

Abstract
Cell-based hepatitis C virus (HCV) replicon systems have provided a means for understanding HCV replication mechanisms and for testing new antiviral agents. We describe here a mathematical model of HCV replication that assumes that the translation of the HCV polyprotein occurs in the cytoplasm, that HCV RNA synthesis occurs in vesicular-membrane structures, and that the strategy of replication involves a double-stranded RNA intermediate. Our results shed light on the intracellular dynamics of subgenomic HCV RNA replication from transfection to steady state within Huh-7 cells. We predict the following: (i) about 6 × 103 ribosomes are involved in generating millions of HCV NS5B-polymerase molecules in a Huh-7 cell, (ii) the observed 10:1 asymmetry of plus- to minus-strand RNA levels can be explained by a higher-affinity (200-fold) interaction of HCV NS5B polymerase-containing replication complexes with HCV minus-strand RNA over HCV plus-strand RNA in order to initiate synthesis, (iii) the latter higher affinity can also account for the observed ∼6:1 plus-strand/minus-strand ratio in vesicular-membrane structures, and (iv) the introduction of higher numbers of HCV plus-strand RNA by transfection leads to faster attainment of steady-state but does not change the steady-state HCV RNA level. Fully permissive HCV replication systems have been developed, and the model presented here is a first step toward building a comprehensive model for complete HCV replication. Moreover, the model can serve as an important tool in understanding HCV replication mechanisms and should prove useful in designing and evaluating new antivirals against HCV.
About 200 million people, roughly 3% of the human population, are infected with hepatitis C virus (HCV) (61). Chronic HCV infection is the main cause of chronic liver disease and cirrhosis, leading to liver transplantation or death (3, 47). State-of-the-art therapy (peg-interferon and ribavirin) elicits long-term responses in only about 50% of treated patients (18, 37, 41), with no effective alternative treatment for nonresponders (56).
Progress toward developing model systems of HCV infection that could enhance efforts to identify inhibitors of HCV replication has been hampered by HCV's limited replication in cell culture and the lack of small animal models (32). In 1999 Lohmann et al. (35) engineered a bicistronic subgenomic HCV replicon system in Huh-7 cells. Since then this system, improved substantially both in Huh-7 cells (34) and in other cell lines (64), has become the standard cell-based assay to study HCV replication mechanisms and to evaluate antiviral agents (51).
The first studies of positive-strand RNA virus replication were done with RNA bacteriophages, e.g., Qβ and MS2 (54). These studies showed that viral RNA amplification depended on an RNA-dependent RNA polymerase-containing RNA replicase that specifically interacts with the incoming viral RNA (plus strand) to synthesize its complementary (minus) strand. Once the minus-strand RNA is synthesized, the amplification of the viral RNA by the replicase begins. Based on these systems, Biebricher et al. (6) quantitatively monitored the kinetics of RNA amplification by Qβ replicase and developed a kinetic model for self-replication of Qβ RNA in vitro (5). The full life cycle of Qβ has been mathematically modeled (16) and provides an important starting point for developing intracellular HCV replication models.
HCV is an enveloped positive-strand RNA virus belonging to the genus Hepacivirus in the family Flaviviridae (32). After HCV enters a cell, the HCV genome is translated, by host ribosomes, into a large polyprotein, about 3,000 amino acids long, that is processed into structural and nonstructural (NS) proteins. A multiprotein viral replicase is assembled from the NS proteins (35) and begins the synthesis of a minus-strand RNA using the positive single-strand RNA (ssRNA) as a template. Once the minus-strand RNA is synthesized, it can remain as a free minus ssRNA or be attached to the positive-strand RNA to form a double-stranded RNA (dsRNA). The newly synthesized minus-strand RNA (either as ssRNA or as part of the dsRNA) then serves as a template for the synthesis of additional plus-strand RNAs. It has not yet been determined whether HCV RNA in cell culture (or in vivo) is replicated using as a template the minus-strand RNA in a dsRNA form, as established for the Kunjin virus (10), or as a ssRNA template, as shown for the Qβ phage (16). In the present study, we make the assumption that after the first minus strand is replicated, amplification of viral RNA occurs via a double-stranded template.
All positive-strand RNA viruses replicate their RNA on intracellular membranes, often in association with spherular invaginations of the target membrane (reviewed in reference 53). RNA replication by Kunjin virus, coronaviruses, brome mosaic virus, and poliovirus induces distinct membrane rearrangements of 50 to 350 nm, including invaginations, double-membrane vesicles, and layered membranes, that serve as compartments or miniorganelles for RNA replication (53). Membrane association of the HCV NS5B polymerase was found to be essential for HCV RNA replication (15, 39), and HCV RNA replication complexes colocalize with vesicular-membrane structures (VMS) that have also been termed “the membranous web” (15, 22). Once formed, these membrane structures appear to be relatively stable, with only limited movement and exchange of viral NS proteins (60a).
The experimental characterization of HCV self-amplification in the replicon system in Huh-7 cells reveals that (i) by 24 to 72 h posttransfection, plus-strand RNAs accumulate to ∼5,000 copies per cell (8, 30, 34, 35, 50); (ii) the plus-strand/minus-strand ratio is about 10:1 (35, 50), which is in agreement with the plus-strand/minus-strand ratio observed in infected hepatocytes in humans (9, 27); (iii) the in vitro replicase activity, prepared from Huh-7 cells harboring subgenomic replicons, is highly resistant to nuclease and protease treatment, with both plus- and minus-strand RNAs being fully nuclease resistant (50); and (iv) <5% of the NS5B polymerase molecules are protease resistant (38, 50), suggesting that the majority of replication occurs in sites, such as the VMS, that are protected from nuclease and protease activity, and where a minority of NS5B polymerase may reside.
Recently, we have developed mathematical models to gain insight into HCV RNA dynamics during primary infection (13), liver transplantation (12, 46), alpha interferon (IFN-α) monotherapy (40), and IFN-α and ribavirin combination therapy (14). However, these models were not designed to gain insight into HCV minus- and plus-strand kinetics within the cell. Based on quantitative data for subgenomic HCV replication in Huh-7 cells, we sought to gain a better understanding of its dynamics from transfection to steady state using a mathematical model.
MATERIALS AND METHODS
Model description.
Our model of subgenomic HCV replication in Huh-7 cells is based on the HCV replication scheme shown in Fig. 1. We assume that translation of the HCV polyprotein occurs in the cytoplasm by host ribosomes (equations 1 to 4), whereas HCV replication (equations 5 to 9) occurs in VMS.
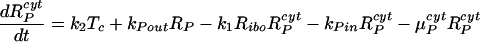 |
(1) |
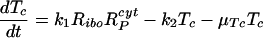 |
(2) |
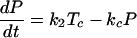 |
(3) |
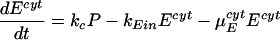 |
(4) |
RPcyt, Tc, P, and Ecyt represent the numbers (in the cytoplasm) of plus-strand HCV RNA molecules, translation complexes, HCV polyprotein molecules, and the enzyme NS5B and associated viral proteins needed for HCV RNA synthesis, respectively. Plus-strand RNA, RPcyt, interacts with host cell ribosomes, Ribo, at an effective rate k1 to form a translation complex, Tc, which degrades at rate μTc. For simplicity, we assume that 10 ribosomes simultaneously translate the same HCV mRNA (59), and Ribo represents a complex of 10 ribosomes that interacts concomitantly with RPcyt to initiate translation. In the Appendix we present a more detailed model that considers the sequential attachment of ribosomes to the HCV mRNA. Viral-polyprotein translation takes an average time of 1/k2, and ribosomes dissociate when the translation of a polyprotein, P, is complete. We assume that the number of ribosome complexes available for HCV RNA translation in the cell (RiboTot) is constant and is a fraction of the total pool of cellular ribosomes. Thus, the number of free ribosome complexes involved in HCV RNA translation is calculated as follows: Ribo = RiboTot − Tc. Free plus-strand RNA molecules, RPcyt, disappear at rate k1 by forming polysomes and reappear at rate k2 when translation is complete. Free plus-strand RNAs in the cytoplasm can be degraded by nucleases at rate μPcyt, lost from the cytoplasm by transport into VMS at rate kPin, and gained by transport out of the VMS at rate kPout. We assume that free viral polyprotein molecules, P, are generated by translation within the cytoplasm at rate k2 per translation complex and are cleaved into separate viral proteins, including the enzymes responsible for HCV RNA synthesis, Ecyt, e.g., NS5B, at rate kc. Lastly, we assume these enzymes may be degraded before reaching the VMS at rate μEcyt, or transported into VMS at rate kEin.
