

Abstract
Group B Streptococcus (GBS) is classified into nine serotypes that vary in capsular polysaccharide (CPS) architecture but share in common the presence of a terminal sialic acid (Sia) residue. This position and linkage of GBS Sia closely resembles that of cell surface glycans found abundantly on human cells. CD33-related Siglecs (CD33rSiglecs) are a family of Sia-binding lectins expressed on host leukocytes that engage host Sia-capped glycans and send signals that dampen inflammatory gene activation. We hypothesized that GBS evolved to display CPS Sia as a form of molecular mimicry limiting the activation of an effective innate immune response. In this study, we applied a panel of immunologic and cell-based assays to demonstrate that GBS of several serotypes interacts in a Sia- and serotype-specific manner with certain human CD33rSiglecs, including hSiglec-9 and hSiglec-5 expressed on neutrophils and monocytes. Modification of GBS CPS Sia by O acetylation has recently been recognized, and we further show that the degree of O acetylation can markedly affect the interaction between GBS and hSiglec-5, -7, and -9. Thus, production of Sia-capped bacterial polysaccharide capsules that mimic human cell surface glycans in order to engage CD33rSiglecs may be an example of a previously unrecognized bacterial mechanism of leukocyte manipulation.
Group B Streptococcus (GBS) is the leading cause of bacterial pneumonia, sepsis, and meningitis in human newborns and is increasingly recognized as a pathogen in adult populations, including diabetics, pregnant women, and the elderly. A critical factor contributing to GBS virulence is its surface capsular polysaccharide (CPS). The CPS forms the outermost layer of the bacterial surface and is typically composed of repeating subunits of four monosaccharides, i.e., glucose, galactose, N-acetylglucosamine, and N-acetylneuraminic acid, polymerized in serotype-specific configurations (10). GBS isolates are categorized as belonging to one of nine different serotypes (Ia, Ib, and II to VIII) based on antibody recognition. Although the physical structure of the CPS of each GBS serotype is unique, all GBS serotypes invariably share a terminal α2,3-linked N-acetylneuraminic acid (Neu5Ac), which is identical to the predominant sialic acid (Sia) found on human cells. It has recently been discovered that GBS modify its CPS Sia by addition of a single O-acetyl group at the seventh carbon position that can subsequently migrate to the eighth or ninth carbon position (23, 24). GBS synthesizes and modifies Sia by using a de novo pathway in which four genes, neuA to neuD, are responsible for Sia synthesis and O acetylation, while cpsK mediates transfer of Sia to the CPS subunit (9, 15, 23, 31, 32).
The Sia-recognizing immunoglobulin (Ig) superfamily lectins (Siglecs) are a family of structurally related type I transmembrane proteins composed of one unique (or two in the case of Siglec-XII) V-set Ig domain responsible for recognizing sialylated glycoconjugates, followed by a variable number of structural C2-set Ig domains (33). The CD33/Siglec-3-related Siglecs (CD33rSiglecs) are a rapidly evolving subgroup of the Siglecs and in humans consist of Siglec-3, -5 to -11, and -XII. CD33rSiglecs, with the exception of Siglec-XII, which lacks a functional Sia-binding domain, are expressed predominantly on the surfaces of leukocytes in a cell type-specific fashion suggesting distinct functional roles (11). The CD33rSiglecs share 50 to 80% sequence similarity, with the highest similarity in the V-set domain involved in Sia recognition and the two intracellular tyrosine signaling motifs. The membrane-proximal signaling domain is an immunoreceptor tyrosine-based inhibitory motif (ITIM) with the canonical sequence (I/L/V)xYxx(L/V), whereas the membrane-distal domain is an ITIM-like motif whose role is less well defined (29, 34). Upon phosphorylation by a Src family kinase, these motifs can recruit protein tyrosine phosphatases SHP-1, SHP-2, and SHIP. Many transmembrane proteins containing these motifs are involved in inhibitory signaling that dampens or counteracts activating signals sent by other immunoreceptors, such as those containing immunotyrosine-activating motifs.
In this study we examined whether CPS Sias of various GBS serotypes can specifically engage human CD33rSiglecs present on human leukocytes involved in control of bacterial infections. Further, we examined how O-acetyl modification of CPS Sia can modify the interaction between GBS and these leukocyte surface receptors. The implications of GBS surface expression of a human-like monosaccharide, Neu5Ac, are then discussed in the context of molecular mimicry and innate immune evasion.
MATERIALS AND METHODS
Bacteria and growth conditions.
GBS wild-type (WT) strains of serotypes Ia (A909), Ib (M709), II (DK23), III (COH1), V (NCTC10/84), and VI (NT-6) are well-characterized isolates from human neonates with invasive infections. Generation of the ΔNeuA and ΔNeuD mutants of serotype III strain COH1 was previously reported (23, 24). GBS were grown in Todd-Hewitt broth (THB), pH 7.5, or on THB agar plates. For antibiotic selection, 10 μg/ml erythromycin or 2.5 μg/ml chloramphenicol was used. Escherichia coli strains were grown in Luria-Bertani broth (LB); antibiotic selection employed 500 μg/ml erythromycin or 50 μg/ml chloramphenicol. For functional assays, unless otherwise noted, bacteria were grown to early exponential phase in THB, washed three times with pyrogen-free phosphate-buffered saline (PBS), resuspended in appropriate buffers, and adjusted to the desired concentrations by using a spectrophotometric method confirmed by pour plate colony counts. For enzymatic removal of CPS Sias, GBS was incubated in sterile PBS with 100 mU/ml Arthrobacter ureaficiens sialidase for 1 h at 37°C and then washed three times with PBS. Trypsin-treated bacteria were incubated with 0.5% trypsin plus EDTA in PBS for 30 min at 37°C and washed five times with PBS. Prior to Chinese hamster ovary (CHO) cell binding experiments using Sia-blocking and non-Sia-blocking antibodies, GBS was preincubated for 10 min in PBS plus 5% normal goat serum and then washed twice.
