate. Arch. Microbiol. 148:202-207. [Google Scholar]
- 35.Mukhopadhyay, A., Z. He, E. J. Alm, A. P. Arkin, E. E. Baidoo, S. C. Borglin, W. Chen, T. C. Hazen, Q. He, H. Y. Holman, K. Huang, R. Huang, D. C. Joyner, N. Katz, M. Keller, P. Oeller, A. Redding, J. Sun, J. Wall, J. Wei, Z. Yang, H. C. Yen, J. Zhou, and J. D. Keasling. 2006. Salt stress in Desulfovibrio vulgaris Hildenborough: an integrated genomics approach. J. Bacteriol. 188:4068-4078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nelson, D. L., and M. M. Cox. 2000. Lehninger principles of biochemistry. Worth Publishers, New York, NY.
- 37.Nemati, M., G. E. Jenneman, and G. Voordouw. 2001. Mechanistic study of microbial control of hydrogen sulfide production in oil reservoirs. Biotechnol. Bioeng. 74:424-434. [DOI] [PubMed] [Google Scholar]
- 38.Noguera, D. R., G. A. Brusseau, B. E. Rittmann, and D. A. Stahl. 1998. A unified model describing the role of hydrogen in the growth of Desulfovibrio vulgaris under different environmental conditions. Biotechnol. Bioeng. 59:732-746. [DOI] [PubMed] [Google Scholar]
- 39.Ouattara, A. S., and V. A. Jacq. 1992. Characterization of sulfate-reducing bacteria isolated from Senegal ricefields. FEMS Microbiol. Ecol. 101:217-228. [Google Scholar]
- 40.Sauer, U., D. R. Lasko, J. Fiaux, M. Hochuli, R. Glaser, T. Szyperski, K. Wuthrich, and J. E. Bailey. 1999. Metabolic flux ratio analysis of genetic and environmental modulations of Escherichia coli central carbon metabolism. J. Bacteriol. 181:6679-6688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schmidt, K., J. Nielsen, and J. Villadsen. 1997. Modeling isotopomer distributions in metabolic networks using isotopomer mapping matrices. Biotechnol. Bioeng. 55:831-840. [DOI] [PubMed] [Google Scholar]
- 42.Schmidt, K., J. Nielsen, and J. Villadsen. 1999. Quantitative analysis of metabolic fluxes in Escherichia coli, using two-dimensional NMR spectroscopy and complete isotopomer models. J. Biotechnol. 71:175-190. [DOI] [PubMed] [Google Scholar]
- 43.Stelling, J., U. Sauer, Z. Szallasi, F. Doyle, and J. Doyle. 2004. Robustness of cellular functions. Cell 118:675-685. [DOI] [PubMed] [Google Scholar]
- 44.Stephanopoulos, G. N., A. A. Aristidou, and J. Nielsen. 1998. Metabolic engineering principles and methodologies. Academic Press, San Diego, CA.
- 45.Stern, J. R., C. S. Hegre, and G. Bambers. 1966. Glutamate biosynthesis in anaerobic bacteria. II. Stereospecificity of aconitase and citrate synthetase of Clostridium kluyveri. Biochemistry 5:1119. [DOI] [PubMed] [Google Scholar]
- 46.Szyperski, T. 1998. 13C-NMR, MS and metabolic flux balancing in biotechnology research. Q. Rev. Biophys. 31:41-88. [DOI] [PubMed] [Google Scholar]
- 47.Teece, M. A., M. L. Fogel, M. E. Dollhopf, and K. H. Nealson. 1999. Isotopic fraction associated with biosynthesis of fatty acids by a marine bacterium under oxic and anoxic conditions. Org. Geochem. 30:1571-1579. [Google Scholar]
- 48.Thauer, R. K. 1988. Citric-acid cycle, 50 years on modifications and an alternative pathway in anaerobic bacteria. Eur. J. Biochem. 176:497-508. [DOI] [PubMed] [Google Scholar]
- 49.Traore, A. S., M. L. Fardeau, C. E. Hatchikian, J. L. Gall, and J. P. Belaich. 1983. Energetics of growth of a defined mixed culture of Desulfovibrio vulgaris and Methanosarcina barkeri: interspecies hydrogen transfer in batch and continuous cultures. Appl. Environ. Microbiol. 46:1152-1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Traore, A. S., C. E. Hatchikian, J. P. Belaich, and J. L. Gall. 1981. Microcalorimetric studies of the growth of sulfate-reducing bacteria: energetics of Desulfovibrio vulgaris growth. J. Bacteriol. 145:191-199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tu, Y. P., and A. G. Harrison. 1998. The b1 ion derived from methionine is a stable species. Rapid Commun. Spectrom. 12:849-851. [Google Scholar]
- 52.Voordouw, G. 2002. Carbon monoxide cycling by Desulfovibrio vulgaris Hildenborough. J. Bacteriol. 184:5903-5911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Voordouw, G., S. M. Armstrong, M. F. Reimer, B. Fouts, A. J. Telang, Y. Shen, and D. Gevertz. 1996. Characterization of 16S rRNA genes from oil field microbial communities indicates the presence of a variety of sulfate-reducing, fermentative, and sulfide-oxidizing bacteria. Appl. Environ. Microbiol. 62:1623-1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wahl, S. A., M. Dauner, and W. Wiechert. 2004. New tools for mass isotopomer data evaluation in 13C flux analysis: mass isotope correction, data consistency checking, and precursor relationships. Biotechnol. Bioeng. 85:259-268. [DOI] [PubMed] [Google Scholar]
- 55.White, D. 1995. The physiology and biochemistry of prokaryotes. Oxford University Press, New York, NY.
- 56.Wiechert, W., and A. A. de Graaf. 1997. Bidirectional reaction steps in metabolic networks. I. Modeling and simulation of carbon isotope labeling experiments. Biotechnol. Bioeng. 55:101-117. [DOI] [PubMed] [Google Scholar]
- 57.Zhao, J., and K. Shimizu. 2003. Metabolic flux analysis of Escherichia coli K12 grown on 13C-labeled acetate and glucose using GC-MS and powerful flux calculation method. J. Biotechnol. 101:101-117. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.