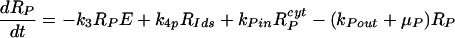 |
(5) |
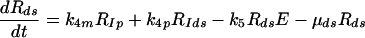 |
(6) |
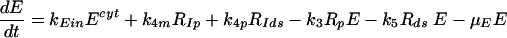 |
(7) |
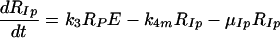 |
(8) |
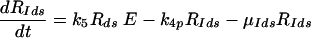 |
(9) |
Equations 5 to 9 describe the kinetics of HCV RNA replication within the VMS, with RP, Rds, and E representing the numbers of plus-strand RNA, dsRNA, and HCV polymerase complexes, respectively, within the VMS. RIp and RIds represent the numbers of plus-strand RNA and dsRNA replicative intermediate complexes, respectively. The RIp complex is composed of a plus strand that serves as a template for the newly synthesized minus-strand RNA, the replication machinery (E), and the nascent complementary minus-strand RNA. The RIds complex contains dsRNA in which the minus strand serves as a template for the newly synthesized plus-strand RNA, the replication machinery E, and the nascent complementary plus-strand RNA. We assume that once the synthesis of the minus strand has ended, the replication complex, RIp, immediately dissociates into dsRNA (consisting of plus- and minus-strand RNAs), Rds, and the replication machinery E. Finally, when the synthesis of the nascent plus strand has ended, the replication complex, RIds, immediately dissociates into three components: the unwound plus strand, the replication machinery (E), and dsRNA. De novo formation of RIp and RIds complexes in VMS occur with rate constants k3 and k5, respectively, and degrade with rate constants μIp and μIds. Synthesis of the nascent plus- and minus-strand RNAs occurs with rate constants k4p and k4m, respectively. Free plus-strand RNA and dsRNA in VMS (RP and Rds) degrade with rate constants μP and μds, respectively. In addition, the polymerase complex degrades or loses activity with rate constant μE within VMS, which is possibly less than the cytoplasmic degradation rate μEcyt. We assume that there are abundant nucleotides and amino acids in the cell so that cellular resources do not limit HCV RNA replication (55). We also assume that if any cellular components are needed to form the replication machinery (E), they, too, are abundant. This should be the case in the stage of subgenomic HCV replication being considered here, when Huh-7 cells are replicating and have not yet reached confluence.
FIG. 1.

Schematic model of subgenomic HCV replication in Huh-7 cells. HCV replication starts once plus-strand RNA, RPcyt, enters the cell during transfection. The plus strand interacts with ribosome complexes to form the translation complex (Tc) with rate constant k1. Once Tc is formed, translation begins and the viral polyprotein (P) is produced at rate k2. After the polyprotein is produced, we assume the ribosome complex dissociates from Tc, leading to a free plus-strand RNA. The resulting polyprotein is cleaved with rate constant kc into separate viral proteins, including the NS5B polymerase (Ecyt) (containing RNA replicase) that is transported into the VMS at rate kEin. The plus-strand RNAs in the cytoplasm (RPcyt) are transported into VMS with rate constant kPin and out of VMS with rate constant kPout. Within the VMS, the association of plus-strand RNA (RP) and NS5B (E) result in the formation of the plus-strand replicative intermediate complex (RIp) that occurs at rate k3RP E. The complementary minus-strand RNA is then formed with rate constant k4m, and the RIp complex is dissociated to dsRNA (Rds) and NS5B polymerase (E). Finally, when dsRNA is present, the formation of the dsRNA replicative intermediate (RIds) occurs at rate k5Rds E and replicates nascent plus-strand RNA at rate k4p per complex. Once the full nascent plus RNA is replicated, the unwound plus RNA is released from the RIds complex (along with Rds and E). Ribosomes are indicated as open black squares, plus-strand RNAs are indicated as black lines, minus-strand RNAs are indicated as dotted black lines, NS5B polymerase are indicated as open black ovals, plus-strand polymerase intermediate (RIp) and dsRNA replicative intermediate (RIds) complexes are colored green. Kinetic rates k1 to k5, kPin, kPout, kc, and kEin are indicated as black arrows. Degradation rates of the NS5B polymerase (in cytoplasm [μEcyt] and in VMS [μE]), plus and dsRNA replicative-intermediate complexes (μIp and μIds, respectively), plus-strand RNAs (in cytoplasm [μPcyt] and in VMS [μP]), translation complex (μTc), and dsRNAs (μds) are indicated as blue arrows. Red arrows represent the dissociation of the replicative-intermediate complexes (RIp and RIds) and translation complexes (Tc) immediately after the full synthesis of the nascent RNA strands and viral polyprotein is finished. The vesicular-membrane structures observed in Huh-7 cells (15, 22), are represented here by just one VMS marked with a large dotted pink oval. The VMS size is not drawn to scale in relation to the Huh-7 cell.
Simulation procedure.
We have computed the solutions to equations 1 to 9 numerically using the Rosenbrock algorithm for solving stiff differential equations, implemented in C++ (48). The model was solved 1,000 times with different (randomly chosen) parameter sets in each run. The results of each run were stored and analyzed for consistency with the biological criteria discussed below. At the time of transfection (t = 0), we assume that RPcyt(0) ≥ 1 replication-competent plus-strand RNAs are introduced into a cell and that all other virus variables are zero (Tc = P = E = RIp = RIds = RP = Rds = 0).
The parameters k4p, k4m, k2, μE, μP, and μds were estimated from the literature as described below. The remaining parameters were chosen (Table 1) so the model reaches a steady state with the following experimental characteristics: (i) plus strands are at a level of about 900 to 5,000 copies per cell (23, 35, 50), (ii) the total plus-strand/minus-strand ratio is about 10:1 (23, 35, 50), (iii) the plus-strand/minus-strand ratio in VMS is ∼6:1 (50), (iv) the steady-state is established about 48 h posttransfection (34, 35, 50), (v) the number of NS5B molecules is between 8 × 105 to 2 × 106 per cell (50), and (vi) a small proportion (<5%) of HCV NS proteins expressed in cells harboring HCV replicons are actively engaged in HCV RNA synthesis (38, 50). Thus, we assume, in our model, that the NS5B polymerase molecules in the cytoplasm represent at least 95% of the total NS5B polymerase molecules in a cell with the rest (<5%) being present in VMS. (vii) Moreover, about half of total plus-strand RNAs in a replicon cell are nuclease resistant (50) and thus localize (in our model) to the VMS. The plus-strand RNAs that are present in the cytoplasm are assumed to be involved in viral polyprotein translation.
TABLE 1.