GBS FITC labeling.
GBS was grown overnight, diluted 1:100 in 50 ml of THB, grown to early exponential phase, pelleted, washed three times with sterile PBS (pH 7.4), and heat killed by incubation at 56°C for 40 min. GBS was then washed in 50 mM carbonate buffer (pH 8.0), resuspended in 5 ml carbonate buffer (pH 8.0) plus 0.1% fluorescein isothiocyanate (FITC), and incubated for 1 h at 37°C. Bacteria were extensively washed in PBS to remove trace amounts of free FITC and then resuspended in assay buffer (20 mM HEPES, 150 mM NaCl, 1% bovine serum albumin [BSA], pH 7.4). FITC-labeled GBS cells (FITC-GBS) were then enumerated using a bacterial cytometer and a fluorescent microscope. FITC-GBS (1 × 106) were resuspended in PBS plus 1% BSA and analyzed by flow cytometry (BD FACSCalibur) to verify that bacterial staining was uniform throughout each sample.
Siglec-Fc binding assay.
Immulon 4 enzyme-linked immunosorbent assay plates were coated with 0.5 mg/ml protein A in coating buffer (50 mM carbonate buffer, pH 9.5) overnight at 4°C. Wells were washed three times with assay buffer (20 mM HEPES, 150 mM NaCl, 1% BSA) and then blocked with assay buffer for 1 h at room temperature. Human CD33rSiglec-Fc chimeras diluted in assay buffer were added to individual wells at 0.5 mg/ml and allowed to adhere for at least 3 h at room temperature. Wells were washed three times with assay buffer, and 1 × 107 FITC-GBS suspended in assay buffer were added to each well and centrifuged. Bacteria were allowed to adhere for 10 min at 37°C. The initial fluorescence intensity was verified, wells were washed to remove unbound bacteria, and the residual fluorescent intensity (excitation, 488 nm; emission, 530 nm) was measured using a CytoFluorII fluorescent plate reader.
Soluble receptors and stable cell expression.
Soluble hSiglec-Fc chimeras were produced as previously described (2). CHO-K1 cells stably expressing either full-length hSiglec-5 or hSiglec-9 were produced by transfecting WT CHO-K1 cells with either pcDNA3.1(+)Neo-hSiglec-5 or pcDNA3.1(+)Hygro-hSiglec-9 by using Fugene-6 transfection reagent according to the manufacturer's directions. The expression plasmid pcDNA3.1(+)Hygro-hSiglec-9 was produced by subcloning human Siglec-9 from pcDNA3.1(+)Neo into pcDNA3.1(+)Hygro. Stable transfectants were selected by growth in Ham's F-12 medium plus 1 mg/ml G418 or 800 μg/ml hygromycin. CHO cells stably expressing hSiglec-9 were further selected using flow-activated cell sorting at the UCSD/Veteran's Administration San Diego Flow Cytometry Core.
CHO cell adhesion assay.
CHO-K1 cells with or without hSiglec expression were grown to 80% confluence in 100-mm cell culture dishes and lifted using PBS plus 2 mM EDTA, and 2.5 × 105 cells were added to each well of 24 well plates and allowed to grow overnight. CHO cells were washed once with 37°C serum-free Ham's F-12 medium and resuspended in Ham's F-12 medium with or without 10 μg/ml antibodies for 10 min at 37°C and 5% CO2. FITC-GBS were added at an multiplicity of infection of 100:1, spun down onto the cell monolayer, and allowed to adhere for 15 min at 37°C and 5% CO2. Nonadherent bacteria were removed by repeated washing, and fluorescent images were acquired with a 5× objective lens, using a Zeiss Axiovert 40 inverted microscope with appropriate fluorescent filters and a charge-coupled device (CCD) camera.
Isolation of human neutrophils.
Venous blood was drawn from healthy volunteers under institutional review board approval, using Vacutainers containing EDTA. Neutrophils were isolated by density gradient centrifugation using Polymorphprep solution (Axis Shield PoC AS, Oslo, Norway) according to the manufacturer's instructions. The neutrophil layer was washed with pyrogen-free PBS without Ca2+ and Mg2+ at 4°C; contaminating erythrocytes were hypotonically lysed. Subsequently, neutrophils were washed twice and finally resuspended in Hanks' balanced salt solution without Ca2+ and Mg2+. Viability of cells exceeded 95% as assessed by trypan blue exclusion.
Human neutrophil and GBS fluorescence microscopy.
FITC-GBS were added to isolated human neutrophils in RPMI 1640 without serum at a multiplicity of infection of 10:1, centrifuged to initiate contact, incubated at 37°C in 5% CO2 for 5 min, and then washed with ice cold PBS plus 1% BSA and spun down at 100 × g for 5 min at 4°C. Neutrophils were fixed using 2% paraformaldehyde in PBS for 10 min at 4°C. Human neutrophil Siglec-9 was labeled using mouse anti-hSiglec-9 antibody Fab fragments at 5 μg/ml in PBS plus 1% BSA for 30 min on ice. Anti-hSiglec-9 Fab fragments were prepared using BD PharMingen mouse anti-hSiglec-9 clone E10-286 and the Pierce ImmunoPure IgG1 Fab and F(ab)2 preparation kit according to the manufacturer's instructions for producing Fab antibody fragments. Neutrophils were washed, goat anti-mouse-phycoerythrin antibody was added in PBS plus 1% BSA, and the cells were incubated at 4°C for 30 min. The cells were washed again and treated with Hoechst stain at a 1:1,000 dilution in PBS for 2 min on ice, and then cells were rewashed and resuspended in 50 μl of PBS plus 1% BSA. Cells were added to glass slides and allowed to dry in the dark at 4°C, and coverslips were mounted using Gelvatol. Images were captured with a DeltaVision Restoration microscope system (Applied Precision Inc., Issaquah, WA) using a Photometrics Sony Coolsnap HQ CCD camera system attached to an inverted, wide-field fluorescence microscope (Nikon TE-200). Optical sections were acquired using a 100× (numerical aperture, 1.4) oil immersion objective in 0.2-μm steps in the z axis, using the attached Applied Precision Inc. motorized stage. The fluorescent markers were excited with a standard mercury arc lamp, and fluorescence was detected using a standard DAPI (4′,6′-diamidino-2-phenylindole)-FITC-rhodamine filter set.
RESULTS
Sialylated GBS bind to many different human Siglecs.
Well-characterized sialylated GBS strains from serotypes Ia, Ib, II, III, V, and VI (CPS structures are shown in Fig. 1) and an Sia-negative serotype III ΔNeuA allelic exchange mutant were tested for their ability to bind to hSiglec-3, -5, -6, -7, -8, -9, -10, and -11. Human Siglec-Fc chimeras were prebound to protein A-coated enzyme-linked immunosorbent assay plates in a high-avidity format, and whole-cell heat-killed FITC-GBS were added to the wells. Each of the GBS serotypes tested was capable of binding to at least one hCD33rSiglec, while the Sia-negative ΔNeuA mutant did not bind to any of the hSiglec constructs tested (Fig. 2). In parallel experiments, similar interactions were observed between live (calcein-AM-labeled) GBS and hSiglec-Fc constructs (data not shown). The representative serotype Ia and Ib WT GBS strains were very effective at binding to many different hSiglecs in this high-avidity assay format. Interestingly, the serotype III strain common in late-onset GBS disease was recognized effectively only by Siglec-9. Furthermore, in contrast to hSiglec-9, to which all GBS serotypes tested bound, not one of the GBS serotypes tested was able to bind to hSiglec-3 or hSiglec-10. Many of the hSiglecs to which GBS bound are located on the cell surfaces of leukocytes involved in innate immunity to bacterial infections. Human neutrophils express Siglec-5 and -9 on their surfaces; human monocytes express Siglec-5, -7, and -9; and human macrophages express Siglec-5 and -11 (1, 14, 25).
FIG. 1.

All GBS serotypes described to date contain a terminal α2,3-linked Sia on their CPSs. The repetitive subunits of the CPS of GBS serotypes Ia, Ib, II, III, and V were previously published as Ia/Ib (18), II (19), III (39), and V (38). The repetitive subunits of each serotype are covalently joined end to end to form the CPS. The structure of the serotype VI repetitive subunit is not known but consists of a 2:2:1 molar ratio of glucose, galactose, and N-acetylneuraminic acid, respectively (36).
FIG. 2.

Binding of various group B Streptococcus serotypes to hCD334rSiglecs. All GBS serotypes bind to at least one hCD33rSiglec, while several serotypes interact with multiple hCD33rSiglecs. FITC-GBS were added to hSiglec-Fc chimera-coated wells, and the efficiency of binding was tested. The values represent the means from at least three separate experiments ± standard deviations.
GBS serotype III binding to hSiglec-9 is Sia specific.
The significant loss of hSiglec-9 binding of the isogenic Sia-negative mutant compared to the WT serotype III parent strain (Fig. 2) suggests a Sia-dependent interaction. Independent verification of Sia dependence was achieved by treatment of WT serotype III FITC-GBS with either trypsin (to cleave bacterial cell surface proteins while leaving the bacterial CPS intact) or an exogenous sialidase (to remove all Sias attached to the surfaces of the bacteria). Consistent with our previous results using the Sia-negative ΔNeuA mutant, the sialidase-treated (“enzymatically Sia-negative”) bacteria had significantly diminished binding to hSiglec-9 (Fig. 3). In contrast, the trypsin-treated serotype III GBS largely retained its ability to bind to hSiglec-9. We thus conclude that serotype III GBS interacts with hSiglec-9 in a Sia-specific manner.
FIG. 3.