Parameter estimates of subgenomic HCV replication in Huh-7 cellsa
| Rate constantb | Reaction definition | Kinetic rate constants (h−1)c:
|
|
|---|---|---|---|
| From the literature and simulations (half-life [h]) | Based on sensitivity analyses (half-life [h]) | ||
| k1 | Tc formation | 1-100† (molecule−1) | 34-83‡ |
| k2# | Nascent NS polyprotein translation | 100* | 100* |
| kc | Viral polyprotein cleavage | 0.2-1† | 0.4-0.8‡ |
| kPin | RPcyt transport into VMS | 0.2† | 0.2† |
| kPout | RP transport into cytoplasm | 0.2† | 0.2† |
| kEin | Ecyt transport into VMS | 4.0 × 10−6 to 4.0 × 10−5† | 1.2 × 10−5 to 3.3 × 10−5‡ |
| k3 | RIp formation | 0.001-0.02† (molecule−1) | 0.01-0.02‡ |
| k4p# | RP synthesis | 1.7* | 1.7* |
| k4m# | Rds synthesis | 1.7* | 1.7* |
| k5 | RIds formation | k5/k3 = 200† | k5/k3 = 200† |
| μpcyt | RPcyt degradation | 0.06-15.0*† (12-0.05) | 2.9-11.3‡ (0.06-0.2) |
| μp | RP degradation | 0.07* (10) | 0.07* (10) |
| μds | Rds degradation | 0.06* (12) | 0.06* (12) |
| μIp | RIp degradation | 0.01-0.06*† (17-12) | 0.02-0.05‡ |
| μIds | RIds degradation | μIds/kEin = 104† | μIds/kEin = 104† |
| μTc | Tc degradation | 0.001-0.02† (700-35) | 0.004-0.015‡ (46-173) |
| μE | E degradation | 0.001-0.06† (700-12) | 0.01-0.05‡ (14-70) |
| μEcyt | Ecyt degradation | 0.06* (12) | 0.06* (12) |
Abbreviations: Tc, translation complex, which is composed of plus-strand RNA and 10 ribosomes; RPcyt, plus-strand RNA in cytoplasm; RP and Rds, free plus-strand RNA and dsRNA in VMS; RIp and RIds, replicative intermediate complexes (NS5B polymerase and template RNA), where the RP and Rds, respectively, serve as templates; E, NS5B polymerase in VMS; Ecyt, NS5B polymerase in cytoplasm; NS, nonstructural.
#, We assume that once the translation or synthesis processes are done, the Tc, RIp, and RIds complexes dissociate immediately.
*, Obtained from the literature as explained in Materials and Methods; †, obtained from simulation as explained in Results; ‡, obtained from sensitivity analysis as explained in Results. Half-life values are indicated in parentheses.
Polysome size and HCV polyprotein elongation rate.
The elongation rate in eukaryotes has been estimated at three to eight amino acids per second per ribosome (33, 43). At three to eight amino acids per second, the subgenomic HCV polyprotein (∼2,000 amino acids) is translated at a mean rate of 10 (range, 5.4 to 14.4) polyproteins per h per ribosome. Wang et al. (59) have shown that at least eight ribosomes were present on an efficient replicon HCV RNA during translation. As shown in the Appendix, one can use a complex model that includes the sequential attachment of multiple ribosomes to each HCV mRNA. However, we found that it is feasible to simplify the situation and use the model described here when the rate constant for the attachment of the first ribosome to free plus-strand RNA is much lower than the rate constant for subsequent ribosomes to attach. This may be reasonable since the rRNA helicases need first to unwind the secondary structure of the mRNA (25), which then might lead to a faster attachment of the subsequent ribosomes. We thus used the simplified model and fixed the polysome size at 10 ribosomes per HCV mRNA, yielding a subgenomic HCV polyprotein translation rate k2 = 100 polyproteins per h per polysome.
HCV plus- and minus-strand RNA synthesis rate.
HCV RNA has been estimated to be synthesized at approximately 150 nucleotides (nt) per min by HCV recombinant NS5B purified from Escherichia coli (42) and up to 180 nt/min in Huh-7 cells (36). Thus, the synthesis rate for subgenomic plus or minus HCV RNA (∼6,300 nt [35]) is k4p = 1.7 RNA molecules per h per replicative intermediate complex. We assume that there is no difference in synthesis rate when the minus strand (as part of dsRNA) or plus strand serves as a template for replication (i.e., k4p = k4m = 1.7).
HCV plus- and minus-strand RNA degradation rates.
Plus- and minus-strand RNAs involved in replication complexes are fully resistant to nuclease treatment (50) and are assumed, in our model, to degrade at a slower rate than free plus-strand RNA in the cytoplasm. Thus, we assume that the degradation half-lives of free plus- and double-strand RNAs (10 and 11.5 h, respectively [23]) estimated in IFN-α-treated Huh-7 cells correspond to the rates of degradation of HCV RNA in VMS, i.e., μP = 0.07 h−1 and μds = 0.06 h−1. In line with that, it is likely that the degradation rate of free plus-strand RNAs in cytoplasm (μpcyt) is significantly faster than the observed overall plus-strand RNA degradation rate of μP = 0.07 h−1 (23).
NS5B polymerase degradation rate in cytoplasm.
More than 95% of NS5B polymerase molecules in vitro were shown to be sensitive to protease treatment and not involved in replicase activity (38, 50). Thus, it is likely that the observed NS5B polymerase half-life, 12 h (44, 45, 59), corresponds to the NS5B polymerase half-life in cytoplasm, i.e., μEcyt = 0.06 h−1.
Parameter estimation.
The remaining parameter values—k1, kPin, kPout, kEin, kc, k3, k5, μPcyt, μE, μIds, μIp, μTc, RiboTot, and RPcyt(0)—are not known and were chosen to be consistent with the establishment of a biologically realistic steady state. We varied each unknown parameter one at a time and tested for consistency with the aforementioned experimental steady-state criteria (see “Simulation procedures” above). Consequently, as explained in Results, we fixed μIds/kEin = 1.0 × 104, k5/k3 = 200, and kPin = kPout = 0.2 h−1 and then estimated a range of values for the rest of the unknown parameters as shown in Table 1.
To test the parameter sensitivity of our simulation results, we used parameter ranges (Table 1) consistent with HCV subgenomic steady-state levels found in replicons. We randomly varied the parameter values 1,000 times [RPcyt(0), RiboTot, k1, kEin, kc, k3, μIp, μE, μPcyt, and μTc] within the ranges given in Table 1. Each simulation was numerically integrated for 300 h and was checked for reaching a steady-state of 900 to 5,000 total plus-strand RNAs and 8 × 105 to 2 × 106 NS5B molecules. To define the approximate time when this steady state was reached, we searched backward from 300 h to the time of transfection for the time when the total plus-strand RNA was within 0.5 log of its steady-state level.
RESULTS
Based on current knowledge of related positive ssRNA viruses and bacteriophage replication mechanisms, we developed a model for subgenomic HCV replication in Huh-7 cells described in Materials and Methods. Quantitative studies of HCV replication both in cell culture and in vitro allowed us to minimize the uncertainty of model parameters and provided experimental data to compare with simulation results. We then estimated unknown host and viral components and kinetic rate constants involved in subgenomic HCV replication within Huh-7 cells. We used our model to explore the biological basis of the plus-strand to minus-strand asymmetry and the great excess of HCV NS proteins found in replicon cells.
Number of ribosomes available for HCV translation.
We found that the number of ribosomes available for HCV translation (RiboTot) is the only parameter that significantly affects the number of NS5B polymerase molecules in the cytoplasm (Fig. 2A). To obtain approximately a million NS5B polymerase molecules (50), we needed to assume that 500 to 1,000 ribosome-HCV RNA complexes (corresponding to 5,000 to 10,000 ribosomes) are involved in HCV translation at steady state.
FIG. 2.