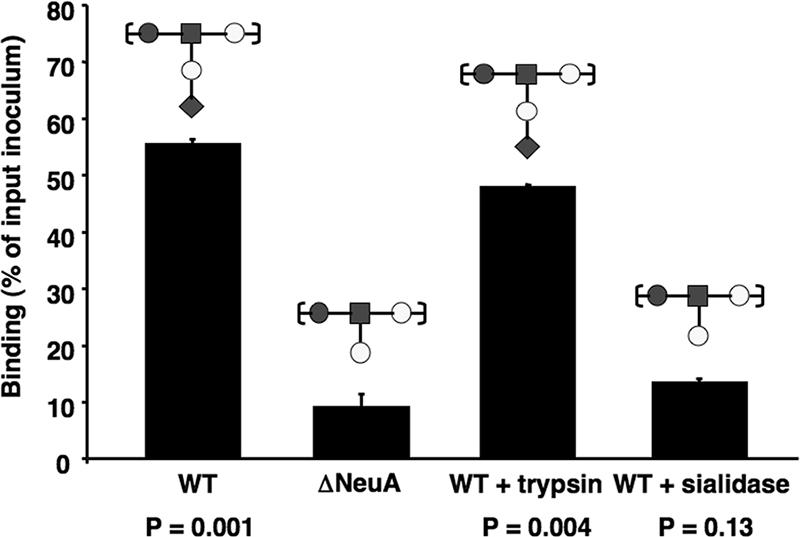
Serotype III GBS engages hSiglec-9 through Sias present on the CPS. FITC-GBS (serotype III) were treated with trypsin in order to degrade bacterial surface proteins or with exogenous sialidase to remove all cell surface sialic acid and tested for their ability to bind hSiglec-9-Fc chimeras in comparison to serotype III ΔNeuA. The graphs show mean percent binding ± standard deviation for one representative experiment that was repeated three times with similar results. P values are from two-tailed t tests.
O acetylation of GBS CPS Sia alters binding to hSiglec-5, -7, and -9.
GBS modifies CPS Sias with O-acetyl groups at carbon positions 7, 8, and 9. It was previously shown that O acetylation of Sias present on synthetic or natural glycan probes affects the binding of some Siglecs (8, 22, 30). To determine whether O acetylation of GBS Sias modifies interaction with hCD33rSiglecs, we utilized an assay format similar to that used for Fig. 2 with three isogenic serotype III strains: (i) the WT parent strain that expresses O acetylation on ∼50% of total Sias; (ii) an isogenic O-acetyltransferase allelic exchange mutant, ΔNeuD, displaying very little Sia and no O acetylation; and (iii) the ΔNeuD mutant transformed with a NeuD expression plasmid harboring a single amino acid mutation in the active site (pNeuD-K123A), which restores sialylation but reduces O acetylation to ∼2% (23). Like the Sia-negative serotype III ΔNeuA mutant, the serotype III ΔNeuD mutant with low Sia expression was also unable to bind to hSiglec-5, -7, and -9 (Fig. 4) or any of the other hCD33rSiglecs tested (data not shown). The restoration of CPS sialylation with greatly diminished O acetylation (complemented strain ΔNeuD plus pNeuD-K123A) altered the patterns of binding to hSiglec-5, -7, and -9 in comparison to that in the WT parent GBS. In the case of hSiglec-5 and -7, removal of bacterial O acetylation increased bacterial binding. However, in the case of hSiglec-9, the removal of O acetylation from CPS Sias had the opposite effect, such that binding of the bacterium was inhibited. All other hCD33rSiglecs tested showed no significant change in binding affinity between the WT highly O-acetylated serotype III GBS parent strain and its low-O-acetylated derivative ΔNeuD with pNeuD-K123A (data not shown).
FIG. 4.
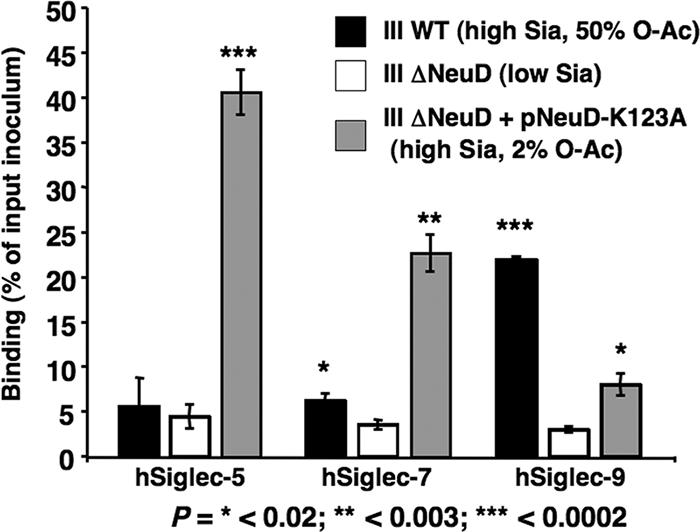
Binding of WT serotype III GBS, its isogenic ΔNeuD mutant, and the ΔNeuD mutant complemented with pNeuD-K123A to hSiglec-5, -7, and -9. Values represent the means from three separate experiments ± standard deviations. P values are from two-tailed t tests comparing each sialylated GBS group with the nonsialylated negative control. O-Ac, O acetylation.
GBS engages CHO cells expressing hSiglec-9 or hSiglec-5 in a Sia- and Siglec-specific manner.
To determine if the binding patterns described above were representative of GBS interactions with cell surface-expressed hCD33rSiglecs, we stably transfected CHO cells with vectors expressing hSiglec-5 and -9 and compared adherence of sialylated and nonsialylated serotype III FITC-GBS by fluorescence microscopy. A low background level of either sialylated or nonsialylated GBS adhered to the surface of untransfected CHO cells (Fig. 5A, first column). Likewise, the nonsialylated ΔNeuA mutant GBS had a relatively low level of adherence to CHO cells whether or not hSiglecs were expressed (Fig. 5A, first row). In contrast, the WT sialylated serotype Ia and III strains had greatly increased interaction with CHO cells expressing specific hSiglecs compared to the untransfected CHO cells (Fig. 5A, second and third rows). As observed using the in vitro binding assays, the serotype Ia GBS strain engaged CHO cells expressing either hSiglec-5 or -9, while the serotype III GBS strain bound only CHO cells expressing hSiglec-9. To verify that this interaction reflects an engagement of the bacterial Sia with the Sia-binding V-set domain of hSiglec-9, we performed additional studies with Sia-blocking and Sia-nonblocking monoclonal antibodies to hSiglec-9. Binding of WT serotype III GBS was inhibited by the addition of monoclonal antibodies that block the Sia-binding pocket of hSiglec-9. In contrast, when Sia-nonblocking antibodies against hSiglec-9 were added, no inhibition of GBS binding was observed (Fig. 5B). Similar results were found in assays using the serotype Ia GBS WT strain (data not shown). Therefore, serotype III GBS interact with cell surface-expressed hSiglec-9 in a manner dependent on both bacterial Sia and the ability of hSiglec-9 to interact with Sia. The results obtained from our in vitro hSiglec-Fc binding studies closely parallel those obtained using full-length hSiglecs expressed on the surfaces of cells, indicating that our in vitro system represents a useful technique for assessing bacterial binding to hCD33rSiglecs.
FIG. 5.

Sialylated GBS adheres to CHO cells expressing hSiglecs in a Sia- and Siglec-specific manner. (A) FITC-labeled serotype III Sia-negative ΔNeuA (top row), sialylated WT serotype Ia (middle row), and sialylated WT serotype III (bottom row) GBS were allowed to adhere to untransfected CHO cells (first column) or to CHO cells expressing either hSiglec-9 (second column) or hSiglec-5 (third column). (B) Adherence of sialylated serotype III FITC-GBS to CHO cells expressing hSiglec-9 in the presence or absence of 10 μg/ml hSiglec-9-specific Sia-blocking or Sia-nonblocking antibodies. All images were captured using an inverted microscope with an appropriate fluorescent filter set and CCD digital camera. Images are representative of three separate experiments with similar results.
Sialylated GBS interacts with hSiglec-9 present on the neutrophil surface.
Neutrophils play an important role in immune defense against GBS infections (16). These specialized leukocytes also express relatively high levels of Siglec-9 and Siglec-5 on their surfaces. To test the ability of GBS to engage Siglecs on the surfaces of purified human neutrophils, polymorphonuclear leukocytes were incubated with sialylated FITC-GBS at 37°C for 5 min, followed by washing with 4°C PBS plus 1% BSA (to prevent internalization). Neutrophil-GBS interactions were visualized using fluorescence deconvolution microscopy with an hSiglec-9 specific Fab antibody fragment and a fluorescently tagged secondary antibody to label Siglec-9. Punctate staining of hSiglec-9 on the surfaces of human neutrophils was observed in the presence or absence of bacteria (Fig. 6). When sialylated GBS was allowed to interact with the neutrophil surface, clear foci of colocalization between hSiglec-9 staining (red) and GBS (green) were observed (Fig. 6).
FIG. 6.

GBS attached to the surface of human neutrophils colocalizes with hSiglec-9. FITC-GBS were added to isolated purified human neutrophils and allowed to interact for 5 min, after which cells were fixed and hSiglec-9 was labeled using fluorescent antibodies. Cells were mounted onto slides, and images were acquired using DeltaVision deconvolution microscopy and a CCD camera. Three images representing the nucleus (A) (blue Hoechst stain), bacteria (B) (green FITC), and hSiglec-9 (C) (red phycoerythrin) are shown separately and then combined in one merged image (D). The areas of colocalization between FITC-GBS (green) and hSiglec-9 (red) appear yellow in the merged image. The experiment was repeated three times, and this image is representative of many GBS-neutrophil interactions.
DISCUSSION
Group B Streptococcus is the leading bacterial pathogen associated with neonatal sepsis and meningitis (12). The adaptive immune system of neonates, especially preterm neonates, is impaired in its ability to produce a significant immunoglobulin response to an infection with an encapsulated bacterium such as GBS (6, 16). The neonate is thus very dependent on its innate immune system to provide protection from GBS infection and disease in concert with passive antibody acquired from the mother. In humans, neutrophils and monocytes/macrophages are important elements of the innate immune defense against invasive bacterial pathogens. These cells express different members of a family of cell surface Sia-recognizing receptors called the CD33rSiglecs (neutrophils, Siglec-5 and Siglec-9; monocytes, Siglec-5, Siglec-7, and Siglec-9; and macrophages, Siglec-5 and Siglec-11) that are capable of sending inhibitory signals to the cells to modulate inflammatory responses.
In this study, we asked whether sialylated GBS engages hCD33rSiglecs that are present on human neutrophils and monocytes/macrophages. We show that all GBS serotypes interact with at least one hCD33rSiglec and that some serotypes engage multiple hCD33rSiglecs. Serotypes Ia and Ib were the most promiscuous, engaging the largest number of hCD33rSiglecs with the greatest strength, whereas serotype II appears to be the least effective binder. We further demonstrate that GBS CPS Sia can engage hSiglec-9 as expressed on the surfaces of CHO cells and human neutrophils. Each serotype tested displays the same terminal α2,3 linkage of Sia; however, due to the way in which the repetitive subunit is polymerized into the CPS, types Ia and Ib will contain the highest density of Sias over a given length of CPS and serotype II will have the fewest (one Sia per two monosaccharides for Ia and Ib versus one Sia per five monosaccharides for II [Fig. 1]). Most glycan-protein interactions have low binding affinities compared to protein-protein interactions, and thus the capacity of types Ia and Ib to form dense arrays of Sias, in addition to having the proper binding epitope, may be essential for creating sufficient avidity for strong binding.