Simulation of subgenomic HCV RNA replication in Huh-7 cells. To explore the impact of each unknown parameter on subgenomic HCV kinetics, from time of transfection (t = 0) to steady state, we varied one or more chosen parameters within a given range, while all other parameters were maintained fixed as follows: k1 = 80 h−1 molecule−1, kPin = 0.2 h−1, kPout = 0.2 h−1, kEin = 1.3 × 10−5 h−1, kc = 0.6 h−1, k2 = 100 h−1, k4p = k4m = 1.7 h−1, k3 = 0.02 h−1 molecule−1, k5 = 4 h−1 molecule−1, μIp = 0.04 h−1, μE = 0.04 h−1, μpcyt = 10 h−1, μp = 0.07 h−1, μds = 0.06 h−1, μIds = 0.13 h−1, μEcyt = 0.06 h−1, μTc = 0.015 h−1, RiboTot = 700 ribosome complexes, and RPcyt(0) = 500 plus-strand RNA copies. (A) Within a range of 1 to 1,000 available ribosome complexes per cell (RiboTot), the model reached steady-state characteristics when 500 < RiboTot < 1,000. We found that the steady-state HCV NS5B polymerase level (green line) increases with higher RiboTot numbers. The total plus- and minus-strand RNAs at steady state are shown as black and red lines, respectively. (B) We chose kEin and μIds within the ranges 5 × 10−6 to 2 × 10−4 h−1and 0.01 to 0.9 h−1, respectively. We found that the ratio (RP + RIp + Rds + RIds)/(RPcyt + Tc), which is the ratio of total plus-strand RNAs inside and outside of the VMS (thick line), and the ratio of total plus-strand RNA to total minus-strand RNA (RPTot/RMTot, thin line) increase with the ratio μIds/kEin between the rate of replicative intermediate degradation and the rate of polymerase transport into the VMS. To obtain a 1:1 ratio of (RP + RIp + Rds + RIds)/(RPcyt + Tc) and a 10:1 ratio of RPTot/RMTot, the ratio μIds/kEin needs to be about 104. A ratio of ∼6 of total plus-strand RNA to total minus-strand RNA in VMS is found with many different μIds and kEin rates (dashed line). (C) We chose k5 and k3 within the ranges 0.8 to 4.0 h−1 and 0.004 to 0.02 h−1, respectively. The ratio of total plus-strand RNA to total minus-strand RNA increases with the ratio k5/k3 of formation rates of minus-strand to plus-strand replication complexes. While k5/k3 = 1 does not allow for a 10:1 total plus-strand-to-minus-strand asymmetry (thick line), and 6:1 total plus-strand-to-minus-strand asymmetry in VMS (thin line), k5/k3 ∼ 200 will. (D) We checked whether the total plus-strand-to-minus-strand asymmetry can be generated with different synthesis rates of plus- and minus-strand RNA, i.e., k4p > k4m. We assumed the same formation rates (k5 = k3 = 0.02 h−1) of plus-strand and double-strand replicative intermediate complexes (RIp and RIds, respectively). Interestingly, although 50- to 500-fold faster synthesis rates for the plus-strand RNA than for the minus-strand RNA generate larger numbers of RIp complexes (thick red line) than of RIds complexes (thick black lines), the total plus-strand RNA (thin black line) is approximately equal to the total minus-strand RNA (thin red line). Free plus-strand RNA (RP) is represented in blue. The curve for Rds superimposes on the curve for RMTot since almost all of the minus-strand RNA is in double-stranded complexes. (E) With 10 subgenomic plus-strand RNAs successfully transfected [RPcyt(0) = 10 per cell (thin lines)], total plus-strand RNA (black line), minus-strand RNA (red line), and NS5B molecules (green line) reach steady state in about 300 h. However, with RPcyt(0) = 500 (thick lines), the steady state is attained in about 50 h. The total plus-strand RNA level at steady state is not affected by different initial RPcyt numbers. (F) We tested how the plus-strand RNA copy numbers (1 to 1,000) at time of transfection, RPcyt(0), affect the time to attain steady state. Only when RPcyt(0) was >7 was a steady state attained, whereas RPcyt(0) at <7 led to elimination of viral RNA (not shown). Higher initial RPcyt numbers leads to faster attainment of steady-state.
1:1 ratio of plus-strand RNA inside and outside the VMS.
Approximately half of the total plus-strand RNAs in a replicon cell is nuclease resistant (50) and thus assumed in our model to localize in the VMS. To obtain a total number of plus-strand RNAs in the cytoplasm (RPcyt + Tc) equal to the total number of plus-strand RNAs in VMS (RP + RIp + RIds + Rds), we needed to set the ratio kPin/kPout of the rates at which plus-strand RNA is transported into and out of the VMS to 1, set kPin = 0.2 h−1, and set the ratio μIds/kEin of the rates at which RIds complexes degrade (μIds) and NS5B molecules are transported into the VMS (kEin) at ∼1.0 × 104 (Fig. 2B). Varying the other unknown parameters over a large range, as given in Table 1, did not significantly affect the 1:1 ratio between total plus-strand RNA in and out of the VMS. We thus fixed μIds/kEin = 1.0 × 104 and kPin/kPout = 0.2 h−1 for the rest of our analysis.
Plus-strand HCV RNA levels at steady state.
We found that kEin and μpcyt are the parameters that most affect the steady-state level of total (RPTot = RPcyt + RP + Tc + RIp + RIds + Rds) plus-strand RNAs in a cell. Nonetheless, ranges of μpcyt (0.06 to 15.0 h−1) and kEin (3.8 × 10−6 to 6.0 × 10−5 h−1) allow a steady state with the observed plus-strand RNA level (not shown).
Plus-strand and minus-strand HCV RNA asymmetry levels.
The overall plus-strand RNA (RPTot = RPcyt + RP + Tc + RIp + RIds + Rds) to minus-strand RNA (RMTot = Rds + RIds) ratio (RPTot /RMTot) increases with the ratio μIds/kEin (Fig. 2B) and the ratio k5/k3 of rates at which the dsRNA and plus-strand replicative intermediate complexes are formed (RIds and RIp, respectively). To obtain an approximate overall 10:1 plus-strand/minus-strand ratio (with the above-estimated parameters values), we need to assume that the formation rate of RIds complexes is ∼200-fold faster than the formation rate of RIp (k5/k3 ∼ 200), and that k3, the rate constant for formation of RIp, is between 0.004 and 0.02 h−1 molecule−1 (Fig. 2C).
If we restrict this analysis to the VMS, the ratio of plus-strand RNA (RPVMS = RP + RIp + RIds + Rds) to minus-strand RNA (RMTot = Rds + RIds) (RPVMS /RMTot) also increases with the ratio k5/k3 (Fig. 2C). To obtain the approximate 6:1 plus-strand/minus-strand ratio in the VMS (50), k5/k3 should again be large: ca. 200 to 500. However, to also attain an overall ratio of 10:1, we fixed k5/k3 at 200 for the rest of our analysis.
Theoretically, the observed RPTot /RMTot ratio can be governed by significantly faster synthesis of the nascent plus-strand RNA relative to the synthesis of nascent minus-strand RNA (i.e., k4p ≫ k4m; assuming that the formation rates of RIp and RIds are equal, k3 = k5). However, even a 500-fold difference between the plus- and minus-strand RNA synthesis rates did not generate a 10:1 RPTot /RMTot ratio (Fig. 2D). It appears that faster synthesis of the plus-strand RNA relative to the minus-strand RNA leads to more free dsRNA, Rds, than free plus-strand RNA, RP, and to higher numbers of plus-strand replicative intermediate complexes than double-strand replicative intermediate complexes (Fig. 2D). The more plus-strand replicative intermediate complexes that are formed, the more minus strands that are synthesized, and this eventually leads to an RPTot/RMTot of ∼1. Thus, faster plus-strand synthesis cannot generate the observed 10:1 ratio (Fig. 2D).
Time to reach steady state.
After achieving a plus-strand/minus-strand ratio of 10:1 and a plus-strand RNA level of 900 to 5,000 copies/cell, we sought to understand how to adjust the remaining parameters [kc, k1, k3, μE, μTc, and RPcyt(0)] so that the time it takes the system to reach steady state is about 48 h. Assuming the fastest plus and minus intermediate complex formation rates consistent with the above-mentioned range, i.e., k3 = 0.02 h−1 molecule−1 and k5 = 4.0 h−1 molecule−1, the number of plus strands at the time of transfection, RPcyt(0), the degradation rate constant of translation complexes (μTc), and the formation rate constant of translation complexes (k1) affect the time it takes to attain steady state. When RPcyt(0) is ∼500 copies/cell, μTc is <0.02 h−1, and k1 is 80 h−1 molecule−1, both strands will reach steady state in about 48 h (Fig. 2E). In Fig. 2F we show an inverse correlation between RPcyt(0) and the time it takes to attain steady state. In addition, it is likely that the degradation rate of free NS5B in VMS (μE) is slower than in cytoplasm (μEcyt = 0.06 h−1) due to the protection of the VMS from protease activity. However, μE within the range 0.01 to 0.06 h−1 and the polyprotein cleavage rate kc within the range 0.1 to 5.0 h−1 do not affect the rate of attainment of the RNA steady state (not shown).