Careful analysis of the specificity of the serotype III GBS interaction with hSiglecs revealed that biochemical and genetic perturbation of bacterial sialylation or Sia O acetylation altered Siglec binding. Cell-based assays confirmed and extended the in vitro studies, showing that interactions between hSiglec-9-transfected CHO cells and GBS required both bacterial sialylation and accessibility of the hSiglec-9 Sia-binding domain for interaction with type III GBS. Furthermore, the presence of O acetylation of CPS Sias altered the binding of GBS to hSiglec-5, -7, and -9. In the context of the GBS CPS, O acetylation blocks binding to hSiglec-5 and -7 while promoting binding to hSiglec-9. Further studies are needed to determine the precise mechanism(s) by which O acetylation modifies hSiglec-GBS binding. It may be that O-acetyl modification of GBS CPS Sias directly changes the binding affinity of the V-set domains for the Sias or, alternatively, alters the three-dimensional architecture of the CPS (21), with secondary effects on the GBS-hSiglec binding interaction.
CD33rSiglecs contain an ITIM and an ITIM-like motif that can be phosphorylated and recruit Src homology phosphatases SHP-1 and SHP-2, which are thought to play a role in downregulation of cell activation (4, 17). Numerous in vitro studies have shown the capacity of CD33rSiglecs to act in an inhibitory fashion, including negative regulation of T-cell signaling (17) and inhibition of NK cell toxicity (27). Therefore, it is possible that the GBS CPS serotypes have evolved to engage hCD33rSiglecs on neutrophils and monocytes/macrophages in order to exert suppressive effects upon the innate immune response. Furthermore, hSiglec-9 can activate accelerated apoptosis in human neutrophils (35), an additional mechanism for avoidance of phagocytic killing. Conversely, it has also recently been shown that many CD33rSiglecs, including hSiglec-9, can act as endocytic receptors (7) that could theoretically aid in the internalization of bound GBS. Thus, evolutionary forces modulating hSiglec-GBS interactions could be operative from both the pathogen and host perspectives.
Sia expression by GBS and other pathogens plays an important role in resistance to host complement-mediated killing, perhaps through binding of host factor H to impair complement deposition on the bacterial surface (3, 26, 37). In addition to the data we present here supporting the interaction between GBS CPS Sias and multiple CD33rSiglecs, it was recently shown that hSiglec-5 expressed on CHO cells can bind to sialylated Neisseria meningitides (20) and that hSiglec-7 present on dendritic cells can bind to an isolate of sialylated Campylobacter jejuni (5). Therefore, it may be that GBS and other bacteria have evolved a shared virulence mechanism of decorating their surfaces with a human-like monosaccharide, the Sia Neu5Ac, which acts as a ligand for both factor H and hCD33rSiglecs. In this fashion, the Sia-expressing bacterial pathogens could simultaneously interfere with complement and cellular components of innate immunity. In the particular case of GBS, clinical disease is seen in the neonate and other special populations, including pregnant women, the elderly, and diabetics (13, 28). Future studies could examine both the patterns of hCD33rSiglec expression and the cellular responses mediated by these receptors in these populations for correlations with increased susceptibility to GBS infection.
Acknowledgments
We thank Nancy Hurtado-Ziola and Pascal Lanctot for the production of the human Siglec-Fc chimeras. We thank Kersi Pestonjamasp and Arrate Mallabiabarrena for their work in connection with the UCSD Cancer Center Digital Imaging Shared Resource, and we thank Judy Nordberg for work in conjunction with the Flow Cytometry Research Core Facility of the VA San Diego Healthcare System, the San Diego Center for AIDS Research, and the VA Research Center for AIDS and HIV Infection.
The San Diego Center for AIDS Research is supported by NIH grant AI 36214. This work was supported by NIH awards R01-HD051796 (to V.N.) and P01-HL57345 (to A.V.) and by an American Heart Association Established Investigator Award (to V.N.).
Footnotes
Published ahead of print on 22 September 2006.
REFERENCES
- 1.Angata, T., S. C. Kerr, D. R. Greaves, N. M. Varki, P. R. Crocker, and A. Varki. 2002. Cloning and characterization of human Siglec-11. A recently evolved signaling that can interact with SHP-1 and SHP-2 and is expressed by tissue macrophages, including brain microglia. J. Biol. Chem. 277:24466-24474. [DOI] [PubMed] [Google Scholar]
- 2.Angata, T., and A. Varki. 2000. Cloning, characterization, and phylogenetic analysis of siglec-9, a new member of the CD33-related group of siglecs. Evidence for co-evolution with sialic acid synthesis pathways. J. Biol. Chem. 275:22127-22135. [DOI] [PubMed] [Google Scholar]
- 3.Aoyagi, Y., E. E. Adderson, J. G. Min, M. Matsushita, T. Fujita, S. Takahashi, Y. Okuwaki, and J. F. Bohnsack. 2005. Role of l-ficolin/mannose-binding lectin-associated serine protease complexes in the opsonophagocytosis of type III group B streptococci. J. Immunol. 174:418-425. [DOI] [PubMed] [Google Scholar]
- 4.Avril, T., H. Floyd, F. Lopez, E. Vivier, and P. R. Crocker. 2004. The membrane-proximal immunoreceptor tyrosine-based inhibitory motif is critical for the inhibitory signaling mediated by Siglecs-7 and -9, CD33-related Siglecs expressed on human monocytes and NK cells. J. Immunol. 173:6841-6849. [DOI] [PubMed] [Google Scholar]
- 5.Avril, T., E. R. Wagner, H. J. Willison, and P. R. Crocker. 2006. Sialic acid-binding immunoglobulin-like lectin 7 mediates selective recognition of sialylated glycans expressed on Campylobacter jejuni lipooligosaccharides. Infect. Immun. 74:4133-4141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bauer, K., M. Zemlin, M. Hummel, S. Pfeiffer, J. Karstaedt, G. Steinhauser, X. Xiao, H. Versmold, and C. Berek. 2002. Diversification of Ig heavy chain genes in human preterm neonates prematurely exposed to environmental antigens. J. Immunol. 169:1349-1356. [DOI] [PubMed] [Google Scholar]
- 7.Biedermann, B., D. Gil, D. T. Bowen, and P. R. Crocker. 2007. Analysis of the CD33-related siglec family reveals that Siglec-9 is an endocytic receptor expressed on subsets of acute myeloid leukemia cells and absent from normal hematopoietic progenitors. Leuk. Res. 31:211-220. [DOI] [PubMed] [Google Scholar]
- 8.Brinkman-Van der Linden, E. C., and A. Varki. 2000. New aspects of siglec binding specificities, including the significance of fucosylation and of the sialyl-Tn epitope. Sialic acid-binding immunoglobulin superfamily lectins. J. Biol. Chem. 275:8625-8632. [DOI] [PubMed] [Google Scholar]
- 9.Chaffin, D. O., L. M. Mentele, and C. E. Rubens. 2005. Sialylation of group B streptococcal capsular polysaccharide is mediated by cpsK and is required for optimal capsule polymerization and expression. J. Bacteriol. 187:4615-4626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cieslewicz, M. J., D. Chaffin, G. Glusman, D. Kasper, A. Madan, S. Rodrigues, J. Fahey, M. R. Wessels, and C. E. Rubens. 2005. Structural and genetic diversity of group B streptococcus capsular polysaccharides. Infect. Immun. 73:3096-3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crocker, P. R. 2005. Siglecs in innate immunity. Curr. Opin. Pharmacol. 5:431-437. [DOI] [PubMed] [Google Scholar]
- 12.Doran, K. S., and V. Nizet. 2004. Molecular pathogenesis of neonatal group B streptococcal infection: no longer in its infancy. Mol. Microbiol. 54:23-31. [DOI] [PubMed] [Google Scholar]
- 13.Edwards, M. S., M. A. Rench, D. L. Palazzi, and C. J. Baker. 2005. Group B streptococcal colonization and serotype-specific immunity in healthy elderly persons. Clin. Infect. Dis. 40:352-357. [DOI] [PubMed] [Google Scholar]
- 14.Floyd, H., J. Ni, A. L. Cornish, Z. Zeng, D. Liu, K. C. Carter, J. Steel, and P. R. Crocker. 2000. Siglec-8. A novel eosinophil-specific member of the immunoglobulin superfamily. J. Biol. Chem. 275:861-866. [DOI] [PubMed] [Google Scholar]
- 15.Haft, R. F., and M. R. Wessels. 1994. Characterization of CMP-N-acetylneuraminic acid synthetase of group B streptococci. J. Bacteriol. 176:7372-7374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Henneke, P., and R. Berner. 2006. Interaction of neonatal phagocytes with group B streptococcus: recognition and response. Infect. Immun. 74:3085-3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ikehara, Y., S. K. Ikehara, and J. C. Paulson. 2004. Negative regulation of T cell receptor signaling by Siglec-7 (p70/AIRM) and Siglec-9. J. Biol. Chem. 279:43117-43125. [DOI] [PubMed] [Google Scholar]
- 18.Jennings, H. J., E. Katzenellenbogen, C. Lugowski, and D. L. Kasper. 1983. Structure of native polysaccharide antigens of type Ia and type Ib group B Streptococcus. Biochemistry 22:1258-1264. [DOI] [PubMed] [Google Scholar]
- 19.Jennings, H. J., K. G. Rosell, E. Katzenellenbogen, and D. L. Kasper. 1983. Structural determination of the capsular polysaccharide antigen of type II group B Streptococcus. J. Biol. Chem. 258:1793-1798. [PubMed] [Google Scholar]
- 20.Jones, C., M. Virji, and P. R. Crocker. 2003. Recognition of sialylated meningococcal lipopolysaccharide by siglecs expressed on myeloid cells leads to enhanced bacterial uptake. Mol. Microbiol. 49:1213-1225. [DOI] [PubMed] [Google Scholar]