To verify the robustness of the estimated-parameter-value ranges, 1,000 combinations of the unknown parameters were chosen randomly within the range given in Table 1. We found that ca. 70% of the 1,000 random parameter sets (Table 1, third column) led to a steady state consistent with the experimental observations criteria given in Materials and Methods (Fig. 3A). Among these 700 parameter sets, only 600 (i.e., 60% of the 1,000 random parameter sets) attain an RNA steady-state by 48 h posttransfection (data not shown). Thus, these 600 sets that fulfill the experimental observations define a new, narrower, range for each parameter (Table 1, fourth column). We then generated 1,000 random parameter sets within the new ranges and found that more than 99% lead to a steady state, in agreement with experimental observations (Fig. 3B). For example, starting from the initial ranges for RiboTot (500 to 1,000 ribosome complexes) and RPcyt(0) (10 to 1,000 plus-strand RNA copies), we found that to be consistent with the set of experimental observation in Materials and Methods, RiboTot and RPcyt(0) should be between 650 to 910 and 340 to 830, respectively. All other new parameter ranges are shown in Table 1, column 4.
FIG. 3.

Sensitivity analyses for the model of subgenomic HCV RNA replication in Huh-7 cells. One thousand parameter sets within the parameter ranges given in Table 1 (column 3) were randomly chosen. (A) Within these parameter ranges we found that: (i) ∼20% of parameter sets led to a total plus-strand RNA steady state of <900 copies/cell or HCV RNA elimination, (ii) ∼8% led to a total plus-strand RNA steady state of >900 copies/cell but NS5B molecules of <8 × 105, (iii) ∼70% (700) of parameter sets generated steady states of between 900 and 5,000 total plus-strand RNAs and 8 × 105 to 4 × 106 NS5B molecules, and (iv) ∼2% led to a total plus-strand RNA steady state of >5,000 copies/cell (not shown). Among these 700 parameter sets only 85% (600 sets) attained an RNA steady-state in 48 h posttransfection (data not shown). (B) Using the interquartile ranges for each parameter in those 600 sets (Table 1, column 4), we found that more than 99% of 1,000 simulations using randomly chosen parameters within these ranges of the parameter led to steady states consistent with the characteristics given in Materials and Methods. Mean values of total plus-strand RNA, minus-strand RNA, and NS5B molecules are shown in black, red, and green filled circles, respectively. Vertical lines represent three standard deviations.
Due to the lack of experimental data on the number of plus strands at the time of transfection and the lack of detailed kinetic information about the growth rate of plus and minus strands from transfection to steady state, we cannot estimate parameter values precisely. However, our analysis provides plausible parameter ranges (Table 1).
Model without VMS.
We sought to understand whether the observed RNA steady-state can be reached without the VMS. We thus simplified our model so that HCV RNA synthesis and viral polyprotein translation occur in the same compartment. Briefly, equations 1 and 5 were summed and kPin, kEin, kPout, and μP were set to zero. In addition, equations 4 and 7 were summed, and kEin and μE were set to zero. To explore the impact of each remaining parameter on subgenomic HCV kinetics, from the time of transfection (t = 0) to steady state, we varied one or more chosen parameters within a given range, while all other parameters were maintained fixed as indicated in the legend to Fig. 2 (except for k1 and RiboTot, which needed to be varied in order to allow RNA amplification to the observed steady-state level). Simulation results of this model within large-parameter-value ranges (e.g., 0.1 < μIds < 1.5 h−1, 0.2 < k1 < 100 h−1 molecule−1, and 2 < RiboTot < 30 ribosome complexes) were in agreement with the observed RNA steady-state levels but were not consistent with the observation of millions of free NS5B molecules in cytoplasm. Typically, we found fewer than 100 free NS5B molecules. The model also predicts that only a few ribosome complexes (RiboTot < ∼30) are needed to attain realistic RNA steady-state levels within large-parameter-value ranges (not shown). This result is reasonable; without a VMS the total plus-strand RNA in the cell can be involved in HCV polyprotein translation. To increase the predicted number of NS5B molecules, one needs to increase the number of available ribosome complexes. However, choosing a RiboTot value of greater than ∼30 led to 10- to 100-fold-higher RNA steady-state levels than are observed experimentally.
DISCUSSION
We have developed a kinetic model of HCV RNA replication in the absence of viral particle formation, as occurs in subgenomic replicon systems. Our results are in agreement with the recent experimental results of Quinkert et al. (50) and provide parameter ranges for the relevant kinetic parameters. Our model estimates that about 7 × 103 ribosomes are involved in generating millions of HCV NS5B-polymerase molecules in a Huh-7 cell and suggests that the observed 10:1 asymmetry of plus-strand to minus-strand RNA levels can be explained by a higher promoter strength or, as we call it here, a higher-affinity (200-fold) interaction of HCV NS5B-polymerase complexes with minus-strand RNA over the plus-strand RNA in order to start HCV RNA synthesis.
In our model, we assumed that plus-strand RNAs initially serve as templates to synthesize the NS5B polymerase and other essential proteins (59). We also assumed that plus-strand RNAs involved in translation cannot be templates for simultaneous RNA synthesis, as was previously shown for poliovirus replication (20). Because it is assumed that the positive-strand RNAs must be used for translation prior to RNA replication (1), HCV might have a mechanism, yet to be determined, to downregulate translation to begin RNA synthesis. However, in the model developed here translation and replication of HCV RNA coexist, although there is a competition between the two processes, with any given HCV RNA strand either being translated or replicated.
For simplicity, we assumed that 10 ribosomes bind to plus-strand RNA simultaneously and that the polysomes dissociate after each viral polyprotein is synthesized, as was previously assumed in models of Qβ replication (16, 26). We show in the Appendix that one can use this simplified model with confidence only if the rate for ribosome attachment to a free plus-strand RNA is lower than the rate constant for subsequent ribosomes to attach. This assumption may be plausible in light of RNA helicase function within a ribosome that needs first to unwind the secondary structure of the mRNA (25), which then might lead to a faster attachment of the next ribosome. In addition, to correctly account for the rate of translation with 10 ribosomes, we assumed the virus polyprotein elongation rate is 10 times faster than the estimated elongation rate per ribosome in eukaryotes (see Materials and Methods).
The total ribosome number and the fraction of available ribosomes for HCV replication in an Huh-7 cell have not yet been defined. However, it is plausible that the number of ribosomes in an Huh-7 cell is between 2 × 104, as estimated in prokaryotic organisms (E. coli [16]), and 6 × 106 ribosomes, as found in eukaryotic cells (49). According to our model, we estimated that approximately 6 × 103 ribosomes are available for HCV replication, corresponding to ca. 0.1% of the total number of ribosomes in a eukaryotic cell.
In the early 1960s, Spiegelman (54) and others questioned how a single-plus-strand virus RNA genome (e.g., Qβ phage) can be amplified among thousands of host mRNA molecules present in a bacterium. It was found that the Qβ replicase has a specific template affinity to the viral genome, thus allowing specific amplification of the Qβ genome. It is possible that other single-stranded RNA viruses (e.g., HCV, poliovirus, and others [53]), which replicate in eukaryotic cells, developed additional mechanisms for efficient RNA amplification by colocalization of their replicative intermediates (including their replicase) in enclosed VMS. In agreement with that, we implement in our model (equations 1 to 9) the existence of VMS and generated with our model the observed subgenomic HCV steady-state characterizations. Interestingly, without the VMS, if more than ∼30 ribosome complexes (i.e., 300 ribosomes) are available to support HCV replication, then significantly higher RNA steady states than we observed can occur. Thus, the VMS might restrain viral amplification and prevent host cell damage. Other possible roles for the VMS include hiding dsRNA molecules from the innate immune system of the host cell (19) and/or serving as a switch for slowing down translation in favor of HCV RNA replication.
Three major species of Flavivirus RNAs have been described in cell-free systems (2, 4, 10, 57): dsRNA, dsRNA and recently synthesized plus- or minus-strand RNAs, and single-stranded plus- and minus-strand RNAs. However, unlike other Flaviviridae (11, 21, 60) double-stranded species (dsRNA and/or dsRNA plus recently synthesized plus- or minus-strand RNA) have not been detected during HCV replication in cell culture (24, 28, 35) or in liver biopsies (7). We predict that dsRNA species and dsRNA plus recently synthesized plus- or minus-strand RNA species are <3% and <10% of the total plus-strand RNA per cell, respectively, which may make it difficult to detect using current assays. Thus, although the exact strategy of HCV replication in Huh-7 cells is not yet firmly established, we assumed a double-stranded strategy in our model. However, our model can be simply modified to involve a single-stranded replication strategy (not shown).
The mechanism by which the asymmetry (∼10:1 ratio) between plus- and minus-strand HCV RNA levels is gained in replicons (35, 50) or in liver cells (9, 27) is still unknown. It might be that host factors, required for synthesizing minus-strand RNA, are responsible for this asymmetry or that there are different rate constants for the production of plus- and minus-strand RNA. It was previously shown by Eigen et al. (16) that this asymmetry can be caused simply by the fact that Qβ replicase has to compete for plus-strand RNA with ribosomes and coat protein, whereas minus-strand RNAs are free for the production of plus-strand RNA. However, since HCV RNA synthesis probably occurs in VMS (15, 22), we do not implement in our model a competition between HCV replicase and ribosomes. Since host factors responsible for this asymmetry have not been identified, only different rate constants for the production of plus- and minus-strand RNAs were examined in our model. Interestingly, our model predicts that whereas different synthesis-rate constants of plus- and minus-strand RNAs do not contribute to the observed asymmetry, a higher-affinity (∼200-fold) interaction of minus-strand RNA over plus-strand RNA with the NS5B polymerase-containing RNA replicase, in order to start HCV RNA synthesis, does contribute significantly. In addition, the latter higher affinity (>200-fold) leads to ∼6:1 plus-strand/minus-strand ratio in VMS, in agreement with recent experimental results (50).
Our prediction for a higher-affinity interaction of the NS5B containing replicase with minus-strand RNA than with plus-strand RNA is in agreement with an observation by Reigadas et al. (52), who showed in vitro that the 3′-terminal region of minus-strand RNA was preferentially bound by purified HCV NS5B polymerase over the 3′ end of plus-strand RNA. However, it is likely the template preferences and the resulting asymmetric RNA replication will involve higher-order RNA interactions, such as those identified in the NS5B coding region (17, 29, 62), which function through a “kissing interaction” with a loop sequence in the 3′NTR. Additional viral and cellular proteins are also likely to participate in determining template preference (38; D. Quinkert et al., 13th Int. HCV Conf., abstr. 212, 2006).
Recent experimental results of Quinkert et al. (50) indicated that each HCV replication complex is composed of multiple copies of HCV NS proteins. These authors suggested that the huge excess of NS proteins is required to build up the viral replication complexes and that only <0.1% of them are required to be enzymatically active. Since in our model we only keep track of the enzymatically active NS5B molecules, our results are in agreement with their observations. In addition, the excess of NS5B molecules seen in our model also implies an excess of other NS viral proteins, since each HCV polyprotein (P) is cleaved into one copy of each HCV NS protein.
According to the model, higher numbers of plus-strand RNAs at the time of transfection lead to faster attainment of the RNA steady state but do not change its magnitude. Thus, the RNA amplification from transfection to steady state is inversely correlated with the number of plus-strand RNAs that enters the cell by transfection, in agreement with observations of Lohmann et al. (34). However, small numbers of transfected RNAs (see the legend to Fig. 2F) may lead to the elimination of HCV RNA. Of note, in the model we do not consider that some of the transfected RNAs might be defective. If this were the case then if a few RNAs were transfected it would be possible that no viral RNA amplification occurred.
In summary, we have developed a mathematical model for subgenomic HCV replication within Huh-7 cell that uses a double-stranded strategy for RNA amplification. Our model suggests a mechanism by which the ratio of plus-strand to minus-strand RNA is regulated and shows that RNA replication occurring in a membrane compartment has advantages for the HCV life cycle. Now that fully permissive HCV replication systems have been developed (31, 58, 63), the next step will be to incorporate virus production and infection to create one comprehensive model of the complete HCV life cycle. The model developed here can serve as an important tool in understanding HCV replication mechanisms and may prove useful in designing and evaluating new antivirals for use against HCV.
APPENDIX
Modeling multiple ribosomes attaching to HCV mRNA.
To explore the sequential attachment of 10 ribosomes to each HCV mRNA (59), we developed the following model:
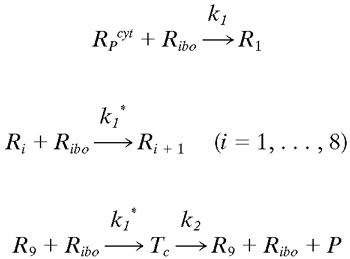 |
Let Ri represents the number of polysomes consisting of i ribosomes attached to an HCV mRNA (plus-strand RNA). Free plus-strand RNA molecules, RPcyt, are converted at rate k1 into R1 due to ribosome attachment. Then Ri = 1, … , 8 disappear at rate k1* by additional ribosome attachment, forming Ri+1. Finally, R9 disappears at rate k1* by forming the translation complex, Tc, and reappears at rate k2 when the translation is complete and the newly synthesized polyprotein, P, and its related ribosome dissociate from the HCV mRNA.
We further assume that once plus-strand RNA attaches to a ribosome and/or becomes a polysome, it will remain in this complexed state until its degradation, with an average rate of μTc. Under these assumptions this model can be converted to the following differential equations:
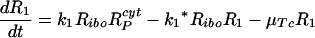 |
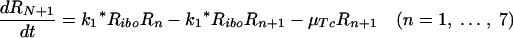 |
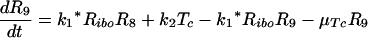 |
These equations can be easily included in our simplified model, equations 1 to 9, necessitating modifying equations 1 and 2 as follows:
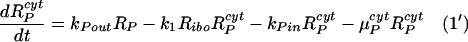 |
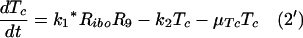 |
In addition, we modified the equation in Materials and Methods for the free ribosomes (Ribo) as follows:
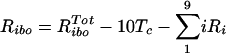 |
We found that if one assumes that the rate constant k1, for the first ribosome to attach is lower than the rate constant k1*, for subsequent ribosomes to attach, then the simplified model and complex model give similar results (not shown). However, when k1 ≥ k1* the model results, within the parameter ranges given in Table 1, are not in agreement with the experimental data (not shown).
Acknowledgments
We thank Volker Lohmann and Tom Oh for critical comments on the manuscript and Amichai Feigenboim for help with the sensitivity computations.
This research was performed under the auspices of the U.S. Department of Energy under contract DE-AC52-06NA25396 and supported by National Institutes of Health (NIH) grant RR06555 (to A.S.P.). C.M.R. is supported by the Greenberg Medical Research Institute, NIH, and the Ellison Medical Foundation. H.D. is partially funded by a fellowship from the Fulbright Foundation. R.M.R. and H.D. are also supported by grant P20-RR18754 from the National Center for Research Resources (NCRR), a component of the NIH.
This study is solely the responsibility of the authors and does not necessarily represent the official views of the NCRR or the NIH.
Footnotes
Published ahead of print on 11 October 2006.
REFERENCES
- 1.Agol, V. I., A. V. Paul, and E. Wimmer. 1999. Paradoxes of the replication of picornaviral genomes. Virus Res. 62:129-147. [DOI] [PubMed] [Google Scholar]
- 2.Ali, N., K. D. Tardif, and A. Siddiqui. 2002. Cell-free replication of the hepatitis C virus subgenomic replicon. J. Virol. 76:12001-12007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alter, M. J., H. S. Margolis, K. Krawczynski, F. N. Judson, A. Mares, W. J. Alexander, P. Y. Hu, J. K. Miller, M. A. Gerber, R. E. Sampliner, et al. 1992. The natural history of community-acquired hepatitis C in the United States. N. Engl. J. Med. 327:1899-1905. [DOI] [PubMed] [Google Scholar]
- 4.Bartholomeusz, A. I., and P. J. Wright. 1993. Synthesis of dengue virus RNA in vitro: initiation and the involvement of proteins NS3 and NS5. Arch. Virol. 128:111-121. [DOI] [PubMed] [Google Scholar]
- 5.Biebricher, C. K., M. Eigen, W. C. Gardiner, Y. Husim, H. C. Keweloh, and A. Obst. 1987. Complex chemical reaction systems, p. 17-38. In J. Warnatz and W. Jager (ed.), Mathematical modeling and simulation. Springer-Verlag, Berlin, Germany.