- 21.Kadirvelraj, R., J. Gonzalez-Outeirino, B. L. Foley, M. L. Beckham, H. J. Jennings, S. Foote, M. G. Ford, and R. J. Woods. 2006. Understanding the bacterial polysaccharide antigenicity of Streptococcus agalactiae versus Streptococcus pneumoniae. Proc. Natl. Acad. Sci. USA 103:8149-8154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kelm, S., R. Brossmer, R. Isecke, H. J. Gross, K. Strenge, and R. Schauer. 1998. Functional groups of sialic acids involved in binding to siglecs (sialoadhesins) deduced from interactions with synthetic analogues. Eur. J. Biochem. 255:663-672. [DOI] [PubMed] [Google Scholar]
- 23.Lewis, A. L., M. E. Hensler, A. Varki, and V. Nizet. 2006. The group B streptococcal sialic acid O-acetyltransferase is encoded by neuD, a conserved component of bacterial sialic acid biosynthetic gene clusters. J. Biol. Chem. 281:11186-11192. [DOI] [PubMed] [Google Scholar]
- 24.Lewis, A. L., V. Nizet, and A. Varki. 2004. Discovery and characterization of sialic acid O-acetylation in group B Streptococcus. Proc. Natl. Acad. Sci. USA 101:11123-11128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lock, K., J. Zhang, J. Lu, S. H. Lee, and P. R. Crocker. 2004. Expression of CD33-related siglecs on human mononuclear phagocytes, monocyte-derived dendritic cells and plasmacytoid dendritic cells. Immunobiology 209:199-207. [DOI] [PubMed] [Google Scholar]
- 26.Marques, M. B., D. L. Kasper, M. K. Pangburn, and M. R. Wessels. 1992. Prevention of C3 deposition by capsular polysaccharide is a virulence mechanism of type III group B streptococci. Infect. Immun. 60:3986-3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nicoll, G., T. Avril, K. Lock, K. Furukawa, N. Bovin, and P. R. Crocker. 2003. Ganglioside GD3 expression on target cells can modulate NK cell cytotoxicity via siglec-7-dependent and -independent mechanisms. Eur. J. Immunol. 33:1642-1648. [DOI] [PubMed] [Google Scholar]
- 28.Palazzi, D. L., M. A. Rench, M. S. Edwards, and C. J. Baker. 2004. Use of type V group B streptococcal conjugate vaccine in adults 65-85 years old. J. Infect. Dis. 190:558-564. [DOI] [PubMed] [Google Scholar]
- 29.Ravetch, J. V., and L. L. Lanier. 2000. Immune inhibitory receptors. Science 290:84-89. [DOI] [PubMed] [Google Scholar]
- 30.Sjoberg, E. R., L. D. Powell, A. Klein, and A. Varki. 1994. Natural ligands of the B cell adhesion molecule CD22 beta can be masked by 9-O-acetylation of sialic acids. J. Cell Biol. 126:549-562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Suryanti, V., A. Nelson, and A. Berry. 2003. Cloning, over-expression, purification, and characterisation of N-acetylneuraminate synthase from Streptococcus agalactiae. Protein Expr. Purif. 27:346-356. [DOI] [PubMed] [Google Scholar]
- 32.Vann, W. F., D. A. Daines, A. S. Murkin, M. E. Tanner, D. O. Chaffin, C. E. Rubens, J. Vionnet, and R. P. Silver. 2004. The NeuC protein of Escherichia coli K1 is a UDP N-acetylglucosamine 2-epimerase. J. Bacteriol. 186:706-712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Varki, A., and T. Angata. 2006. Siglecs—the major subfamily of I-type lectins. Glycobiology 16:1R-27R. [DOI] [PubMed] [Google Scholar]
- 34.Vely, F., S. Olivero, L. Olcese, A. Moretta, J. E. Damen, L. Liu, G. Krystal, J. C. Cambier, M. Daeron, and E. Vivier. 1997. Differential association of phosphatases with hematopoietic co-receptors bearing immunoreceptor tyrosine-based inhibition motifs. Eur. J. Immunol. 27:1994-2000. [DOI] [PubMed] [Google Scholar]
- 35.von Gunten, S., S. Yousefi, M. Seitz, S. M. Jakob, T. Schaffner, R. Seger, J. Takala, P. M. Villiger, and H. U. Simon. 2005. Siglec-9 transduces apoptotic and nonapoptotic death signals into neutrophils depending on the proinflammatory cytokine environment. Blood 106:1423-1431. [DOI] [PubMed] [Google Scholar]
- 36.von Hunolstein, C., S. D'Ascenzi, B. Wagner, J. Jelinkova, G. Alfarone, S. Recchia, M. Wagner, and G. Orefici. 1993. Immunochemistry of capsular type polysaccharide and virulence properties of type VI Streptococcus agalactiae (group B streptococci). Infect. Immun. 61:1272-1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wessels, M. R., P. Butko, M. Ma, H. B. Warren, A. L. Lage, and M. C. Carroll. 1995. Studies of group B streptococcal infection in mice deficient in complement component C3 or C4 demonstrate an essential role for complement in both innate and acquired immunity. Proc. Natl. Acad. Sci. USA 92:11490-11494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wessels, M. R., J. L. DiFabio, V. J. Benedi, D. L. Kasper, F. Michon, J. R. Brisson, J. Jelinkova, and H. J. Jennings. 1991. Structural determination and immunochemical characterization of the type V group B Streptococcus capsular polysaccharide. J. Biol. Chem. 266:6714-6719. [PubMed] [Google Scholar]
- 39.Wessels, M. R., V. Pozsgay, D. L. Kasper, and H. J. Jennings. 1987. Structure and immunochemistry of an oligosaccharide repeating unit of the capsular polysaccharide of type III group B Streptococcus. A revised structure for the type III group B streptococcal polysaccharide antigen. J. Biol. Chem. 262:8262-8267. [PubMed] [Google Scholar]