- 6.Biebricher, C. K., M. Eigen, and R. Luce. 1981. Kinetic analysis of template-instructed and de novo RNA synthesis by Qβ replicase. J. Mol. Biol. 148:391-410. [DOI] [PubMed] [Google Scholar]
- 7.Blight, K., R. Trowbridge, and E. J. Gowans. 1996. Absence of double-stranded replicative forms of HCV RNA in liver tissue from chronically infected patients. J. Viral Hepat. 3:29-36. [DOI] [PubMed] [Google Scholar]
- 8.Blight, K. J., J. A. McKeating, and C. M. Rice. 2002. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J. Virol. 76:13001-13014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang, M., O. Williams, J. Mittler, A. Quintanilla, R. L. Carithers, Jr., J. Perkins, L. Corey, and D. R. Gretch. 2003. Dynamics of hepatitis C virus replication in human liver. Am. J. Pathol. 163:433-444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chu, P. W., and E. G. Westaway. 1985. Replication strategy of Kunjin virus: evidence for recycling role of replicative form RNA as template in semiconservative and asymmetric replication. Virology 140:68-79. [DOI] [PubMed] [Google Scholar]
- 11.Cleaves, G. R., T. E. Ryan, and R. W. Schlesinger. 1981. Identification and characterization of type 2 dengue virus replicative intermediate and replicative form RNAs. Virology 111:73-83. [DOI] [PubMed] [Google Scholar]
- 12.Dahari, H., A. Feliu, M. Garcia-Retortillo, X. Forns, and A. U. Neumann. 2005. Second hepatitis C replication compartment indicated by viral dynamics during liver transplantation. J. Hepatol. 42:491-498. [DOI] [PubMed] [Google Scholar]
- 13.Dahari, H., M. Major, X. Zhang, K. Mihalik, C. M. Rice, A. S. Perelson, S. M. Feinstone, and A. U. Neumann. 2005. Mathematical modeling of primary hepatitis C infection: noncytolytic clearance and early blockage of virion production. Gastroenterology 128:1056-1066. [DOI] [PubMed] [Google Scholar]
- 14.Dixit, N. M., J. E. Layden-Almer, T. J. Layden, and A. S. Perelson. 2004. Modeling how ribavirin improves interferon response rates in hepatitis C virus infection. Nature 432:922-924. [DOI] [PubMed] [Google Scholar]
- 15.Egger, D., B. Wolk, R. Gosert, L. Bianchi, H. E. Blum, D. Moradpour, and K. Bienz. 2002. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J. Virol. 76:5974-5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eigen, M., C. K. Biebricher, M. Gebinoga, and W. C. Gardiner. 1991. The hypercycle: coupling of RNA and protein biosynthesis in the infection cycle of an RNA bacteriophage. Biochemistry 30:11005-11018. [DOI] [PubMed] [Google Scholar]
- 17.Friebe, P., J. Boudet, J. P. Simorre, and R. Bartenschlager. 2005. Kissing-loop interaction in the 3′ end of the hepatitis C virus genome essential for RNA replication. J. Virol. 79:380-392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fried, M. W., M. L. Shiffman, K. R. Reddy, C. Smith, G. Marinos, F. L. Goncales, Jr., D. Haussinger, M. Diago, G. Carosi, D. Dhumeaux, A. Craxi, A. Lin, J. Hoffman, and J. Yu. 2002. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N. Engl. J. Med. 347:975-982. [DOI] [PubMed] [Google Scholar]
- 19.Gale, M., Jr., and E. M. Foy. 2005. Evasion of intracellular host defense by hepatitis C virus. Nature 436:939-945. [DOI] [PubMed] [Google Scholar]
- 20.Gamarnik, A. V., and R. Andino. 1998. Switch from translation to RNA replication in a positive-stranded RNA virus. Genes Dev. 12:2293-2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gong, Y., R. Trowbridge, T. B. Macnaughton, E. G. Westaway, A. D. Shannon, and E. J. Gowans. 1996. Characterization of RNA synthesis during a one-step growth curve and of the replication mechanism of bovine viral diarrhoea virus. J. Gen. Virol. 77(Pt. 11):2729-2736. [DOI] [PubMed] [Google Scholar]
- 22.Gosert, R., D. Egger, V. Lohmann, R. Bartenschlager, H. E. Blum, K. Bienz, and D. Moradpour. 2003. Identification of the hepatitis C virus RNA replication complex in Huh-7 cells harboring subgenomic replicons. J. Virol. 77:5487-5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guo, J. T., V. V. Bichko, and C. Seeger. 2001. Effect of alpha interferon on the hepatitis c virus replicon. J. Virol. 75:8516-8523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guo, J. T., Q. Zhu, and C. Seeger. 2003. Cytopathic and noncytopathic interferon responses in cells expressing hepatitis C virus subgenomic replicons. J. Virol. 77:10769-10779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang, W., Y. Hou, and M. Inouye. 1997. CspA, the major cold-shock protein of Escherichia coli, is an RNA chaperone. J. Biol. Chem. 272:196-202. [DOI] [PubMed] [Google Scholar]
- 26.Kim, H., and J. Yin. 2004. Energy-efficient growth of phage Qβ in Escherichia coli. Biotechnol. Bioeng. 88:148-156. [DOI] [PubMed] [Google Scholar]
- 27.Lanford, R. E., D. Chavez, F. V. Chisari, and C. Sureau. 1995. Lack of detection of negative-strand hepatitis C virus RNA in peripheral blood mononuclear cells and other extrahepatic tissues by the highly strand-specific rTth reverse transcriptase PCR. J. Virol. 69:8079-8083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lanford, R. E., B. Guerra, H. Lee, D. R. Averett, B. Pfeiffer, D. Chavez, L. Notvall, and C. Bigger. 2003. Antiviral effect and virus-host interactions in response to alpha interferon, gamma interferon, poly(I)-poly(C), tumor necrosis factor alpha, and ribavirin in hepatitis C virus subgenomic replicons. J. Virol. 77:1092-1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee, H., H. Shin, E. Wimmer, and A. V. Paul. 2004. cis-Acting RNA signals in the NS5B C-terminal coding sequence of the hepatitis C virus genome. J. Virol. 78:10865-10877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin, K., A. D. Kwong, and C. Lin. 2004. Combination of a hepatitis C virus NS3-NS4A protease inhibitor and alpha interferon synergistically inhibits viral RNA replication and facilitates viral RNA clearance in replicon cells. Antimicrob. Agents Chemother. 48:4784-4792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lindenbach, B. D., M. J. Evans, A. J. Syder, B. Wolk, T. L. Tellinghuisen, C. C. Liu, T. Maruyama, R. O. Hynes, D. R. Burton, J. A. McKeating, and C. M. Rice. 2005. Complete replication of hepatitis C virus in cell culture. Science 309:623-626. [DOI] [PubMed] [Google Scholar]
- 32.Lindenbach, B. D., and C. M. Rice. 2001. Flaviviridae: the viruses and their replication., p. 991-1041. In D. M. Knipe and P. M. Howley (ed.), Fields virology, 4th ed. Lippincott-Raven Publishers, Philadelphia, PA.
- 33.Lodish, H. F., and M. Jacobsen. 1972. Regulation of hemoglobin synthesis: equal rates of translation and termination of α- and β-globin chains. J. Biol. Chem. 247:3622-3629. [PubMed] [Google Scholar]
- 34.Lohmann, V., S. Hoffmann, U. Herian, F. Penin, and R. Bartenschlager. 2003. Viral and cellular determinants of hepatitis C virus RNA replication in cell culture. J. Virol. 77:3007-3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lohmann, V., F. Korner, J. Koch, U. Herian, L. Theilmann, and R. Bartenschlager. 1999. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285:110-113. [DOI] [PubMed] [Google Scholar]
- 36.Ma, H., V. Leveque, A. De Witte, W. Li, T. Hendricks, S. M. Clausen, N. Cammack, and K. Klumpp. 2005. Inhibition of native hepatitis C virus replicase by nucleotide and non-nucleoside inhibitors. Virology 332:8-15. [DOI] [PubMed] [Google Scholar]
- 37.Manns, M. P., J. G. McHutchison, S. C. Gordon, V. K. Rustgi, M. Shiffman, R. Reindollar, Z. D. Goodman, K. Koury, M. Ling, and J. K. Albrecht. 2001. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet 358:958-965. [DOI] [PubMed] [Google Scholar]
- 38.Miyanari, Y., M. Hijikata, M. Yamaji, M. Hosaka, H. Takahashi, and K. Shimotohno. 2003. Hepatitis C virus non-structural proteins in the probable membranous compartment function in viral genome replication. J. Biol. Chem. 278:50301-50308. [DOI] [PubMed] [Google Scholar]
- 39.Moradpour, D., V. Brass, E. Bieck, P. Friebe, R. Gosert, H. E. Blum, R. Bartenschlager, F. Penin, and V. Lohmann. 2004. Membrane association of the RNA-dependent RNA polymerase is essential for hepatitis C virus RNA replication. J. Virol. 78:13278-13284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Neumann, A. U., N. P. Lam, H. Dahari, D. R. Gretch, T. E. Wiley, T. J. Layden, and A. S. Perelson. 1998. Hepatitis C viral dynamics in vivo and the antiviral efficacy of interferon-alpha therapy. Science 282:103-107. [DOI] [PubMed] [Google Scholar]
- 41.National Institutes of Health. 2002. National Institutes of Health consensus development conference statement. Management of Hepatitis C:10-12. National Institutes of Health, Bethesda, MD.
- 42.Oh, J. W., T. Ito, and M. M. Lai. 1999. A recombinant hepatitis C virus RNA-dependent RNA polymerase capable of copying the full-length viral RNA. J. Virol. 73:7694-7702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Palmiter, R. D. 1973. Ovalbumin messenger ribonucleic acid translation: comparable rates of polypeptide initiation and elongation on ovalbumin and globin messenger ribonucleic acid in a rabbit reticulocyte lysate. J. Biol. Chem. 248:2095-2106. [PubMed] [Google Scholar]
- 44.Pause, A., G. Kukolj, M. Bailey, M. Brault, F. Do, T. Halmos, L. Lagace, R. Maurice, M. Marquis, G. McKercher, C. Pellerin, L. Pilote, D. Thibeault, and D. Lamarre. 2003. An NS3 serine protease inhibitor abrogates replication of subgenomic hepatitis C virus RNA. J. Biol. Chem. 278:20374-20380. [DOI] [PubMed] [Google Scholar]
- 45.Pietschmann, T., V. Lohmann, G. Rutter, K. Kurpanek, and R. Bartenschlager. 2001. Characterization of cell lines carrying self-replicating hepatitis C virus RNAs. J. Virol. 75:1252-1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Powers, K. A., R. M. Ribeiro, K. Patel, S. Pianko, L. Nyberg, P. Pockros, A. J. Conrad, J. McHutchison, and A. S. Perelson. 2006. Kinetics of hepatitis C virus reinfection after liver transplantation. Liver Transpl. 12:207-216. [DOI] [PubMed] [Google Scholar]
- 47.Poynard, T., V. Ratziu, Y. Benhamou, P. Opolon, P. Cacoub, and P. Bedossa. 2000. Natural history of HCV infection. Baillieres Best Pract. Res. Clin. Gastroenterol. 14:211-228. [DOI] [PubMed] [Google Scholar]
- 48.Press, W. H., S. A. Teukolsky, W. T. Vetterling, and B. P. Flannery. 2002. Numerical recipes in C++, p. 734-747. Cambridge University Press, Cambridge, United Kingdom.
- 49.Princiotta, M. F., D. Finzi, S. B. Qian, J. Gibbs, S. Schuchmann, F. Buttgereit, J. R. Bennink, and J. W. Yewdell. 2003. Quantitating protein synthesis, degradation, and endogenous antigen processing. Immunity 18:343-354. [DOI] [PubMed] [Google Scholar]
- 50.Quinkert, D., R. Bartenschlager, and V. Lohmann. 2005. Quantitative analysis of the hepatitis C virus replication complex. J. Virol. 79:13594-13605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Randall, G., and C. M. Rice. 2001. Hepatitis C virus cell culture replication systems: their potential use for the development of antiviral therapies. Curr. Opin. Infect. Dis. 14:743-747. [DOI] [PubMed] [Google Scholar]
- 52.Reigadas, S., M. Ventura, L. Sarih-Cottin, M. Castroviejo, S. Litvak, and T. Astier-Gin. 2001. HCV RNA-dependent RNA polymerase replicates in vitro the 3′ terminal region of the minus-strand viral RNA more efficiently than the 3′ terminal region of the plus RNA. Eur. J. Biochem. 268:5857-5867. [DOI] [PubMed] [Google Scholar]
- 53.Schwartz, M., J. Chen, W. M. Lee, M. Janda, and P. Ahlquist. 2004. Alternate, virus-induced membrane rearrangements support positive-strand RNA virus genome replication. Proc. Natl. Acad. Sci. USA 101:11263-11268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Spiegelman, P. 1970. Extracellular evolution of replicating molecules, p. 927. In F. O. Schmitt (ed.), The neurosciences. The Rockefeller University, New York, NY.
- 55.Stuyver, L. J., T. R. McBrayer, P. M. Tharnish, A. E. Hassan, C. K. Chu, K. W. Pankiewicz, K. A. Watanabe, R. F. Schinazi, and M. J. Otto. 2003. Dynamics of subgenomic hepatitis C virus replicon RNA levels in Huh-7 cells after exposure to nucleoside antimetabolites. J. Virol. 77:10689-10694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tan, S. L., A. Pause, Y. Shi, and N. Sonenberg. 2002. Hepatitis C therapeutics: current status and emerging strategies. Nat. Rev. Drug Discov. 1:867-881. [DOI] [PubMed] [Google Scholar]
- 57.Uchil, P. D., and V. Satchidanandam. 2003. Characterization of RNA synthesis, replication mechanism, and in vitro RNA-dependent RNA polymerase activity of Japanese encephalitis virus. Virology 307:358-371. [DOI] [PubMed] [Google Scholar]
- 58.Wakita, T., T. Pietschmann, T. Kato, T. Date, M. Miyamoto, Z. Zhao, K. Murthy, A. Habermann, H. G. Krausslich, M. Mizokami, R. Bartenschlager, and T. J. Liang. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11:791-796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang, C., J. Pflugheber, R. Sumpter, Jr., D. L. Sodora, D. Hui, G. C. Sen, and M. Gale, Jr. 2003. Alpha interferon induces distinct translational control programs to suppress hepatitis C virus RNA replication. J. Virol. 77:3898-3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wengler, G., and H. J. Gross. 1978. Studies on virus-specific nucleic acids synthesized in vertebrate and mosquito cells infected with flaviviruses. Virology 89:423-437. [DOI] [PubMed] [Google Scholar]
- 60a.Wolk, B., B. Buchele, H. E. Blum, D. Moradpour, and C. M. Rice. 2004. 11th Int. Symp. HCV, abstr. O-22.
- 61.World Health Organization. 2000. Hepatitis C—global prevalence (update). Wkly. Epidemiol. Rec. 75:18-19. [PubMed] [Google Scholar]
- 62.You, S., D. D. Stump, A. D. Branch, and C. M. Rice. 2004. A cis-acting replication element in the sequence encoding the NS5B RNA-dependent RNA polymerase is required for hepatitis C virus RNA replication. J. Virol. 78:1352-1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhong, J., P. Gastaminza, G. Cheng, S. Kapadia, T. Kato, D. R. Burton, S. F. Wieland, S. L. Uprichard, T. Wakita, and F. V. Chisari. 2005. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. USA 102:9294-9299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhu, Q., J. T. Guo, and C. Seeger. 2003. Replication of hepatitis C virus subgenomes in nonhepatic epithelial and mouse hepatoma cells. J. Virol. 77:9204-9210. [DOI] [PMC free article] [PubMed] [Google Scholar]